Various Techniques for the Synthesis of 2-Nitrophenylamino-1,4-naphthoquinone Derivatives

Abstract
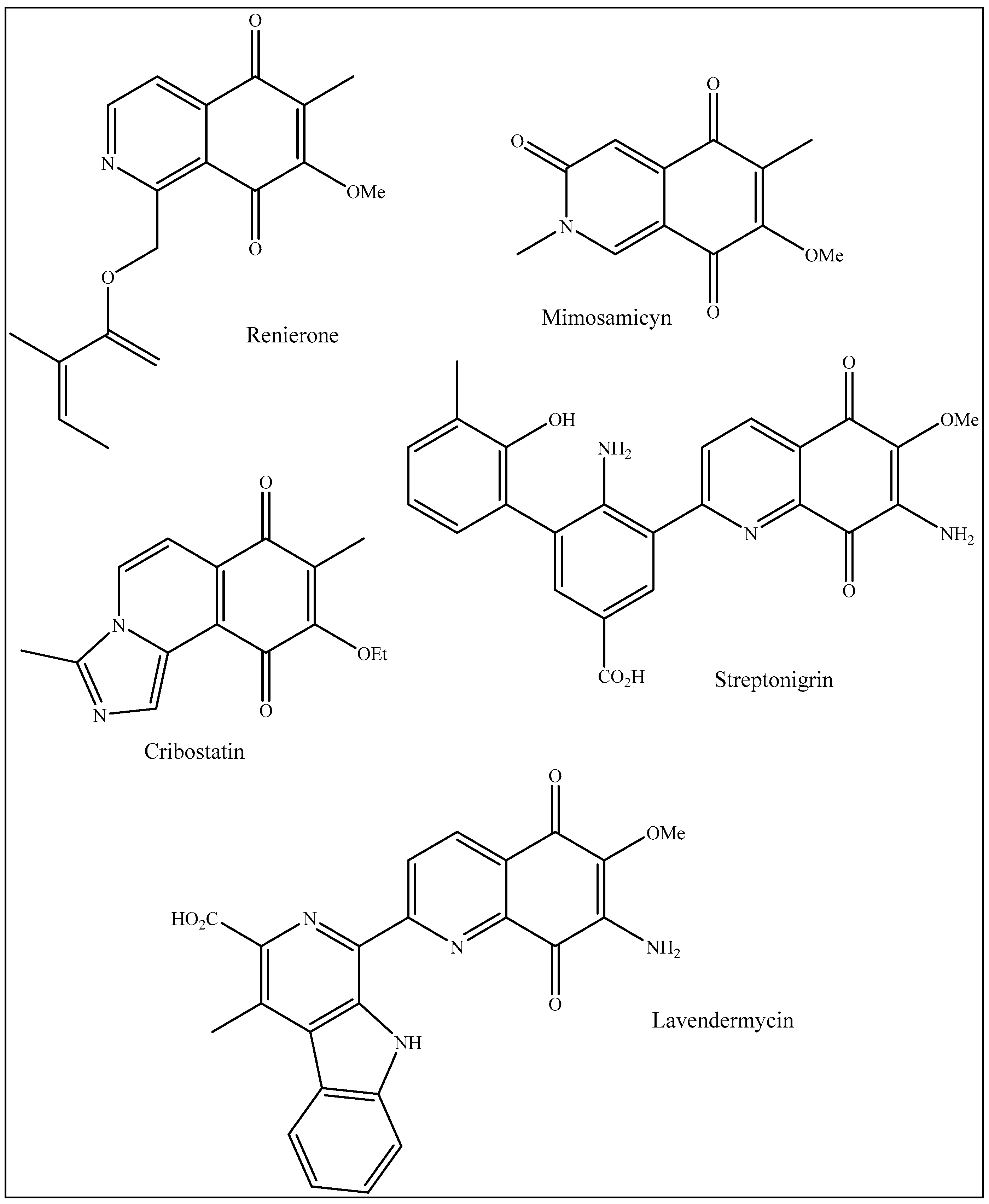
:1. Introduction
2. Material and Methods
2.1. Materials and Instrumentation
2.2. Different Methodologies Reported in the Literature Were Performed with Slight Modifications [35,36,37,38,39]
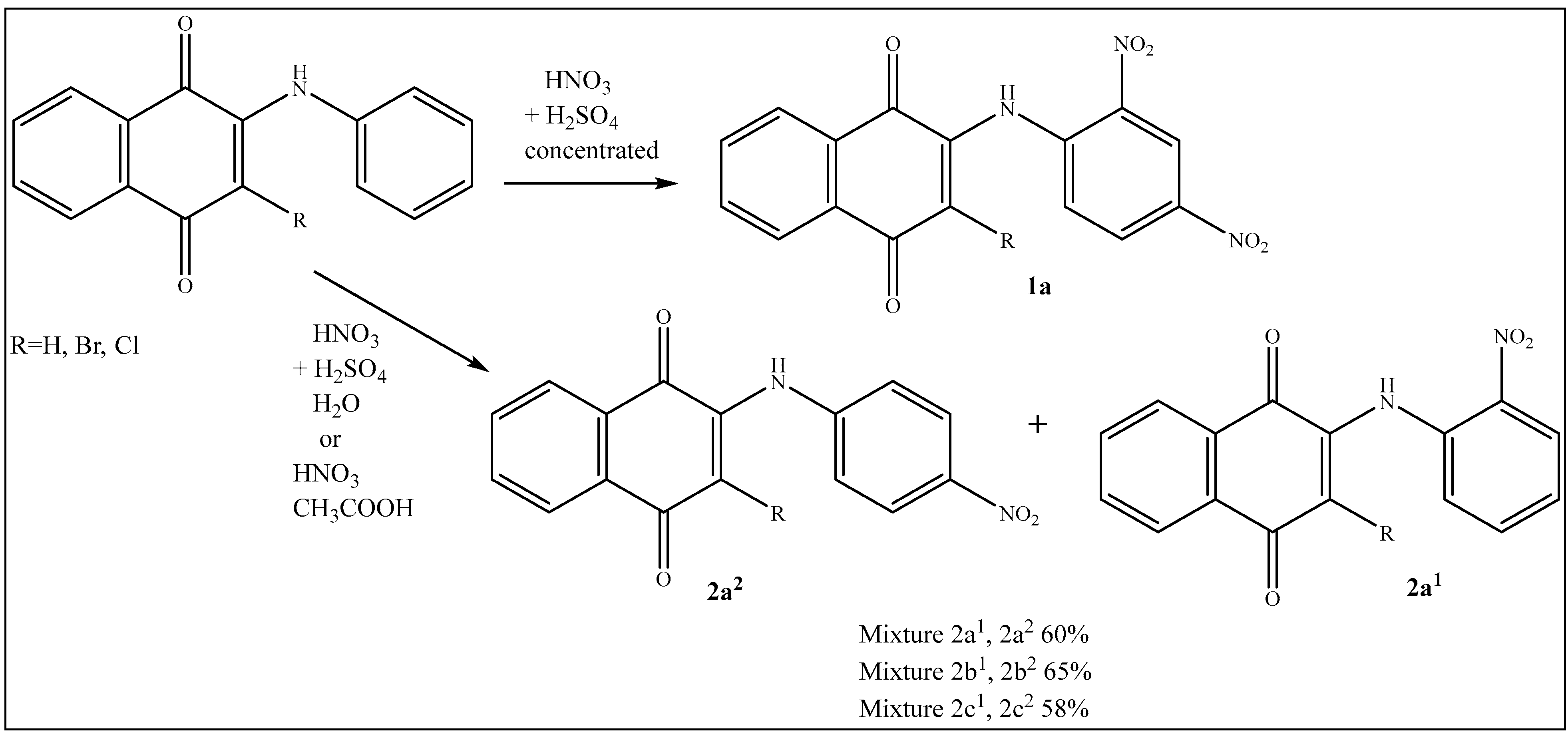
2.3. Preparation of 2a1, 2a2, 2b1, 2b2, 2c1, 2c2
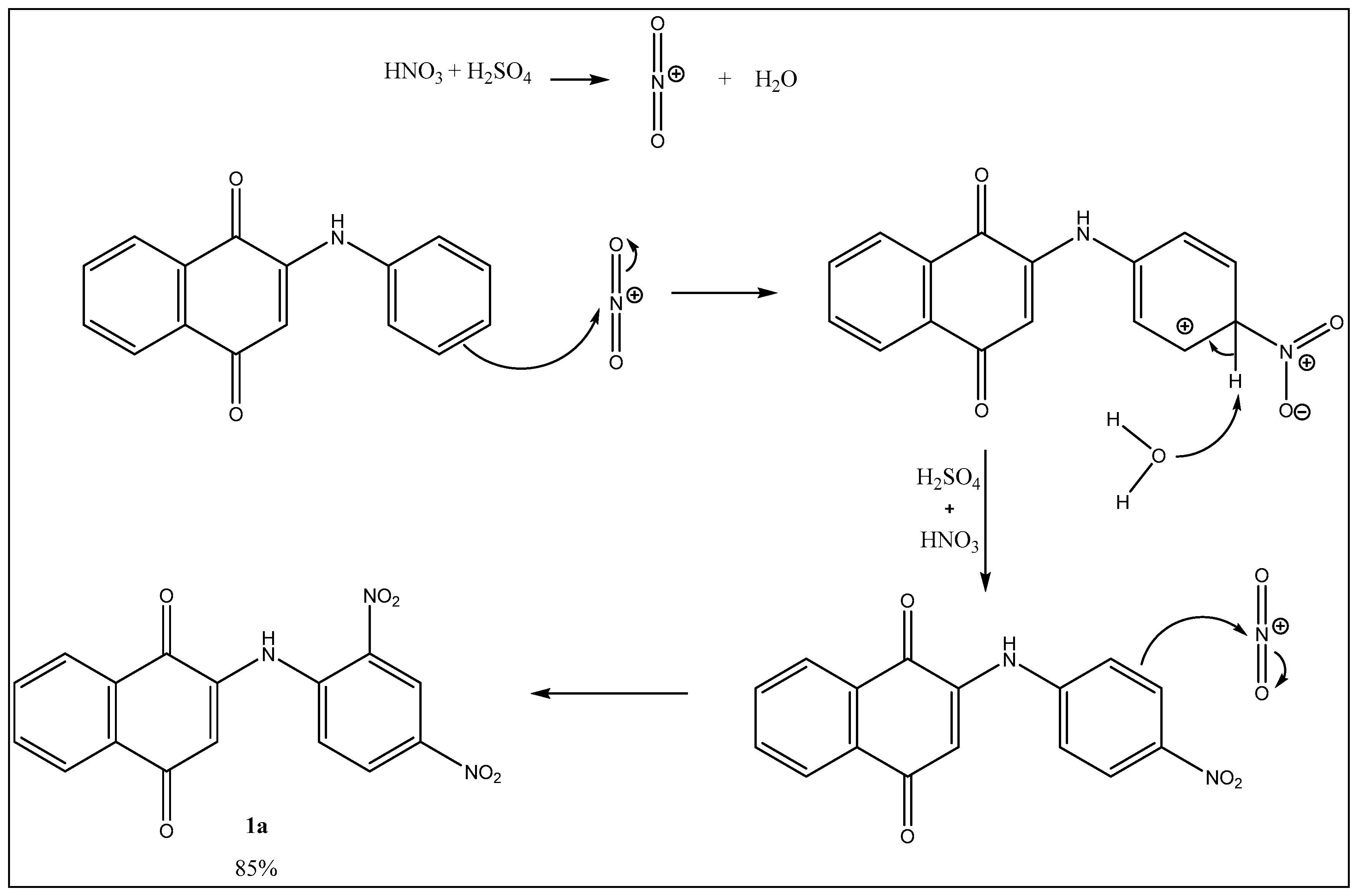
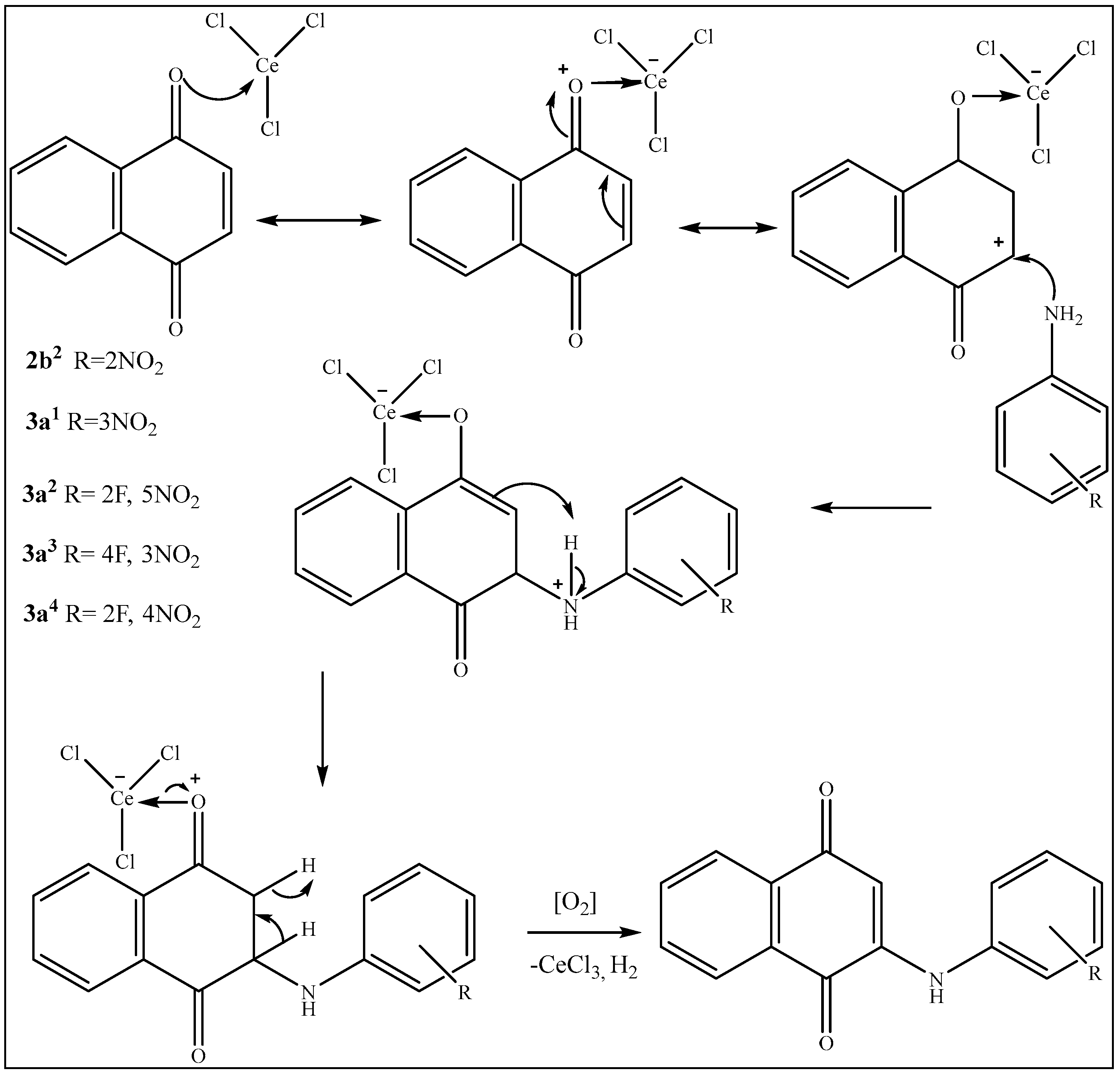
2.4. Preparation of Nitro-Phenylamino Naphthoquinone Derivatives 2a1, 2a2, 3a1, 3a2, 3a3, 3a4; by Michael Addition to 1,4-Naphtoquinone with Nitrated Anilines
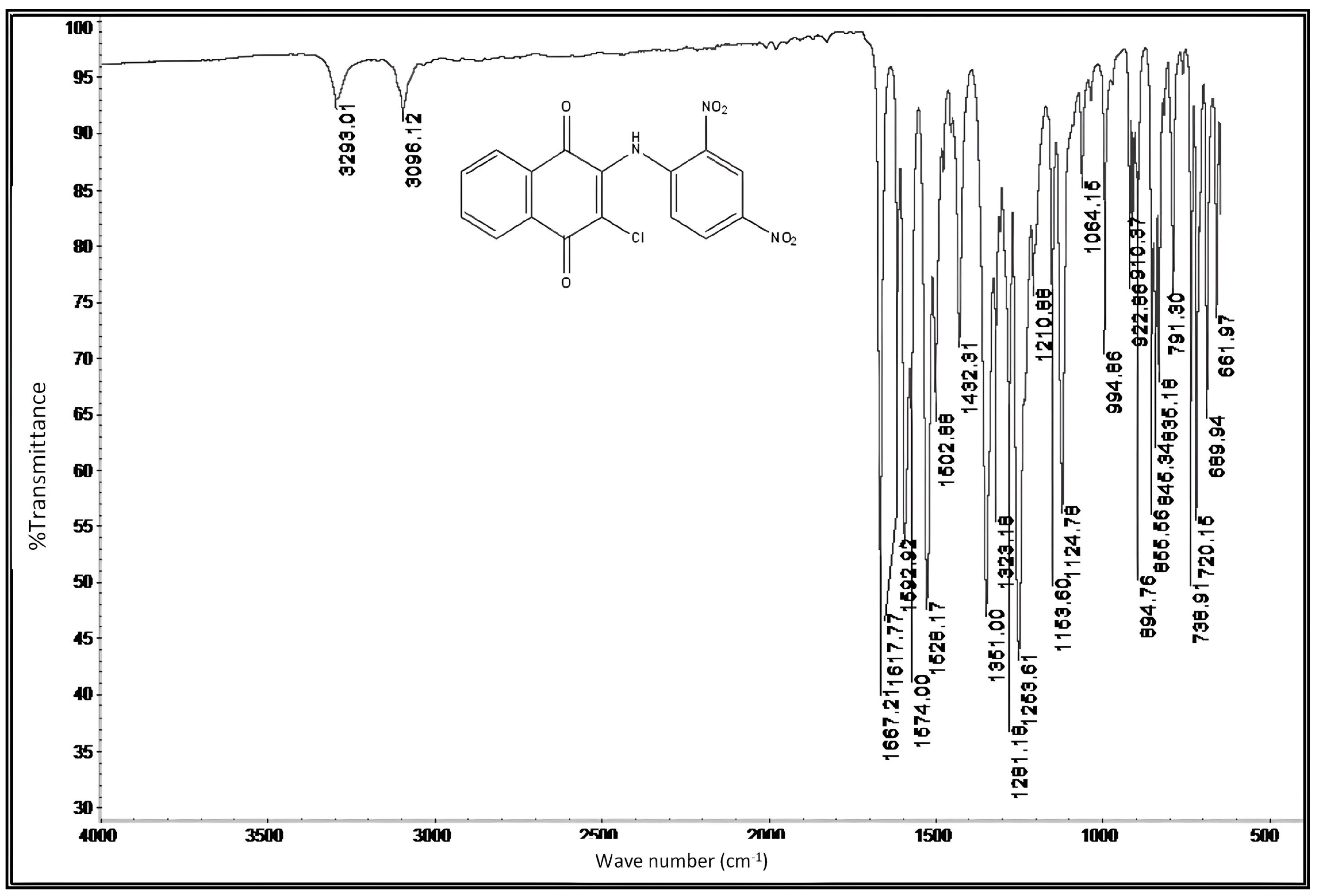
2.5. Characterization
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Da Silva, M.N.; Ferreira, V.F.; De Souza, M.C.B.V. Um Panorama atual da química e da farmacologia de Naftoquinonas, com enfase na ß-Lapachona e derivados. Quim. Nova 2003, 26, 407–416. [Google Scholar] [CrossRef]
- Akiyoshi, T.; Matzno, S.; Sakai, M.; Okamura, N.; Matsuyama, K. The potential of vitamin K3 as an anticancer agent against breast cancer that acts via the mitochondria-related apoptotic pathway. Cancer Chemother. Pharmcol. 2009, 65, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Chaves-Carballo, K.; Lamoureux, G.V.; Perez, A.L.; Bella, A.; Cechinel, V. Novel one-pot synthesis of a library of 2-aryloxy-1,4-naphthoquinone derivatives. Determination of antifungal and antibacterial activity. RSC Adv. 2022, 29, 18507–18523. [Google Scholar]
- Ahmad, T.; Suzuki, Y.J. Juglone in oxidative stress and cell signaling. Antioxidants 2019, 8, 91. [Google Scholar] [CrossRef]
- Badolato, M.; Carullo, G.; Caroleo, M.C.; Cione, E.; Aiello, F.; Manetti, F. Discovery of 1, 4-naphthoquinones as a new class of antiproliferative agents targeting GPR55. ACS Med. Chem. Lett. 2019, 10, 402–406. [Google Scholar] [CrossRef]
- Liu, Z.; Shen, Z.; Xiang, S.; Sun, Y.; Cui, J.; Jia, J. Evaluation of 1,4-naphthoquinone derivatives as antibacterial agents: Activity and mechanistic studies. Front. Environ. Sci. Eng. 2023, 17, 31. [Google Scholar] [CrossRef]
- Epifano, F.; Genovese, S.; Fiorito, S.; Mathieu, V.; Kiss, R. Lapachol and its congeners as anticancer agents: A review. Phytoche. Rev. 2014, 13, 37–49. [Google Scholar] [CrossRef]
- Majdi, C.; Duvauchelle, V.; Meffre, P.; Benfodda, Z. An overview on the antibacterial properties of juglone, naphthazarin, plumbagin and lawsone derivatives and their metal complexes. Biomed. Pharmacother. 2023, 162, 114690. [Google Scholar] [CrossRef]
- Hsieh, Y.J.; Lin, L.C.; Tsai, T.H. Determination and identification of plumbagin from the roots of Plumbago zeylanica L. by liquid chromatography with tandem mass spectrometry. J. Chromatogr. 2005, 1083, 141–145. [Google Scholar] [CrossRef]
- Son, T.G.; Camandola, S.; Arumugam, T.V.; Cutler, R.G.; Telljohann, R.S.; Mughal, M.R.; Moore, T.A.; Luo, W.; Yu, Q.S.; Johnson, D.A.; et al. Plumbagin, a novel Nrf2/ARE activator, protects against cerebral ischemia. J. Neurochem. 2010, 112, 1316–1326. [Google Scholar] [CrossRef]
- Campora, M.; Francesconi, V.; Schenone, S.; Tasso, B.; Tonelli, M. Journey on Naphthoquinone and Anthraquinone Derivatives: New Insights in Alzheimer’s Disease. Pharm. J. 2021, 14, 33. [Google Scholar] [CrossRef] [PubMed]
- Nakhate, K.T.; Bharne, A.P.; Verma, V.S.; Aru, D.N.; Kokare, D.M. Plumbagin ameliorates memory dysfunction in streptozotocin induced Alzheimer’s disease via activation of Nrf2/ARE pathway and inhibition of β-secretase. Biomed. Pharmacother. 2018, 101, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Syed, F.A.; Singh, S.; Hadi, S.M. Prooxidant activity of resveratrol in the presence of copper ions: Mutagenicity in plasmid DNA. Toxicol. Lett. 2005, 159, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.S.; Ahmad, A.; Hadi, S.M. Anti-oxidant, pro-oxidant properties of tannic acid and its binding to DNA. Chem. Biol. Interact. 2000, 125, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Kavaliauskas, P.; Opazo, F.S.; Acevedo, W.; Petraitiene, R.; Grybaitė, B.; Anusevičius, K.; Petraitis, V. Synthesis, biological activity, and molecular modelling studies of naphthoquinone derivatives as promising anticancer candidates targeting COX-2. Pharm. J. 2022, 15, 541. [Google Scholar] [CrossRef]
- Da Silva Júnior, E.N.; de Melo, I.M.; Diogo, E.B.; Costa, V.A.; de Souza Filho, J.D.; Valença, W.O.; Camara, C.A.; de Oliveira, R.N.; de Araujo, A.S.; Emery, F.S.; et al. On the search for potential anti-Trypanosoma cruzi drugs: Synthesis and biological evaluation of 2-hydroxy-3-methylamino and 1,2,3-triazolic naphthoquinoidal compounds obtained by click chemistry reactions. Eur. J. Med. Chem. 2012, 52, 304–312. [Google Scholar] [CrossRef]
- Neves, A.P.; Pereira, M.X.; Peterson, E.J.; Kipping, R.; Vargas, M.D.; Silva-Jr, F.P.; Carneiro, J.W.; Farrell, N.P. Exploring the DNA binding/cleavage, cellular accumulation and topoisomerase inhibition of 2-hydroxy-3-(aminomethyl)-1,4-naphthoquinone Mannich bases and their platinum(II) complexes. J. Inorg. Biochem. 2013, 119, 54–64. [Google Scholar] [CrossRef]
- de Araujo, M.V.; David, C.C.; Neto, J.C.; de Oliveira, L.A.; da Silva, K.C.J.; Dos Santos, J.M.; Alexandre-Moreira, M.S. Evaluation on the leishmanicidal activity of 2-N, N′-dialkylamino-1, 4-naphthoquinone derivatives. Exp. Parasitol. 2017, 176, 46–51. [Google Scholar] [CrossRef]
- Wolstenholm, G.E.W.; O’Conner, C.M. Quinones in Electron Transport; Churchill: London, UK, 1961. [Google Scholar]
- López López, L.I.; Nery Flores, S.D.; Silva Belmares, S.Y.; Sáenz Galindo, A. Naphthoquinones: Biological properties and synthesis of lawsone and derivatives-a structured review. Vitae 2014, 21, 248–258. [Google Scholar] [CrossRef]
- Valle-Bourroueta, G.; Ugalde-Saldívar, V.M.; Gómez, M.; Ortiz- Frade, L.A.; González, I.; Frontana, C. Magnetic interactions as a stabilizing factor of semiquinone species of lawsone by metal complexation. Electrochim. Acta 2010, 55, 9042–9050. [Google Scholar] [CrossRef]
- Pettit, R.K.; Fakoury, B.R.; Knight, J.C.; Weber, C.A.; Pettit, G.R.; Cage, G.D.; Pon, S.J. Antibacterial activity of the marine sponge constituent cribrostatin. J. Med. Microbiol. 2004, 53, 61–65. [Google Scholar] [CrossRef]
- Verniest, G.; Wang, X.P.; De Kimpe, N.; Padwa, A.J. Heteroaryl cross-coupling as an entry toward the synthesis of lavendamycin analogues: A model study. Org. Chem. 2010, 75, 424–433. [Google Scholar] [CrossRef]
- Bai, X.; Liu, Y.; Wang, H.; Zhang, H. Natural Products from the Marine Sponge Subgenus Reniera. Molecules 2021, 26, 1097. [Google Scholar] [CrossRef]
- Kesteleyn, B.; De Kimpe, N. Synthesis of mimosamycin. J. Org. Chem. 2000, 65, 635–639. [Google Scholar] [CrossRef]
- Noriega, S.; Cardoso-Ortiz, J.; López-Luna, A.; Cuevas-Flores, M.D.R.; Flores De La Torre, J.A. The Diverse Biological Activity of Recently Synthesized Nitro Compounds. Pharm. J. 2022, 15, 717. [Google Scholar] [CrossRef]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed]
- Morawska, K.; Jedlińska, K.; Smarzewska, S.; Metelka, R.; Ciesielski, W.; Guziejewski, D. Analysis and DNA interaction of the profluralin herbicide. Environ. Chem. Lett. 2019, 17, 1359–1365. [Google Scholar] [CrossRef]
- Ribeiro, T.A.; Machado-Ferreira, E.; Guimarães, L.F.; Cavaleiro, J.; Britto, A.M.A.; Redua, N.; Soares, C.A.G. Novel cytotoxic amphiphilic nitro-compounds derived from a synthetic route for paraconic acids. Colloids Surf. A Physicochem. Eng. Asp. 2021, 626, 126984. [Google Scholar] [CrossRef]
- Rice, A.M.; Long, Y.; King, S.B. Nitroaromatic antibiotics as nitrogen oxide sources. Biomolecules 2021, 11, 267. [Google Scholar] [CrossRef]
- Tavera-Hernández, R.; Jiménez-Estrada, M.; Alvarado-Sansininea, J.J.; Nieto-Camacho, A.; López-Muñoz, H.; Sánchez-Sánchez, L.; Escobar, M.L. Synthesis of chrysin, quercetin and naringin nitroderivatives: Antiproliferative, anti-inflammatory and antioxidant activity. Lett. Drug Des. Discov. 2021, 18, 795–805. [Google Scholar] [CrossRef]
- Toro, P.M.; Acuña, A.; Mallea, M.; Lapier, M.; Moncada-Basualto, M.; Cisterna, J.; Brito, I.; Klahn, H. Condensation and Substitution Products Obtained in Reactions of Isomeric Bromo-Nitrofuraldehydes with Ferrocenylamine: Electrochemistry and Anti-Parasitic Evaluation. J. Organomet. Chem. 2019, 901, 120946. [Google Scholar] [CrossRef]
- Adedapo, A.D.; Ajayi, A.M.; Ekwunife, N.L.; Falayi, O.O.; Oyagbemi, A.; Omobowale, T.O.; Adedapo, A.A. Antihypertensive effect of Phragmanthera incana (Schum) Balle on NG-nitro-L-Arginine methyl ester (L-NAME) induced hypertensive rats. J. Ethnopharmacol. 2020, 257, 112888. [Google Scholar] [CrossRef] [PubMed]
- Ghatge, S.; Yang, Y.; Moon, S.; Song, W.Y.; Kim, T.Y.; Liu, K.H.; Hur, H.G. A novel pathway for initial biotransformation of dinitroaniline herbicide butralin from a newly isolated bacterium Sphingopyxis sp. strain HMH. J. Hazard. Mater. 2021, 402, 123510. [Google Scholar] [CrossRef] [PubMed]
- Calandra, J.C.; Adams, E.C., Jr. The preparation of amine derivatives of 2-chloro-1, 4-naphthoquinone. J. Ame. Chem. Soc. 1950, 72, 4804. [Google Scholar] [CrossRef]
- Queiroz, J.F.D.; Carneiro, J.W.D.M.; Sabino, A.A.; Sparrapan, R.; Eberlin, M.N.; Esteves, P.M. Electrophilic aromatic nitration: Understanding its mechanism and substituent effects. J. Org. Chem. 2006, 71, 6192–6203. [Google Scholar] [CrossRef]
- Capobianco, A.; Landi, A.; Peluso, A. Is Aromatic Nitration Spin Density Driven? Chemistry 2021, 3, 1286–1301. [Google Scholar] [CrossRef]
- Liljenberg, M.; Stenlid, J.H.; Brinck, T. Mechanism and regioselectivity of electrophilic aromatic nitration in solution: The validity of the transition state approach. J. Mol. Model. 2018, 24, 1–3. [Google Scholar] [CrossRef]
- Pratt, Y.T. Quinolinequinones. VI. Reactions with aromatic amines1. J. Org. Chem. 1962, 27, 3905–3910. [Google Scholar] [CrossRef]
- Gigante, B.; Prazeres, A.O.; Marcelo-Curto, M.J. Mild and selective nitration by “Claycop”. J. Org. Chem. 1994, 60, 3445–3447. [Google Scholar] [CrossRef]
- Leyva, E.; Baines, K.M.; Espinosa-González, C.G.; Magaldi-Lara, D.A.; Loredo-Carrillo, S.E.; De Luna-Méndez, T.A.; López, L.I. 2-(Fluoro-) and 2-(methoxyanilino)-1, 4-naphthoquinones. Synthesis and mechanism and effect of fluorine substitution on redox reactivity and NMR. J. Fluor. Chem. 2015, 180, 152–160. [Google Scholar] [CrossRef]
- Patel, S.S.; Patel, D.B.; Patel, H.D. Synthetic protocols for aromatic nitration: A review. ChemistrySelect 2021, 6, 1337–1356. [Google Scholar] [CrossRef]
- Urben, P. Bretherick’s Handbook of Reactive Chemical Hazards; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Millen, D.J. 509. Vibrational spectra of ionic forms of oxides and oxy-acids of nitrogen. Part IV. Raman spectral evidence of ionisation in crystalline nitronium salts. The constitution of solid dinitrogen pentoxide. Note on the spectrum of the perchlorate ion. J. Chem. Soc. (Resumed.) 1950, 2606–2612. [Google Scholar] [CrossRef]
- Win, T.; Yerushalmi, S.; Bittner, S. Direct nitration of 3-arylamino-2-chloro-1,4-naphthoquinones. Synthesis 2005, 10, 1631–1634. [Google Scholar] [CrossRef]
- Kim, E.K.; Bockman, T.M.; Kochi, J.K. Electron-transfer mechanism for aromatic nitration via the photoactivation of EDA (electron donor-acceptor) complexes. Direct relationship to electrophilic aromatic substitution. J. Am. Chem. Soc. 1993, 115, 3091–3104. [Google Scholar] [CrossRef]
- Leyva, E.; Baines, K.M.; Espinosa-González, C.G.; López, L.I.; Magaldi-Lara, D.A.; Leyva, S. Synthesis of novel 2-(fluoroanilino)-3-(2, 4-dinitroanilino) derivatives of 1, 4-naphthoquinone. Tetrahedron Lett. 2015, 56, 5248–5251. [Google Scholar] [CrossRef]
- Baghernejad, B.; Heravi, M.M.; Oskooie, H.A.; Be-heshtiha, Y.S. Synthesis and biological evaluation of some chrysene derivatives an efficient and regioselective nitration of phenols using NH4NO3, KHSO4. Gazi Univ. J. Sci. 2009, 3, 169–173. [Google Scholar]
- Olah, G.A.; Kunh, S.J.; Flood, H.; Evans, J.C. Aromatic substitution. XIII. 1a comparison of nitric acid and mixed acid nitration of alkylbenzenes and benzene with nitronium salt nitrations. J. Am. Chem. Soc. 1962, 84, 3687–3693. [Google Scholar] [CrossRef]
- Fisher, J.W. The chemistry of dinitrogen pentoxide in nitro compounds: Recent advances in synthesis and chemistry. In Advances in Chemistry Series; Feuer, H., Arnold, A.T., Eds.; VCH Publishers, Inc.: New York, NY, USA, 1990. [Google Scholar]
- Loupy, A. Solvent-free reactions. In Modern Solvents in Organic Synthesis; Springer: Berlin/Heidelberg, Germany, 1999; pp. 153–207. [Google Scholar]
- Loupy, A.; Petit, A.; Hamelin, J.; Texier-Boullet, F.; Jacquault, P.; Mathe, D. New solvent-free organic synthesis using focused microwaves. Synthesis 1998, 9, 1213–1234. [Google Scholar] [CrossRef]
- Beheshti, S.; Kianmehr, E.; Yahyaee, M.; Tabatabai, K. Facile and efficient selective mono-nitration of phenols under solvent-free conditions. Bull. Korean Chem. Soc. 2006, 27, 1056–1058. [Google Scholar] [CrossRef]
- Lampman, G.M.; Pavia, D.L.; Kriz, G.S. Introduction to Spectroscopy, 4th ed.; Wiley-Intrescience Publication: Hoboken, NJ, USA, 2010. [Google Scholar]
- Lisboa, C.S.; Santos, G.V.; Vaz, G.B.; De Lucas, N.C.; Eberlin, N.M.; Garden, J.S. C-H Functionalization of 1,4-naphthoquinone by oxidative coupling with anilines in the presence of a catalytic quantity of copper(II) acetate. J. Org. Chem. 2011, 76, 5264–5273. [Google Scholar] [CrossRef]
- Leyva, E.; López, L.I.; Loredo-Carrillo, S.E.; Rodríguez-Kessler, M.; Montes-Rojas, A. Synthesis, spectral and electrochemical characterization of novel 2-(fluoroanilino)-1, 4-naphthoquinones. J. Fluor. Chem. 2011, 132, 94–101. [Google Scholar] [CrossRef]
- Leyva, E.; Loredo-Carrillo, S.E.; López, L.I. Catalytic, ultrasonic, and microwave-assisted synthesis of naphthoquinone derivatives by intermolecular and intramolecular N-arylation reactions. In Green Sustainable Process for Chemical and Environmental Engineering and Science; Elsevier: Amsterdam, The Netherlands, 2021; pp. 231–264. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Synthesized Compound | Reagent 0.9 mmol or 0.5 mmol | Nitration | Products (Yield%) |
|---|---|---|---|
| 1a | 2-(phenylamino)-1,4-naphthoquinone | 7 mL H2SO4 conc. 1.5 mL HNO3 conc. | 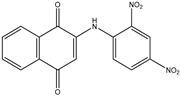 |
| 85% | |||
| 1b | 3-chloro-2-(phenylamino)-1,4-naphthoquinone | 7 mL H2SO4 conc. 1.5 mL HNO3 conc. |  |
| 90% | |||
| 1c | 3-bromo-2-(phenylamino)-1,4-naphthoquinone | 7 mL H2SO4 conc. 1.5 mL HNO3 conc. | 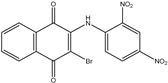 70% |
| 2a1 | 2-(phenylamino)-1,4-naphthoquinone | 7 mL H2SO4 conc. 1.5 mL HNO3 conc. 3 mL of distilled H2O or 7 mL HNO3 conc. 3 mL of acetic acid |  |
| 2a2 | 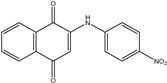 60% | ||
| 2b1 | 3-chloro-2-(phenylamino)-1,4-naphthoquinone | 7 mL H2SO4 conc. 1.5 mL HNO3 conc. 3 mL of distilled H2O or 7 mL HNO3 conc. 3 mL of acetic acid | 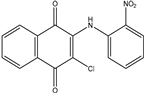 |
| 2b2 | 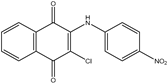 65% | ||
| 2c1 | 3-bromo-2-(phenylamino)-1,4-naphthoquinone | 7 mL H2SO4 conc. 1.5 mL HNO3 conc. 3 mL of distilled H2O or 7 mL HNO3 conc. 3 mL of acetic acid |  |
| 2c2 | 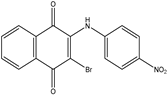 58% |
| Reagent 0.5 mmol | Nitration | Products |
|---|---|---|
| 2-(phenylamino)-1,4-naphthoquinone | 2.4 or 1.2 g of Cu (NO3)2 30 mL of anhydride acetic acid or 15 mL of carbon tetrachloride and 3.7 of acetic anhydride. The reaction mixture was allowed to stir at room temperature (25 °C) and was monitored until no starting material was observed by TLC (hexane 30% and ethyl acetate 70%) (the reaction took place for approximately one hour). | Mixture 2a1, 2a2 |
| 3-chloro-2-(phenylamino)-1,4-naphthoquinone | Mixture 2b1, 2b2 | |
| 3-bromo-2-(phenylamino)-1,4-naphthoquinone | Mixture 2c1, 2c2 | |
| 2-(phenylamino)-1,4-naphthoquinone | 2 g of Ca(NO3)2•4H2O with 20 mL of acetic acid to dissolve the salt. The mixture was heated to 100 °C and refluxed for one hour. Subsequently, the product was precipitated by adding water. | Mixture 2a1, 2a2 |
| 3-chloro-2-(phenylamino)-1,4-naphthoquinone | Mixture 2b1, 2b2 | |
| 3-bromo-2-(phenylamino)-1,4-naphthoquinone | Mixture 2c1, 2c2 |
| Product | Rf | Yield (%) |
|---|---|---|
| 1a | 0.5 | 85 |
| 2a1 | 0.42 | 60 (mixture) |
| 2a2 | 0.45 | 60 (mixture) |
| Naphthoquinone | Nitrating Reagent | Products (Yield%) |
|---|---|---|
| 3-chloro-2-(phenylamine)-1,4-naphthoquinone | Copper Nitrate | 2a2 (22%) |
| 3-bromo-2-(phenylamine)-1,4-naphthoquinone | Calcium Nitrate | 2b2 (18%) |
| 2-(phenylamine)-1,4-naphthoquinone | Copper Nitrate | Several products |
| Products | Yield (%) | ||
|---|---|---|---|
| Without Catalysis | CeCl3 | FeCl3 | |
| 2a1 | --- | --- | 23.9 |
| 3a1 | 13.8 | 77.0 | 73.5 |
| 2a2 | --- | 52.8 | 53.0 |
| 1a | --- | --- | --- |
| 3a2 | 6.1 | 65.5 | 61.0 |
| 3a3 | --- | 70.7 | 51.6 |
| 3a4 | --- | 22.0 | 23.0 |
| Products | Yield (%) | |
|---|---|---|
| CeCl3 | FeCl3 | |
| 2a1 | --- | --- |
| 3a1 | 23.3 | 28.7 |
| 2a2 | 18.1 | 10.8 |
| 1a | --- | --- |
| 3a2 | 35.3 | 30.7 |
| 3a3 | 32.3 | 31.6 |
| 3a4 | --- | --- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leyva, E.; Loredo-Carrillo, S.E.; Aguilar, J. Various Techniques for the Synthesis of 2-Nitrophenylamino-1,4-naphthoquinone Derivatives. Reactions 2023, 4, 432-447. https://doi.org/10.3390/reactions4030026
Leyva E, Loredo-Carrillo SE, Aguilar J. Various Techniques for the Synthesis of 2-Nitrophenylamino-1,4-naphthoquinone Derivatives. Reactions. 2023; 4(3):432-447. https://doi.org/10.3390/reactions4030026
Chicago/Turabian StyleLeyva, Elisa, Silvia E. Loredo-Carrillo, and Johana Aguilar. 2023. "Various Techniques for the Synthesis of 2-Nitrophenylamino-1,4-naphthoquinone Derivatives" Reactions 4, no. 3: 432-447. https://doi.org/10.3390/reactions4030026
APA StyleLeyva, E., Loredo-Carrillo, S. E., & Aguilar, J. (2023). Various Techniques for the Synthesis of 2-Nitrophenylamino-1,4-naphthoquinone Derivatives. Reactions, 4(3), 432-447. https://doi.org/10.3390/reactions4030026