1. Introduction
Germ cell tumors (GCTs) are midline neoplasms that occur most frequently in the gonads and constitute a common malignancy in men aged 15–35 years. Primary extragonadal GCTs are rare, and the most frequent sites include mediastinum, Central Nervous System (CNS), and retroperitoneum [
1]. The CNS is the second most common site of extragonadal GCTs after the mediastinum [
2]. The CNS GCTs account for approximately 3–5% of all intracranial tumors and occur more commonly in children and adolescents, with an incidence peak between 10 and 12 years of age [
2,
3,
4].
Based on clinicopathologic features, GCTs are historically classified as germinomatous (GGCTs) and nongerminomatous germ cell tumors (NGGCTs) [
5]. CNS GGCTs are histologically identical to the gonadal respective and sensitive to radiotherapy (RT) and chemotherapy (CMT). However, GGCTs located in the CNS have lower cure rates and a poorer prognosis compared to those arising from the gonads [
5].
The primary co-occurrence of gonadal and extragonadal CNS GCTs has rarely been reported in the literature [
1,
6,
7,
8,
9,
10,
11,
12]. Therefore, a common opinion on the underlying etiopathogenetic mechanism is still lacking among the authors [
11]. In the present study, we report a case of metachronous CNS NGGCT in a 34-year-old patient treated for a testicular NGGCT 16 years before, and we conduct a systematic review of the literature.
To the best of our knowledge, this is the 7th case described in the literature of co-occurrent testicular NGGCTs and extragonadal CNS GCTs, with a particularly long time interval between the onset of the two lesions.
The description of our findings, together with the review of related cases, might improve current knowledge on this rare condition and contribute to shed more light on the etiology and molecular mechanisms.
2. Materials and Methods
2.1. Institutional Cases Review
We reviewed data from 29 consecutive patients with a diagnosis of CNS GCTs evaluated at one Institution over the past 23 years. Patients were included in our study if they (1) gave consent to the use of their information for research purposes and (2) had at least a primary co-occurrent gonadal GCT. Eligibility was ascertained for one patient with a primary testicular NGGCT and a primary CNS NGGCT. Then, a literature search for other reported cases of primary co-occurrent testicular NGGCTs and extragonadal CNS GCTs was performed.
2.2. Literature Review
2.2.1. Literature Search
The systematic review was conducted according to the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) guidelines [
13]. A comprehensive literature search on the databases PubMed, Ovid MEDLINE, and Ovid EMBASE was designed and conducted by an experienced librarian with input from the authors. The keywords “germ cell tumors”, “central nervous system”, “cerebral”, “intracranial”, “gonadal”, “testicular” and “extragonadal” were used in “AND” and “OR” combinations. Boolean operators and Medical Subject Heading (MeSH) terms were used to find studies. Only articles published between 2000 and 2023 were included in the present review. The first literature search was performed on the 8th of September 2023, and it was updated on the 10th of December 2023.
Studies were selected based on the following inclusion criteria: (1) English language, (2) case series or case reports, (3) studies reporting on the primary co-occurrence of testicular NGGCTs and extragonadal CNS GCTs confirmed by histopathological examination. The exclusion criteria were: (1) studies reporting on testicular seminomas, metastases, or other histotypes different from NGGCTs, (2) extragonadal GCTs in locations other than the CNS, (3) studies reporting on other primary CNS tumor histotypes or metastases and (4) absence of a definitive histopathological diagnosis of GCT.
The studies were identified and imported into Endnote X9, where duplicates were automatically deleted. The list of the studies has been reviewed by two independent and experienced researchers (F.T. and C.F.S.), with knowledge in neurosurgical field. L.D., a third author, resolved all disagreements. Lastly, all papers have been read over before being included in the present review.
2.2.2. Risk of Bias Assessment
Newcastle-Ottawa Scale (NOS) was used to assess the quality of the selected studies [
14]. Higher scores in the NOS scale means that studies have a better quality. Scores greater than or equal to 7 identifies high-quality studies. F.T. and C.F.S. performed independent quality evaluation and discrepancies were solved by L.D. papers re-examination.
2.3. Data Extraction
For the institutional case and each study included in our systematic review, we extracted the following information: baseline demographic and medical history data (age at presentation and gender, etiopathogenetic factors), initial diagnosis, the precise location of the CNS GCT, testicular and cerebral tumors histotypes, treatment strategy, outcome and follow-up period.
2.4. Objectives
We aim to describe a case of co-occurrence of testicular and extragonadal CNS GCTs and to perform a literature review of similar cases. Moreover, we speculated on the pathogenesis and molecular mechanisms that underlie the co-occurrence of GCTs in multiple sites.
3. Results
3.1. Institutional Case Report
A 34-year-old male patient was admitted to the Hospital for progressive asthenia, loss of appetite, widespread muscle pain, polydipsia, and polyuria. His medical history was positive for a left testicular GCT, diagnosed when he was 18 years old. The testicular tumor was managed and treated in another Hospital, where he underwent left orchiectomy after complete tumor staging. The histhopathology confirmed a mixed germinal tumor composed of 95% embryonal carcinoma, 3% yolk sac tumor, and 2% immature teratoma. Subsequent adjuvant chemotherapy was administered. The regular clinical and instrumental follow-up assessments after the end of treatment have not documented disease recurrence so far.
The patient was taken to the Emergency Room and underwent a Computer Tomography (CT) scan that revealed a hyperdense suprasellar mass with initial hydrocephalus.
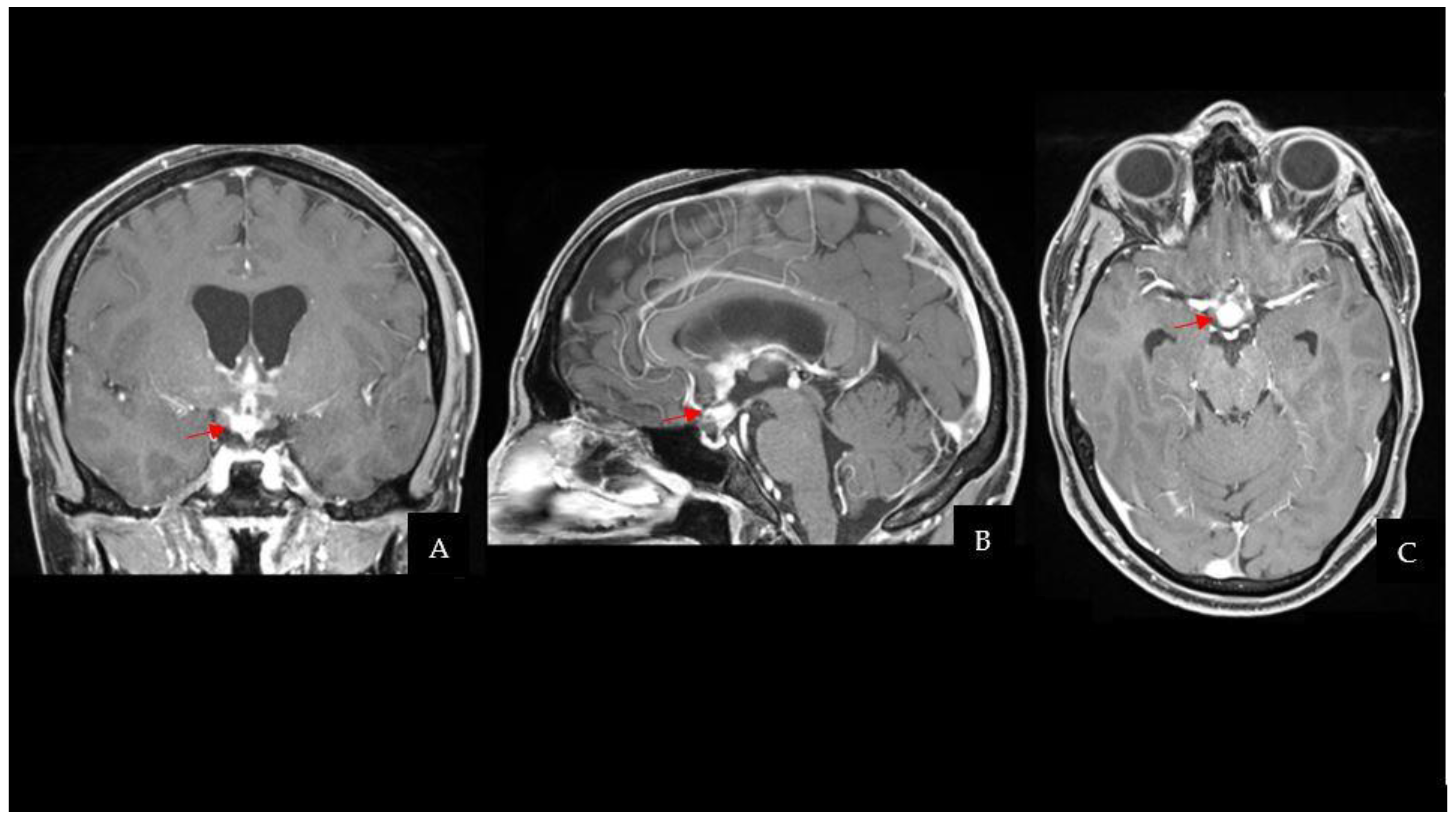
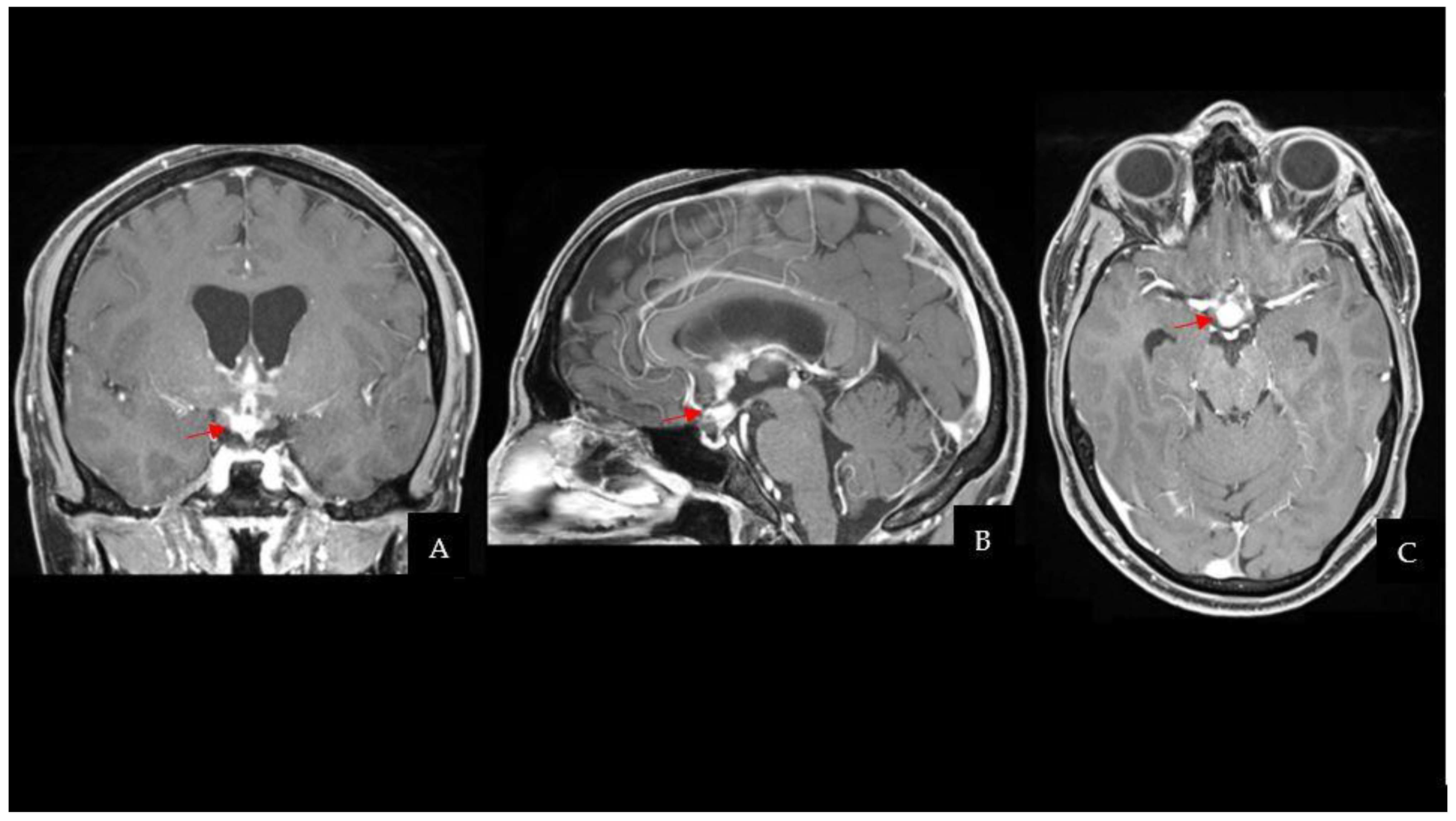
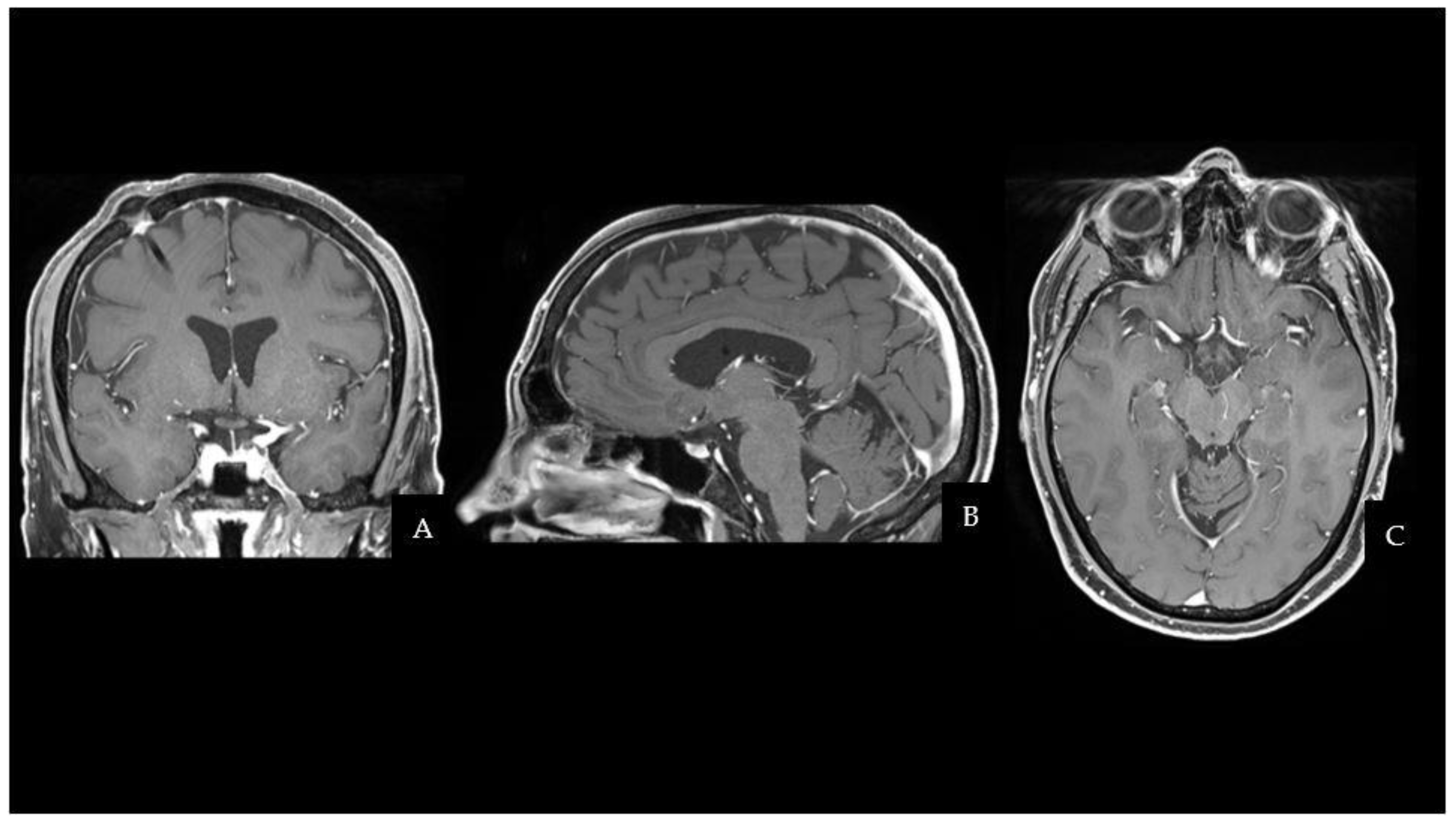
The CT scan was followed by a cerebral magnetic resonance imaging (MRI) scan that showed a pseudonodular suprasellar lesion, isointense with grey matter on both T1- and T2-weighted images, with heterogeneous enhancement after contrast administration. The lesion extended from the pituitary peduncle to the hypothalamic region and the optic tracts, with a maximum craniocaudal extension of 3 cm.
Taking into account the patient past medical history of testicular GCT, neuroradiological findings were considered compatible with a CNS GCT (
Figure 1). However, pituitary adenoma, craniopharyngioma, metastases, sellar region lymphoma and meningioma had to be ruled out. Hence, a neurosurgical hospitalization for further diagnostic investigations was indicated.
A subsequent total-body computed tomography (CT) scan for tumor staging was negative for other sites of disease. An ophthalmology assessment diagnosed a left eye temporal hemianopsia. The serum and cephalo-spinal fluid (CSF) alpha-fetoprotein (AFP), beta-human chorionic gonadotropin (HCG), and carcinoembryonic antigen (CEA) were negative.
Due to the neuroimaging findings, the clinical features, and the absence of tumor markers in the serum and CSF, the patient underwent a surgical biopsy of the lesion to obtain a tumor specimen for pathological diagnosis and treatment plan.
3.1.1. Surgical Procedure
The patient’s position on the operatory table was supine, with the neck slightly flexed (10°). A right frontal linear incision was made behind the hail line. A precoronaric burr hole was performed under neuronavigation guidance. After dura opening and corticectomy, the rigid ventriculoscope was introduced up to the right ventricle. A white-pinkish lesion of the lateral wall of the right lateral ventricle was evident. Multiple samples were intraoperatively taken and sent for histopathological examination. Considering the involvement of the septum pellucidum, we decided to perform a septostomy. The ostomy was made, puncturing the avascular zone of the septum pellucidum between the anterior and the posterior septal veins with the monopolar electrode and endoscopic scissors. The hole was then enlarged using grasping forceps until the flow through it was evident. After an accurate control of the hemostasis, the endoscope was retracted, and a 12 mm Ommaya reservoir was placed.
3.1.2. Postoperative Course
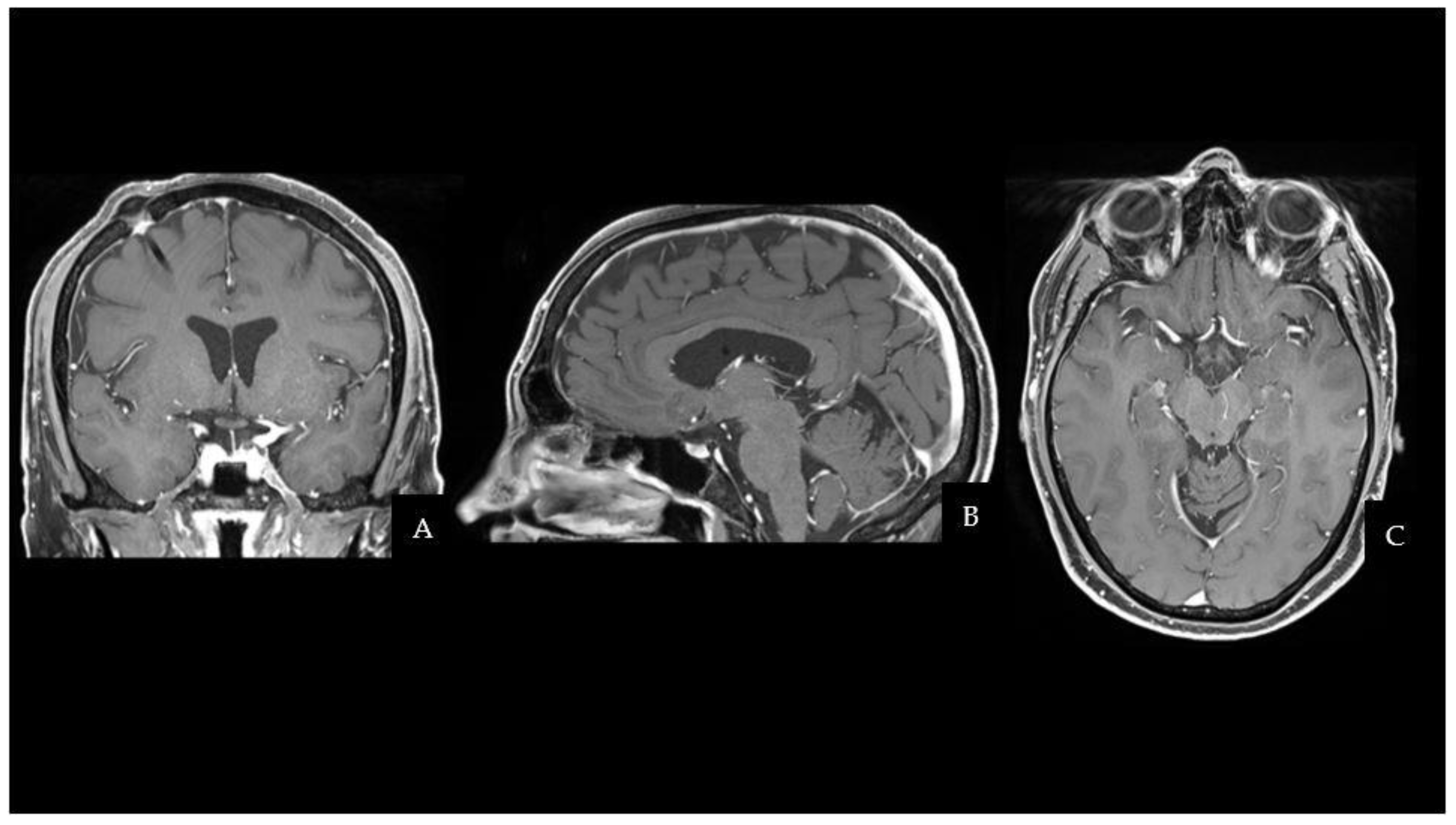
The patient fully recovered after the procedure and had no neurological complications. Even though the histopathological characteristics were different from the previous testicular NGGCT, the morphologic and immunophenotypic examination confirmed the lesion to be a mixed GCT (Oct4+, SALL4+, CD117+; Ki67≃ 10%). One month after surgery, the patient started two cycles of cisplatin, etoposide, and ifosfamide (PEI) chemotherapy, followed by 18 sessions of conventional radiation therapy (CRT). MRI scans at 3 and 6 months after the end of treatment did not document disease recurrence. The two-year follow-up brain MRI is shown in
Figure 2 and has not demonstrated any sign of regrowth in the surgical field. Apart from permanent Diabetes Insipidus, the patient does not present any endocrine disorders or neurological sequelae. Despite medical advice, the patient and his family did not undergo any genetic analysis to investigate cancer predisposition syndromes.
3.2. Literature Review
3.2.1. Literature Search
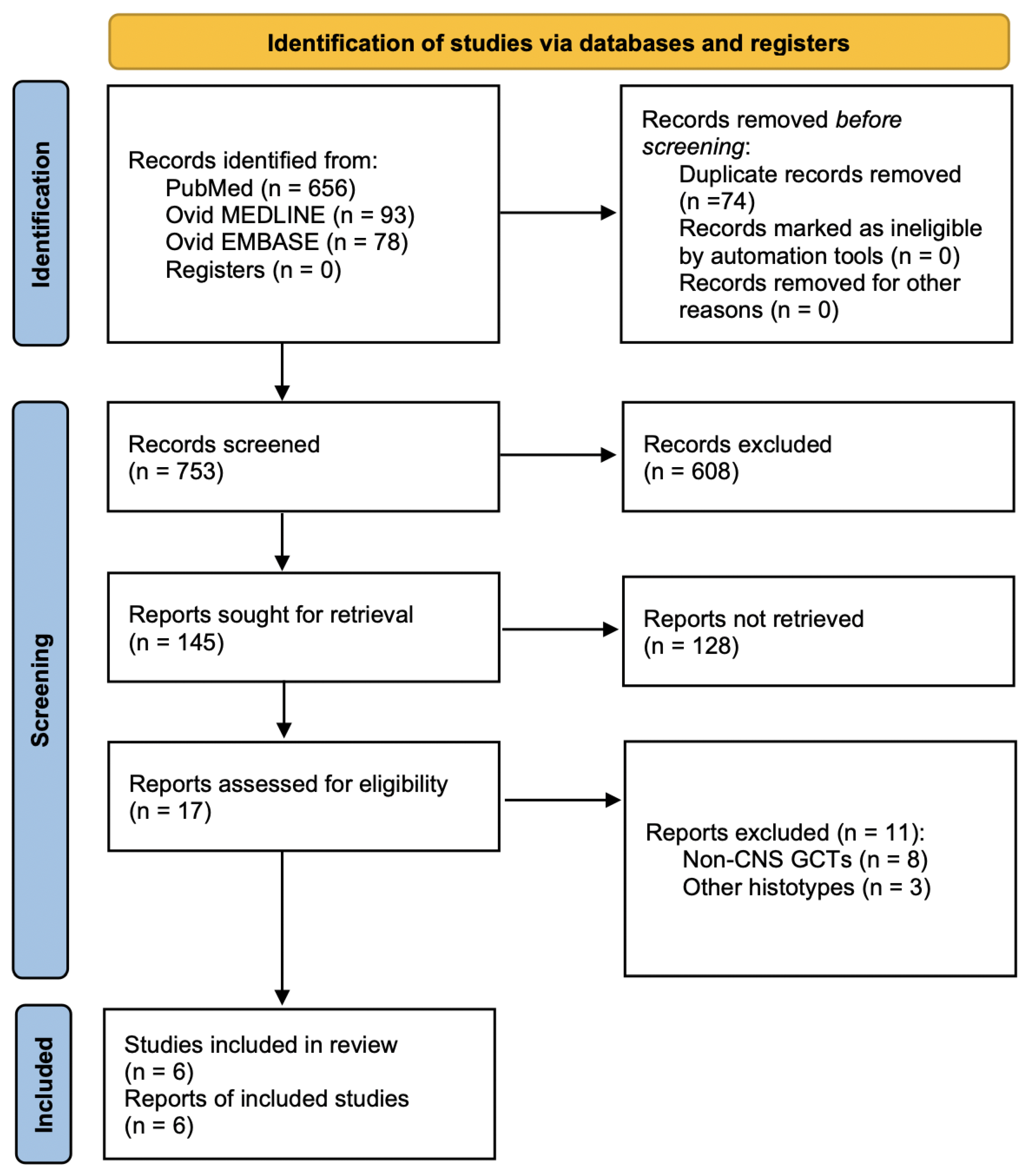
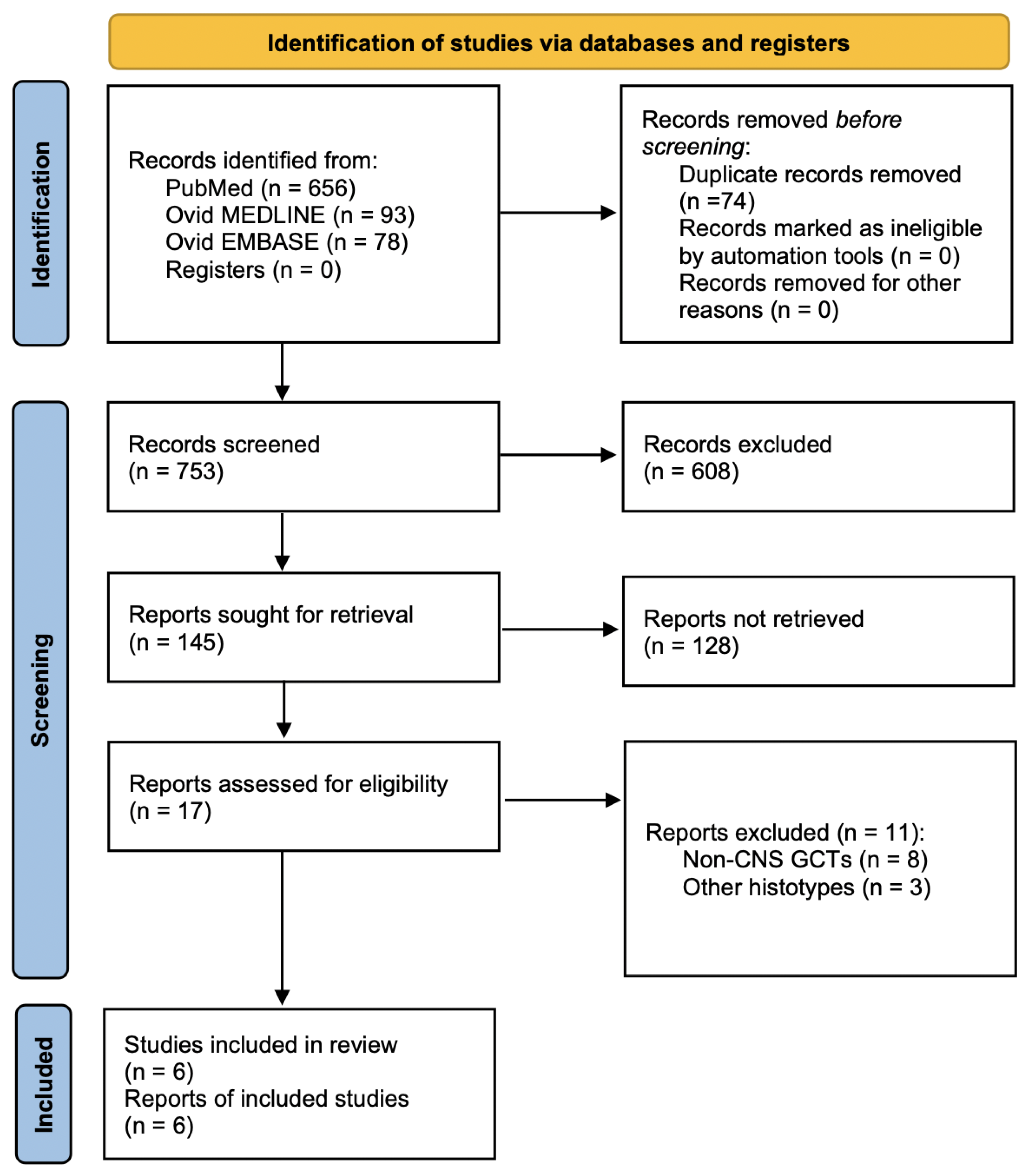
A total of 753 papers were identified after duplicate removal. After title and abstract analysis, 17 articles were identified for full-text analysis. Eligibility was ascertained for six articles [
11,
15,
16,
17,
18,
19]. The remaining 11 articles were excluded for the following reasons: (1) studies including extragonadal non-CNS GCTs (8 articles) and (2) studies describing other CNS tumor histotypes (3 articles). The PRISMA flowchart used is depicted in
Figure 3, as per guidelines for reporting systematic reviews [
13]. Including the case report presented here, a total of 7 patients with a primary co-occurrence of testicular NGGCTs and extragonadal CNS GCTs have been identified.
3.2.2. Baseline Data, Treatment, and Outcomes
Table 1 summarizes the main characteristics of the case reports found in the literature, including the case presented in the present article. Five articles described cases of metachronous GCTs (71%), and two authors reported on synchronous GCTs (29%). The average age at presentation was 17 years old. In 5 cases (71%), the tumor first diagnosed was cerebral, and in 2 (29%) cases was testicular. The latency between the two diagnoses varied from days in synchronous tumors to 16 years in metachronous GCTs.
The cerebral histotypes were mixed GCT (3 cases; 43%), pure germinoma (3 cases, 43%), and one yolk sac tumor (14%). The lesions were located in different CNS midline structures: 5 (72%) in the pineal region, of which one extended to the fourth ventricle, and one had multifocal extensions, one (14%) in the suprasellar region with extension from the pituitary peduncle to the hypothalamus and the last one (14%) in the right basal ganglia. Testicular tumor histotypes were three mixed GCTs (43%), two embryonal carcinomas (29%), one testicular mature teratoma (14%), and one carcinoma in situ (14%).
In all cases, testicular tumors were treated with orchiectomy followed by adjuvant chemotherapy; only in one case a bilateral testicular biopsy was performed before unilateral left orchiectomy and adjuvant chemotherapy. The treatment of CNS GCTs was more heterogeneous: gross total resection (GTR) was performed in 3 cases (43%), biopsy in 3 patients (43%), and sub-total resection (STR) in two of them (29%); in one case, STR followed the biopsy. Adjuvant treatment was always undertaken; chemotherapy (CMT) was administered in three cases (43%), radiotherapy (RT) in one (14%), and combined RT and CMT treatment in three (43%).
One patient was undertaking RT when the article was published; two authors did not specify the follow-up duration; in the remaining 4 cases, the follow-up time varied from 6 months to 3 years, with an average of 23 months. One patient died 2 years after the diagnosis of synchronous CNS and testicular GCTs; 4/7 (57%) patients had a full recovery with a Karnofsky performance status (KPS) of 90; one patient was alive and tumor-free after 2 years without a specific KPS value, and one presented a residual cognitive impairment and Parinaud’s syndrome at the 3rd-year control.
3.2.3. Etiopathogenesis Data
Among our series, two out of seven cases (29%) were syndromic, one suffering from Down Syndrome and the other from Testicular Dysgenesis Syndrome (TDS).
The patient affected by TDS had a synchronous CNS Germinoma and a testicular Carcinoma in situ, while the child affected by Down syndrome was treated for a metachronous testicular embryonal carcinoma, diagnosed 3 years after a CNS germinoma. The other 5 patients (71%) were non-syndromic.
Regarding genetic drivers, Silva et al. reported that aberrant chromosomes and loss of tumor suppressor genes might play a role in the malignant transformation of mismigrated primordial germ cells and the gain of resistance against apoptotic death [
11].
4. Discussion
GCTs represent a heterogeneous group of neoplasms, which can be subclassified as germinoma or non-germinomatous germ cell tumors (NGGCT). Histopathological entities are germinoma, yolk sac tumour, embryonal carcinoma, choriocarcinoma, teratoma (mature or immature) and mixed tumours of these subtypes [
2].
In Europe and America, GCTs represent approximately 4% of primary pediatric brain tumors, with an incidence peak from 10 to 12 years of age [
2,
5]. In Eastern countries, the incidence of CNS GCTs is around 15% of childhood CNS tumors. Males have a higher incidence of GCTs than females [
2].
Gonadal and extragonadal GCTs involve the midline structures of the body. The most common extragonadal locations are represented by the mediastinum, retroperitoneum, and brain, followed by the pineal gland and sacrococcygeal area. Isolated cases of involvement of other sites have been reported [
2,
3,
4].
Prognosis is highly dependent on both tumor location and histopathological subtypes. In fact, outcomes for patients affected by CNS NGGCTs are generally worse compared to those with CNS GGCTs, as NGGCTs are more aggressive and less sensitive to chemotherapy and radiotherapy [
5].
The co-occurrence of synchronous or metachronous gonadal NGGCTs and CNS GCTs has been rarely described in the literature.
We herein report a case of metachronous GCTs involving testis and suprasellar region. In the present case, the intracranial lesion and the testicular one were both histopathologically mixed germ cell tumors. However, the long-time interval between the onset of the two malignancies (16 years), together with the remission of the primary CNS tumor in the absence of other sites of metastatic dissemination, increased the likelihood of independent origin of the two tumors.
Our literature review identified six additional cases of primary co-occurrence of testicular NGGCTs and extragonadal CNS GCTs [
11,
15,
16,
17,
18,
19]. Apart from one case [
18], all patients were younger than 20 years of age at initial diagnosis [
11,
15,
16,
17,
19]. Differently from our patient, in the majority of metachronous tumor cases the initial diagnosis was cerebral, and the pineal region was the most common CNS tumor location.
The coexistence of primary neoplasms that appear to have the same origin but different histopathological characteristics has generated discussions about their etiopathogenesis. Some authors previously postulated that the co-occurrence of GCT in multiple sites might result from the malignant transformation of totipotent primordial germ cells that failed to migrate to the urogenital ridge during embryogenic development, being sequestrated in midline structures. Other authors have hypothesized that oncogenic factors acting on both gonadal and extragonadal germ cells may lead to the occurrence of two primary GCTs [
11,
20,
21].
The mutational landscape of CNS GCTs has been extensively described, focusing on multiple gene pathways disrupted in CNS GCTs and abnormal cell processes. However, the molecular mechanism that underlies the malignant transformation of the germ cell is not entirely understood [
11].
Table 2 summarizes the various etiopathogenesis reported in the literature, with a list of syndromes associated with testicular NGGCTs and extragonadal CNS GCTs co-occurrence and the disrupted somatic pathways identified in GCTs.
4.1. Gene and Pathways Mutations
A particularly high frequency of mutations involving genes encoding MAPK (mitogen-activated protein kinase) and/ or PI3K (Phosphatidylinositol-4-Phosphate 3-Kinase)/AKT (Protein Kinase B)/ mTOR (mammalian target of rapamycin) pathway components have been identified in GCTs [
21,
22].
These genetic alterations have been demonstrated to guarantee the survival and transformation of mismigrated primordial germ cells.
PTEN (Phosphatase and tensin homolog), PIK3C2B (Phosphatidylinositol-4-Phosphate 3-Kinase Catalytic Subunit Type 2 Beta), PIK3R2 (Phosphoinositide-3-Kinase Regulatory Subunit 2), and mTOR are the most frequently mutated gene in the PI3K pathway [
21,
22], whilst MAPK alterations are the earliest and fundamental events in the development of CNS GCTs [
21,
22]. The KIT mutation is the most frequent across all subtypes of CNS GCTs. Ichimura et al. demonstrated that the KIT and KITLG are essential for the survival of mismigrated PGCs. Normally, the down-regulation of KITLG induces apoptotic death in non-migrated cells, but oncogenic mutations of KIT determine a ligand-independent activation and anomalous survival of mismigrated PGCs [
22].
Other genetic candidates in the tumorigenesis are the suppressor genes Retinoblastoma 1 (RB1), those encoding for Netrin receptor DCC (Deleted in Colorectal Cancer) DCC Non-metastatic (NME), and finally, adenosine diphosphate-ribosylation factor (ARF) [
11]. Indeed, loss of tumor suppressor genes, such as Rb1, DCC, and NME, leads to tumor progression in germ cell cancers.
In addition, mutations of the p53 oncogene have been identified in some CNS GCTs, as well as gene amplification of MDM2 (Murine Double Minute 2). The MDM2 protein is a negative regulator of p53, that binds p53 and inhibits its transcriptional activity, promoting its nuclear export and degradation [
11]. The overexpression of MDM2 in GCTs inhibits p53 function as G1-S checkpoint controller and apoptosis inducer. MDM2 function is controlled by ARF, whose mutation results in MDM2 accumulation and p53 functional impairment.
Moreover, specific small, single-stranded, non-coding RNA molecules, named microRNAs, are known to be dysregulated in malignant GCTs. MicroRNAs play important roles in posttranscriptional gene regulation, modulating the expression of protein-coding genes and, as a result, critical cell processes. Malignant GCTs are characterized by increased expression levels of microRNA members of the miR-371~373 and miR-302/367 clusters, and their serum and CSF levels have been shown to be raised in patients with both extracranial and CNS GCTs [
20].
4.2. Aberrant Chromosome: 12p Gain
An increased copy number of 12p is implicated in the oncogenesis of GCTs and is found in postpuberal malignant gonadal and extragonadal GCTs, as an isochromosome of the short arm of the chromosome 12 or as tandem duplications in situ or transposed elsewhere in the genome [
11]. The 12p gain is predominantly observed in non-germinomatous GCTs, and it is more common in testicular GCTs (77–88%) than in CNS GCT (20–58%) [
21].
Overexpression of the CCND2 (Cyclin D2) gene located at chromosome band 12p13 activates the CDK4/6 (cyclin-dependent kinases 4 and 6) and facilitates progression from the G1 phase of the cell cycle to the S phase, giving cells protection against apoptotic death. This leads to the re-initiation of the cell cycle and consequent genomic instability [
11].
Intriguingly, amplification of 12p is lacking in most GCT arising in infants, which display different genetic aberrations such as imbalances of chromosome 1 and loss of 6q. In fact, it is known that prepuberal and postpuberal GCTs have different genetic features despite identical histopathological characteristics [
11].
4.3. Syndromic Associations
An increased risk of GCTs has been reported in patients affected by Testicular Dysgenesis Syndrome (TDS) and Down Syndrome.
Testicular dysgenesis syndrome is a male condition characterized by hypospadias, cryptorchidism, poor semen quality, and testicular cancer. It is thought to be the result of a hormonal imbalance during fetal life, related to genetic effects and environmental factors, that affects gonadal development and embryonal programming [
15,
17,
23].
The association of GCTs with Down Syndrome is frequently reported in the literature, with a fivefold higher incidence than in the general population. Hashimoto et al. described a case of a 17-year-old boy with Trisomy 21, affected by gonadal embryonal carcinoma and CNS Germinoma DS [
15].DS is known to predispose to the development of GCTs through various mechanisms. l [
15]. However, the specific pathogenetic drivers are yet to be understood.
4.4. Limitations
Since retrospective studies were mainly collected in this literature review, all the limitations inherent to retrospective studies have to be considered. Because of the small number of studies responding to the inclusion criteria, no formal statistical comparison was performed.
The patient from our Institution and most cases included in the literature review did not undergo genetic analysis to investigate germline cancer predisposition, which narrowed the chances to improve the current knowledge on the oncogenic process. However, as far as we are aware, we reported the 7th case of primary co-occurrence of CNS GCTs and testicular NGGCTs, and we provide a comprehensive synopsis of the molecular pathways involved in the development of this rare condition.
The risk of bias assessment was avoided using the NOS. Selection criteria, comparability of the study and outcome evaluation were evaluated using this quality assessment scale. The choice to use the NOS assessment permitted to select articles reporting reliable studies which provided robust evidences concerning the etiopathogenetic mechanisms of the coexistence of primary testicular NGGCTs and extragonadal CNS GCTs.


 ,
, 




{kind=link}
{kind=link}
{kind=link}