Abstract
Toxic gases emitted by industries and vehicles cause environmental pollution and pose significant health risks which are becoming increasingly dangerous. Therefore, the detection of the toxic gases is crucial. The development of gas sensors with high sensitivity and fast response based on nanomaterials has garnered significant interest. In this work, we studied the adsorption behavior of
wheel structures of pristine and nitrogen functionalized borophene quantum dots for major hazardous environmental gases, such as NO2, CO2, CO, and NH3. The self-consistent-charge density-functional tight-binding method (SCC-DFTB) method was performed to investigate structural geometries, the most favorable adsorption sites, charge transfer, total densities of states, and electronic properties of the structures before and after adsorption of the gas molecules. Based on calculated results, it was found that the interaction between the borophene quantum dots and the gas molecules was chemisorption. The functionalized nitrogen atom contributed to impurity states, leading to higher adsorption energies of the functionalized borophene quantum dots compared to the pristine ones. Total densities of states revealed insights into electronic properties of gas molecules adsorbed on borophene quantum dots. The nitrogen-doped borophene quantum dots demonstrated excellent performance as a sensing material for hazardous environmental gases, especially CO2.
1. Introduction
Hazardous gases (such as COx, NOx, and NH3) produced by industrial activities and vehicles cause respiratory diseases in humans, contribute to the greenhouse effect, and damage the environment [1]. For the safety of human health and environmental protection, gas sensor technology is crucial for the detection of these gases. Due to unique properties of nanomaterials, two-dimensional materials such as graphene, silicene, Mxene, phosphorene, and transition metal dichalcogenides have attracted significant interest in various studies [2,3,4,5,6,7,8]. For gas sensing applications, graphene exhibits great performance due to its excellent mobility and high surface-to-volume ratio [9,10,11]. However, the absence of a band gap in graphene makes it difficult to control charge in the sheet [9,12]. Borophene, a two-dimensional material composed of monolayer of boron atoms, has been successfully synthesized on silver surface under ultrahigh vacuum conditions [13,14]. Borophene has gained widespread attraction for research in nanotechnology because of its unique electronic, optical, thermal, magnetic, and mechanical properties [15,16,17,18,19]. Boron possesses three valence electrons in 2s22p1 orbitals which can form sp2 hybridization [9,15] and the bonding between boron atoms is highly complex with polymorphisms [13]. This complexity leads to various physical and chemical properties [20,21], which distinguish it from other two-dimensional materials like graphene and transition metal dichalcogenides. Owing to a variety of atomic arrangements, there have been reports on theoretical and experimental studies of borophene structures with several stable structures including triangular and hexagonal planar or quasi-planar structure motifs [17,22,23,24,25,26]. An anionic cluster () is a highly symmetric planar wheel-like structure of borophene composed of a single boron atom at the center of a ring [27,28]. The structure provides double aromaticity and high electronic stability resulting from the bonding between the central boron atom and the B8 ring through three delocalized σ and three delocalized π bonds [27]. Doping with metal atoms can modify the structures and properties of borophene with small planar or quasi-planar structures tailored for specific applications [27,29,30,31,32]. The structural diversity offers enormous potential to tailor the properties of borophene for applications in areas ranging from electronics [33,34], energy storage [35,36], gas sensing [9,37,38], etc.
Focusing on gas sensing applications, the extraordinary geometries, high surface-to- volume ratio, and excellent electronic properties of borophene allow it to be used as gas sensors. Several theoretical studies have reported on the adsorption of gas molecules by borophene. For examples, Huang et al. [37] calculated adsorption energies, charge transfer, and electronic structures of borophene with buckled and line-defective phases for adsorption of NH3, NO, NO2, and CO using density functional theory (DFT). Shukla et al. [9] used DFT calculation combined with non-equilibrium Green’s function (NEGF) to study adsorption behavior of monolayer borophene for gas molecules of CO, CO2, NO, NO2, and NH3. Ta et al. [38] theoretically investigated the potential for the adsorption of five main hazardous gases including CO, NO, NH3, NO2, and CO2 on β12 borophene by using van der Waals (vdW) density functionals (vdW-DFs). These studies showed that borophene could be developed as sensing materials for gas sensors to detect toxic gases. However, the mentioned studies focused on gas adsorption of pristine borophene sheets with different structures.
Recently, borophene quantum dots (BQDs) have been explored for various applications, including solar cell [39], biosensors [40,41], catalysts [42], supercapacitors [43], and biomedical applications [44]. For instance, Liu et al. reported the preparation of BQDs/BC2N heterostructures for an ultrasensitive humidity sensor. The sensor showed ultra-high sensitivity, high selectivity, fast response and recovery times, and good flexibility over a wide detection range (11–97% RH) by using the Grotthuss chain reaction sensing mechanism via hydrogen bonding and multilayer physisorption [45]. The BQDs exhibit quantum confinement effects, where their electronic and optical properties are influenced by the size and shape of the dots [40,46]. However, research on the application of pristine and chemically functionalized BQDs for gas sensing application is still limited.
In the present study, we theoretically investigate the adsorption behaviors of NO2, CO2, CO, and NH3 gas molecules on wheel structures of pristine and nitrogen functionalized BQDs using self-consistent charge density functional tight-binding (SCC-DFTB) method. We examine adsorption energies, optimal adsorption sites, charge transfer, density of states (DOS), and electronic properties to understand the interaction between these gas molecules and the pristine and nitrogen functionalized BQDs.
2. Calculation Methods
The SCC-DFTB method is derived from second-order expansion of the DFT Kohn–Sham total energy in terms of the charge density fluctuations [47,48,49]. The Kohn–Sham approach simplifies the complex problem of interacting electrons by mapping it onto a system of non-interacting electrons that reproduce the same electron density. Although the non-interacting system is easier to solve, it still provides the correct ground-state electron density for the original interacting system. In the DFTB approach, the traditional self-consistency between the effective potential and the charge density is replaced by a simpler self-consistency in the distribution of Mulliken charges. The method is optimized for high accuracy by using a minimal basis set to represent the single-electron Kohn–Sham-like eigenstates. This allows electronic interactions to be precomputed within a two-center model using an effective Hamiltonian derived from a carefully chosen reference density [50]. Based on Kohn–Sham orbitals, the total energy for the SCC-DFTB method is expressed as [51,52]:
where is the Kohn–Sham orbitals, is unperturbed Hamiltonian, and are the induced charges on the atoms A and B, respectively, is the second derivative with respect to its total charges (Coulombic-like interaction potential), and denotes repulsive potential for the pair of atoms A and B.
The benchmark of the SCC-DFTB method is a balance between accuracy and computational efficiency with reduced computational costs compared to standard DFT [47,49]. For large systems, the SCC-DFTB calculations were 2–3 orders of magnitude faster than DFT [47,51]. It should be noted that that calculation time depends on both the number of atoms and the complexity of the structures being simulated. While the system in this work may not be considered large in absolute terms, SCC-DFTB still ensures faster computation times. The SCC-DFTB method simulates electronic properties, energies, and molecular structures of molecules and materials, and provides results in good agreement with DFT methods [53]. For gas adsorption, the SCC-DFTB method can be used to investigate interaction between gas molecules and sensing materials [54,55,56]. The benchmark of the SCC-DFTB method has been performed for theoretical studies of boron clusters [52,57].
In this work, wheel-like structures of pristine and nitrogen functionalized BQDs ( and BQDs) were built. For nitrogen functionalization, a nitrogen atom was at the center of the ring and bonded with nine surrounding boron atoms. It should be noted that this configuration is theoretically feasible because nitrogen can bond to boron forming a stable structure. However, experimental implementation can be challenging due to the precision required to control the doping process and ensure that the nitrogen atom is properly positioned at the center of the B9 ring. Techniques like molecular beam epitaxy or chemical vapor deposition which allow for atomic-level precision may be required making the process more complex but achievable with advanced fabrication methods. To reduce the impact of edge effects, we applied periodic boundary conditions along with a large simulation box to investigate the QDs. This approach ensures that the QDs’ electronic structure, adsorption phenomena, and charge transfer effects are accurately accounted for providing more reliable insights compared to calculations using a single unit cell. The matsci-0-3 parameter set was used for calculation in O-N-C-B-H system. In addition, the matsci-0-3 parameter set well describes the interaction of covalent organic system, especially small organic molecules containing nitrogen, oxygen, carbon, and hydrogen [49,51,58,59]. The SCC-DFTB command code and its relative parameters are demonstrated in Source Code S1 of Supporting Information. To investigate the most favorable adsorption sites, gas molecules including NO2, CO2, CO, and NH3 were placed at different distances with orientation configurations (parallel and perpendicular) above the surfaces of and BQDs. Adsorption strengths between the structures and gas molecules are described in the term of adsorption energies (Ead). The adsorption energy is calculated as the following equation:
where , and are the total energies of or BQDs with gas molecules, BQDs, and gas molecules, respectively. A negative value of Ead indicates that gas molecules are adsorbed and more negative values are more favorable adsorption sites.
To study charge transfers from/to gas molecules resulting from the adsorption, net charge transfer is defined as the charge difference of gas molecules before and after adsorption [60]. The net charge transfer (Q) is obtained from Equation (3):
where and are the charges of gas molecules adsorbed on the surfaces of BQDs structures and isolated gas molecules, respectively.
Total density of states (DOS) was calculated to analyze the influence of gas molecule adsorption on the electronic properties of BQDs. The k-point sampling density was set to for the DOS studies [37].
3. Results and Discussion
3.1. Structural and Electronic Properties of and BQDs
Figure 1a shows the optimized structures of and BQDs with side and top views. These structures were optimized to obtain the most stable structure. During the optimization process, the initial geometries of the BQD structures were input into the SCC-DFTB framework. The energy minimization was performed iteratively by adjusting atomic positions with the forces on each atom being calculated at each step. The optimization continued until the system reached a stable configuration where the forces on the atoms were below a predefined threshold and the total energy converged. An example of SCC-DFTB optimization command code is provided in Source Code S1 of the Supporting Information.
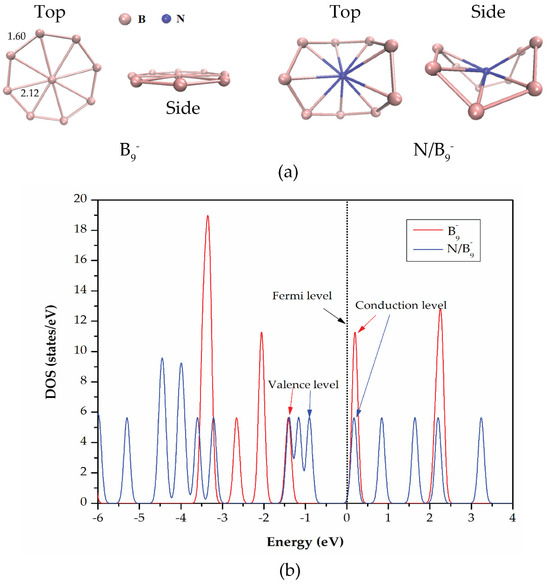
Figure 1.
(a) Side and top views of the optimized structures of and BQDs and (b) DOS of the structures.
For the BQDs, it was found that the bond lengths between the central boron atom and each boron atom of the B8 ring are ~2.12 Å. The structural diameter and the bond lengths between boron atoms on the B8 ring side are ~4.22 Å and ~1.60 Å, respectively. The bond lengths of our optimized BQD structures show good agreement with previously published studies [27,32]. In addition, the structures of all planar boron clusters have been experimentally confirmed to consist of an outer boron atom ring and one or more inner atoms [27]. The central boron atom contributes all three valence electrons to the delocalized bonding [27]. The boron atoms in the ring side contribute their two valence electrons to two-center, two-electron (2c–2e) boron–boron bonds of the ring, while the remaining valence electron participates in delocalized σ and π bonding with the central boron atom [27,61,62]. For nitrogen functionalization, a nitrogen atom (blue color) was placed at the center of the ring. It can be obviously seen that the structural distortion occurred which corresponds to the previous work [63]. The structure is buckled where some boron atoms move inward or outward with respect to the central nitrogen atom. The distortion arises from the inherent nature of borophene with boron atoms forming triangles or hexagons in which nitrogen doping can induce structural distortions. The bond lengths between the boron atoms on the ring side and the central nitrogen atom are changed to be in the range of 1.88–2.00 Å.
To study electronic properties of and BQDs, we calculated energy gap (), , , Fermi energy (), and DOS as presented in Table 1 and Figure 1b. It was found that decreases significantly from 2.72 eV to 0.68 eV after nitrogen functionalization. The large decrease of results from a change in energy levels of the conduction band edge () and the valence band edge (), as shown in Table 1. The conduction band edge shifts downward and the valence band edge shifts upward. This result reveals that nitrogen functionalization at the center of the boron ring introduces impurity states within the band structure of BQDs. The interaction between these impurity states and an additional electron, which occupy in the conduction band or near the conduction band edge in the structure, modifies the energy levels near the Fermi level resulting in the reduced energy gap [64,65]. Figure 1b shows the calculated DOS of and BQDs. It should be noted that the DOS describes the distribution of electronic states as a function of energy. It can be clearly seen that new DOS peaks (blue lines) around the vicinity of the Fermi level appeared after nitrogen functionalization. This confirms that nitrogen functionalization creates new localized states due to the difference in electronic configuration of nitrogen atoms compared to boron atoms [66], which indicates the presence of electrons or holes. The values from Table 1 correspond well with those shown in Figure 1b.

Table 1.
Electronic parameter of and BQDs.
3.2. Adsorption of NO2, CO2, CO, and NH3 Gas Molecules on BQDs
To investigate the adsorption behavior of BQDs for the detection of NO2, CO2, CO, and NH3 gas molecules, the different adsorption structures were constructed and fully optimized. Each gas molecule was placed on the top of the BQD structures with both parallel and perpendicular orientations. Each atom of these gas molecules was pointed toward a boron atom of the structures including N-B, C-B, O-B, and H-B to find the most favorable adsorption sites. Figure 2 shows side and top views of some optimized BQD structures with gas molecules on the top of the structures in parallel and perpendicular orientations. The typical calculated adsorption parameters are summarized in Table 2. Based on overview of the calculated Ead values, it is found that the highest to lowest Ead values are found for NO2, CO, CO2, and NH3, respectively. In each case, the Ead values are higher than adsorption energies of borophene sheets for these toxic gases [9,37]. This result reveals that quantum confinement effects and high surface to volume ratio of the BQDs improve the adsorption performance for gas sensing. For charge transfer calculation, the most values of charge transfer are found to be positive which is consistent with published work [38]. Also, large charge transfer is found for all gas molecules. The positive (negative) sign of charge transfer shows that gas molecules accept (donate) electrons [38].
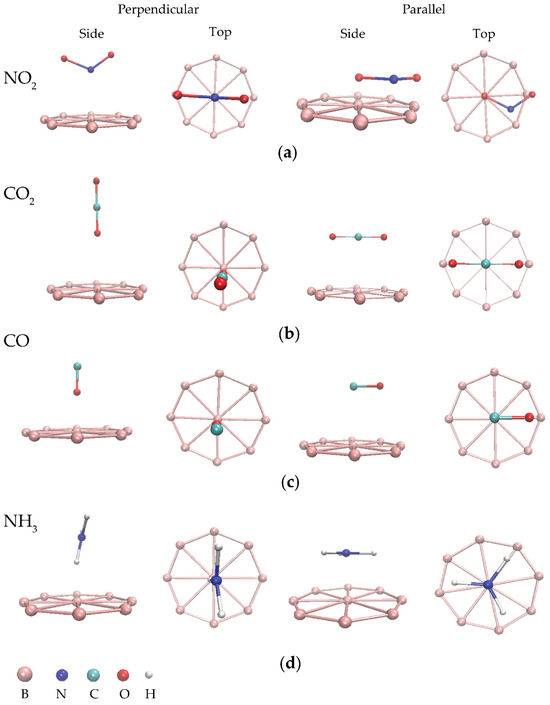
Figure 2.
Side and top views of some optimized BQDs for adsorption of (a) NO2, (b) CO2, (c) CO, and (d) NH3 with perpendicular and parallel orientations.
Table 2.
Typical calculated adsorption parameters for each gas molecule absorbed on the BQD structures.
To consider the adsorption sites of each gas molecule, the most favorable adsorption sites for NO2, CO, CO2, and NH3 were identified as B-O (perpendicular), B-C (parallel), B-C (parallel), and B-N (perpendicular), respectively. Interestingly, both CO and CO2 prefer B-C adsorption sites with parallel orientation. In case of energy gap, it can be obviously seen that the energy gap decreases for all adsorption sites of all gas molecules, which suggests that conductivity of the BQDs increases after gas adsorption. Based on the results of Table 2, high adsorption energies, large charge transfer, and short interaction distances reveal that the interaction between gas molecules and the BQDs is a result of chemisorption [9,37,38]. Additionally, further adsorption sites on the BQDs were investigated, as presented in Figure S1 and Table S1 of the Supporting Information. However, the results from these additional sites were less favorable due to weaker adsorption energies and interactions, resulting in less stable configurations. Therefore, only the perpendicular and parallel orientations were selected for this study.
3.3. Adsorption of NO2, CO2, CO, and NH3 Gas Molecules on BQDs
Figure 3 displays some optimized BQD structures for adsorption of NO2, CO2, CO, and NH3 gas molecules at different interaction distances. The calculated adsorption parameters are presented in Table 3. Each atom of gas molecules was placed on the top of the BQD structures pointing to the nitrogen atom with parallel and perpendicular orientation. According to the results of Table 3, the results shows that the Ead values of the BQDs for all gas molecules at all adsorption sites obviously increased in comparison with the Ead values of the BQDs. The favorable adsorption sites for NO2, CO, and CO2 are parallel orientation, except for NH3, which preferred perpendicular orientation. The highest Ead values of CO2, CO, NO2, and NH3 at their most favorable adsorption sites are −16.57, −12.08, −11.64, and −7.22 eV, respectively. There are two reasons that can describe this result. First, the functionalized nitrogen atom introduces impurity states leading to strong interaction. Second, the buckling of the BQDs allows gas atoms bonding with more than one surrounding boron atoms [37]. Looking at charge transfer, it is found that the most charge transfer is positive and lower than that of the BQDs. This demonstrates that the trend in charge transfer of each gas molecule is still from the structures to gas molecules. It is attributed that the lower charge transfer results from strong interaction (high Ead values), localized electronic states from impurities and buckled surface of the BQDs, which affects pathway of charge transfer. The interaction distances for all adsorption sites are in the range of chemical bonding indicating chemisorption [38].
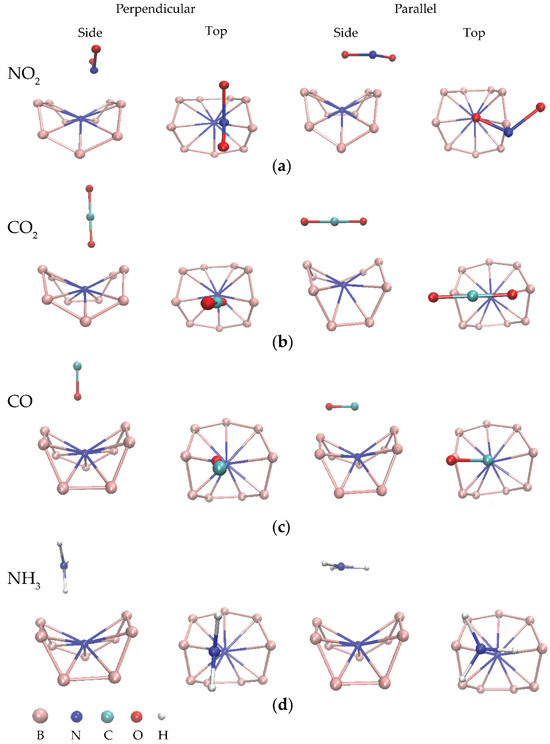
Figure 3.
Side and top views of some optimized BQDs for adsorption of (a) NO2, (b) CO2, (c) CO, and (d) NH3 with perpendicular and parallel orientations.
Table 3.
Typical calculated adsorption parameters for each gas molecule absorbed on the BQD structures.
For energy gap, the trend in slightly increases energy gap of the BQDs after adsorption of NO2, CO2, and NH3, whereas the energy gap of CO adsorbs on the structure tend to be decreased. We will explain this result in the section of DOS (in the Section 3.4). A comparison of adsorption energies with other two-dimensional materials such as graphene, for instance, exhibited weak adsorption energies for NO2, CO2, CO, and NH3 gas molecules [9]. The adsorption energies of NO2, CO and NH3 gas molecules on borophene sheets were found to be −1.75, −1.24, and −1.45 eV, respectively [37]. In case of MoS2, the adsorption energies were −0.44 and −0.33 eV for CO and CO2, respectively [9]. Based on the results, nitrogen functionalization at the center of the BQD ring could improve the adsorption behavior of BQDs for NO2, CO2, CO, and NH3 gas molecules.
3.4. Total Densities of State (DOS)
DOS calculations were performed to gain insights into the interactions between gas molecules and both and BQDs structures. Figure 4 demonstrates the calculated DOS of the most favorable adsorption sites for each gas molecule. It was observed that sharp DOS peaks appeared near the Fermi level for all gas molecules adsorbed on the BQDs indicating strong bonding [38], which corresponds to higher adsorption energies than the BQDs with gas molecules. In both cases, DOS peaks near the Fermi level suggest charge transfer from the BQDs to the gas molecules [9]. Among all these gases, only CO contributes states near the Fermi level, specifically at the top of valence band, which might improve the conductivity [37]. This confirms the decreasing energy gap in case of CO adsorbed on the BQDs, which is the reason for Section 3.3. To consider in details of each case as shown in Figure 4a–d, new local states appeared near the conduction band edge for NO2, CO2, and NH3 on BQDs, while in case of CO- BQD, a new local state appeared below the Fermi level at −0.4 eV. This suggests the existence of localized states from the impurities interacting with gas molecules.
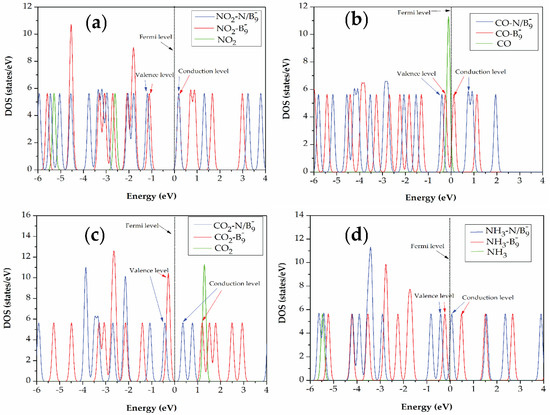
Figure 4.
Calculated DOSs for (a) NO2, (b) CO, (c) CO2, and (d) NH3 adsorbed on and BQDs structures. The Fermi level is set to zero with dashed vertical lines.
4. Conclusions
In summary, we have studied the adsorption behavior of and BQDs structures for NO2, CO, CO2, and NH3 gas molecules using SCC-DFTB method. Structural geometries, the most favorable adsorption sites and electronic properties were investigated. After nitrogen functionalization, the structural and electronic properties of BQDs were changed. The structure was buckled and its energy gap was decreased due to the impurity states. Based on the adsorption results, the adsorption energies of the structures were found to be higher than those of borophene sheets. Additionally, the adsorption energies of the BQDs were higher than those of the BQDs. The BQDs favored NO2 adsorption while the BQDs favored CO2 with parallel orientation. The calculated charge transfer indicated that there was a transfer of charge from the BQDs structures to the gas molecules. In case of DOS analysis, new local states formed around the Fermi level after gas adsorption. According to high adsorption energies, DOS and interaction distances, the interaction between gas molecules and the and BQDs was found to be chemisorption. Our calculation suggests that nitrogen-doped BQDs are promising candidates for detecting hazardous gases in the environment. We believe that this work will provide valuable guidance on future studies in both simulations and experiments.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/jcs8100397/s1, Table S1: Typical calculated adsorption parameters for each gas molecule absorbed on the BQD structures with bridge and hollow sites; Figure S1: Some optimized BQDs for adsorption of (a) NO2, (b) CO2, (c) CO and (d) NH3 with bridge and hollow sites; Source Code S1: An example of DFTB command code for full optimization.
Author Contributions
Conceptualization, K.T.; methodology, K.T.; simulation, K.T.; formal analysis, K.T. and C.W.; investigation, K.T. and C.W.; data curation, K.T.; writing—original draft preparation, K.T.; writing—review and editing, C.W.; validation, C.W.; visualization, K.T.; supervision, C.W.; funding acquisition, K.T. and C.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Thailand Science Research and Innovation (TSRI), RDI PCRU, grant number TSRI 2567/45. C.W. would like to thank the Kasetsart University Research and Development Institute (KURDI) for funding, under the grant number FF(KU)51.67.
Data Availability Statement
The original data presented in the study are included in the article; further inquiries can be directed to the corresponding author.
Acknowledgments
The authors acknowledge Laboratory for Multiscale Innovative Technologies (LMIT), Faculty of Science, Kasetsart University for computer servers.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Seesaard, T.; Kamjornkittikoon, K.; Wongchoosuk, C. A comprehensive review on advancements in sensors for air pollution applications. Sci. Total Environ. 2024, 951, 175696. [Google Scholar] [CrossRef] [PubMed]
- Chaloeipote, G.; Wongchoosuk, C. Flexible humidity sensor based on PEDOT:PSS/Mxene nanocomposite. Flex. Print. Electron. 2024, 9, 015015. [Google Scholar] [CrossRef]
- Seekaew, Y.; Kamlue, S.; Wongchoosuk, C. Room-temperature ammonia gas sensor based on Ti3C2Tx MXene/Graphene Oxide/CuO/ZnO Nanocomposite. ACS Appl. Nano Mater. 2023, 6, 9008–9020. [Google Scholar] [CrossRef]
- Aufray, B.; Kara, A.; Vizzini, S.; Oughaddou, H.; Léandri, C.; Ealet, B.; Le Lay, G. Graphene-like silicon nanoribbons on Ag(110): A possible formation of silicene. Appl. Phys. Lett. 2010, 96, 183102. [Google Scholar] [CrossRef]
- Liu, H.; Neal, A.T.; Zhu, Z.; Luo, Z.; Xu, X.; Tománek, D.; Ye, P.D. Phosphorene: An unexplored 2D semiconductor with a high hole mobility. ACS Nano 2014, 8, 4033–4041. [Google Scholar] [CrossRef]
- Pumera, M.; Sofer, Z.; Ambrosi, A. Layered transition metal dichalcogenides for electrochemical energy generation and storage. J. Mater. Chem. A 2014, 2, 8981–8987. [Google Scholar] [CrossRef]
- Chen, X.; Hu, J.; Chen, P.; Yin, M.; Meng, F.; Zhang, Y. UV-light-assisted NO2 gas sensor based on WS2/PbS heterostructures with full recoverability and reliable anti-humidity ability. Sens. Actuators B Chem. 2021, 339, 129902. [Google Scholar] [CrossRef]
- Pak, Y.; Lim, N.; Kumaresan, Y.; Lee, R.; Kim, K.; Kim, T.H.; Kim, S.M.; Kim, J.T.; Lee, H.; Ham, M.H.; et al. Palladium nanoribbon array for fast hydrogen gas sensing with ultrahigh sensitivity. Adv. Mater. 2015, 27, 6945–6952. [Google Scholar] [CrossRef]
- Shukla, V.; Wärma, J.; Jena, N.K.; Grigoriev, A.; Ahuja, R. Toward the realization of 2D borophene based gas sensor. J. Phys. Chem. C 2017, 121, 26869–26876. [Google Scholar] [CrossRef]
- Yuan, W.; Shi, G. Graphene-based gas sensors. J. Mater. Chem. A 2013, 1, 10078. [Google Scholar] [CrossRef]
- Wang, T.; Huang, D.; Yang, Z.; Xu, S.; He, G.; Li, X.; Hu, N.; Yin, G.; He, D.; Zhang, L. A review on graphene-based gas/vapor sensors with unique properties and potential applications. Nano-Micro Lett. 2016, 8, 95–119. [Google Scholar] [CrossRef] [PubMed]
- Castro Neto, A.H.; Guinea, F.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 109–162. [Google Scholar] [CrossRef]
- Mannix, A.J.; Zhou, X.-F.; Kiraly, B.; Wood, J.D.; Alducin, D.; Myers, B.D.; Liu, X.; Fisher, B.L.; Santiago, U.; Guest, J.R.; et al. Synthesis of borophenes: Anisotropic, two-dimensional boron polymorps. Science 2015, 350, 1513–1516. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Zhang, J.; Zhong, Q.; Li, W.; Li, S.; Li, H.; Cheng, P.; Meng, S.; Chen, L.; Wu, K. Experimental realization of two-dimensional boron sheets. Nat. Chem. 2016, 8, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Kaneti, Y.V.; Benu, D.P.; Xu, X.; Yuliarto, B.; Yamauchi, Y.; Golberg, D. Borophene: Two-dimensional boron monolayer: Synthesis, properties, and potential applications. Chem. Rev. 2022, 122, 1000–1051. [Google Scholar] [CrossRef]
- Hou, C.; Tai, G.; Liu, Y.; Liu, X. Borophene gas sensor. Nano Res. 2022, 15, 2537–2544. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Penev, E.S.; Yakobson, B.I. Two-dimensional boron: Structures, properties and applications. Chem. Soc. Rev. 2017, 46, 6746–6763. [Google Scholar] [CrossRef]
- Sergeeva, A.P.; Popov, I.A.; Piazza, Z.A.; Li, W.L.; Romanescu, C.; Wang, L.S.; Boldyrev, A.I. Understanding boron through size-selected clusters: Structure, chemical bonding, and fluxionality. Acc. Chem. Res. 2014, 47, 1349–1358. [Google Scholar] [CrossRef]
- Hou, C.; Tai, G.A.; Wu, Z.H.; Hao, J.Q. Borophene: Current status, challenges and opportunities. ChemPlusChem 2020, 85, 2186–2196. [Google Scholar] [CrossRef]
- Sabokdast, S.; Horri, A.; Azar, Y.T.; Momeni, M.; Tavakoli, M.B. Detection of nucleobases on borophene nanosheet: A DFT investigation. Bioelectrochemistry 2021, 138, 107721. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Lü, T.-Y.; Wang, H.-Q.; Feng, Y.P.; Zheng, J.-C. Review of borophene and its potential applications. Front. Phys. 2019, 14, 33403. [Google Scholar] [CrossRef]
- Yang, X.; Ding, Y.; Ni, J. Ab initio prediction of stable boron sheets and boron nanotubes: Structure, stability, and electronic properties. Phys. Rev. B 2008, 77, 041402. [Google Scholar] [CrossRef]
- Liu, Y.; Penev, E.S.; Yakobson, B.I. Probing the synthesis of two-dimensional boron by first-principles computations. Angew. Chem. 2013, 52, 3156–3159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, Y.; Gao, G.; Yakobson, B.I. Two-dimensional boron monolayers mediated by metal substrates. Angew. Chem. 2015, 127, 13214–13218. [Google Scholar] [CrossRef]
- Penev, E.S.; Bhowmick, S.; Sadrzadeh, A.; Yakobson, B.I. Polymorphism of two-dimensional boron. Nano Lett. 2012, 12, 2441–2445. [Google Scholar] [CrossRef]
- Piazza, Z.A.; Hu, H.S.; Li, W.L.; Zhao, Y.F.; Li, J.; Wang, L.S. Planar hexagonal B36 as a potential basis for extended single-atom layer boron sheets. Nat. Commun. 2014, 5, 3113. [Google Scholar] [CrossRef]
- Romanescu, C.; Galeev, T.R.; Li, W.L.; Boldyrev, A.I.; Wang, L.S. Transition-metal-centered monocyclic boron wheel clusters (M©Bn): A new class of aromatic borometallic compounds. Acc. Chem. Res. 2013, 46, 350–358. [Google Scholar] [CrossRef]
- Zhai, H.J.; Alexandrova, A.N.; Birch, K.A.; Boldyrev, A.I.; Wang, L.S. Hepta- and octacoordinated boron in molecular wheels of eight- and nine-atom boron clusters: Observation and confirmation. Angew. Chem. Int. Ed. 2003, 42, 6004–6008. [Google Scholar] [CrossRef]
- Kumar, S.; Singh, M.; Sharma, D.K.; Auluck, S. Enhancing gas adsorption properties of borophene by embedding transition metals. Comput. Condens. Matter 2020, 22, e00436. [Google Scholar] [CrossRef]
- Zhang, P.; Xu, X.; Song, E.; Hou, X.; Yang, X.; Mi, J.; Huang, J.; Stampfl, C. Transition metal-doped α-borophene as potential oxygen and hydrogen evolution electrocatalyst: A density functional theory study. Catal. Commun. 2020, 144, 106090. [Google Scholar] [CrossRef]
- Xu, X.; Hou, X.; Lu, J.; Zhang, P.; Xiao, B.; Mi, J. Metal-doped two-dimensional borophene nanosheets for the carbon dioxide electrochemical reduction reaction. J. Phys. Chem. C 2020, 124, 24156–24163. [Google Scholar] [CrossRef]
- Li, W.L.; Romanescu, C.; Galeev, T.R.; Piazza, Z.A.; Boldyrev, A.I.; Wang, L.S. Transition-metal-centered nine membered boron rings: M©B9 and M©B9− (M = Rh, Ir). J. Am. Chem. Soc. 2012, 134, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-J.; Altalhi, T.; Yang, J.-H.; Yakobson, B.I. Semiconducting ά-boron sheet with high mobility and low all-boron contact resistance: A first-principles study. Nanoscale 2021, 13, 8474–8480. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-L.; Yang, J.-H.; Xiang, H.; Gong, X.-G. Fully boron sheet-based field effect transistors from first-principles: Inverse design of semiconducting boron sheets. J. Phys. Chem. Lett. 2021, 12, 576–584. [Google Scholar] [CrossRef]
- Jiang, H.R.; Lu, Z.; Wu, M.C.; Ciucci, F.; Zhao, T.S. Borophene: A promising anode material offering high specific capacity and high rate capability for lithium-ion batteries. Nano Energy 2016, 23, 97–104. [Google Scholar] [CrossRef]
- Jena, N.K.; Araujo, R.B.; Shukla, V.; Ahuja, R. Borophane as a bencmate of graphene: A potential 2D material for anode of Li and Na-ion batteries. ACS Appl. Mater. Interfaces 2017, 9, 16148–16158. [Google Scholar] [CrossRef]
- Huang, C.S.; Murat, A.; Babar, V.; Montes, E.; Schwingenschlögl, U. Adsorption of the gas molecules NH3, NO, NO2, and CO on borophene. J. Phys. Chem. C 2018, 122, 14665–14670. [Google Scholar] [CrossRef]
- Ta, L.T.; Hamada, I.; Morikawa, Y.; Dinh, V.A. Adsorption of toxic gases on borophene: Surface deformation links to chemisorptions. RSC Adv. 2021, 11, 18279. [Google Scholar] [CrossRef]
- Zhao, A.; Han, Y.; Che, Y.; Liu, Q.; Wang, X.; Li, Q.; Sun, J.; Lei, Z.; He, X.; Liu, Z.H. High-quality borophene quantum dot realization and their application in a photovoltaic device. J. Mater. Chem. A 2021, 9, 24036–24043. [Google Scholar] [CrossRef]
- Gogoi, D.; Hazarika, C.; Neog, G.; Mridha, P.; Bora, H.K.; Das, M.R.; Szunerits, S.; Boukherroub, R. Borophene quantum dots as novel peroxidase-mimicking nanozyme: A dual-mode assay for the detection of oxytetracycline and tetracycline antibiotics. ACS Appl. Mater. Interfaces 2024, 16, 14645–14660. [Google Scholar] [CrossRef]
- Joshi, D.J.; Malek, N.I.; Jung Park, T.; Kailasa, S.K. Ultrasonication-assisted synthesis of fluorescent borophene quantum dots for sensing of dehydroepiandrosterone biomarker. J. Mol. Liq. 2023, 385, 122294. [Google Scholar] [CrossRef]
- Yashwanth, H.J.; Hareesh, K.; Rondiya, S.R.; Choudhary, R.J.; Dhole, S.D. The borophene quantum dots scaffolded TiO2 nanocomposite as an efficient photo electrocatalyst for water splitting application. Appl. Surf. Sci. 2024, 646, 158910. [Google Scholar] [CrossRef]
- Ranjan, P.; Lee, J.M.; Kumar, P.; Vinu, A. Borophene: New sensation in flatland. Adv. Mater. 2020, 1, 2000531. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Sadique, M.A.; Kaushik, A.; Ranjan, P.; Khan, R.; Srivastava, K.A. Borophene as an emerging 2D flatland for biomedical applications: Current challenges and future prospects. J. Mater. Chem. B 2022, 10, 1146–1175. [Google Scholar] [CrossRef]
- Liu, X.; Hou, C.; Liu, Y.; Chen, S.; Wu, Z.; Liang, X.; Tai, G. Borophene and BC2N quantum dot heterostructures: Ultrasensitive humidity sensing and multifunctional applications. J. Mater. Chem. A 2023, 11, 24789–24799. [Google Scholar] [CrossRef]
- Wang, H.; An, D.; Wang, M.; Sun, L.; Li, Y.; Li, H.; Li, N.; Hu, S.; He, Y.B. Crystalline borophene quantum dots and their derivative boron nanospheres. Mater. Adv. 2021, 2, 3269–3273. [Google Scholar] [CrossRef]
- Liang, R.; Swanson, J.M.J.; Voth, G.A. Benchmark study of the SCC-DFTB approach for a biomolecular proton channel. J. Chem. Theory Comput. 2014, 10, 451–462. [Google Scholar] [CrossRef]
- Porezag, D.; Frauenheim, T.; Kohler, T.; Seifert, G.; Kaschner, R. Construction of tight-binding-like potentials on the basis of density-functional theory: Application to carbon. Phys. Rev. B 1995, 51, 12947. [Google Scholar] [CrossRef]
- Elstner, M.; Porezag, D.; Jungnickel, G.; Elsner, J.; Haugk, M.; Frauenheim, T.; Suhai, S.; Seifert, G. Self-consistent-charge density-functional tight-binding method for simulation of complex materials properties. Phys. Rev. B 1998, 58, 7260. [Google Scholar] [CrossRef]
- Frauenheim, T.; Seifert, G.; Elstner, M.; Niehaus, T.; Köhler, C.; Amkreutz, M.; Sternberg, M.; Hajnal, Z.; Di Carlo, A.; Suhai, S. Atomistic simulations of complex materials: Ground-state and excited-state properties. J. Phys. Condens. Matter 2002, 14, 3015. [Google Scholar] [CrossRef]
- Timsorn, K.; Wongchoosuk, C. Adsorption of NO2, HCN, HCHO and CO on pristine and amine functionalized boron nitride nanotubes by self-consistent charge density functional tight-binding method. Mater. Res. Express 2020, 7, 055005. [Google Scholar] [CrossRef]
- Stock, B.G.; Bezugly, V.; Kunstmann, J.; Cuniberti, G.; Frauenheim, T.; Niehaus, T.A. SCC-DFTB parametrization for boron and boranes. J. Chem. Theory Comput. 2012, 8, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Gahrouei, M.M.; Vlastos, N.; D’Souza, R.; Odogwu, E.C.; Oliveira, L.D.S. Benchmark investigation of SCC-DFTB against standard and hybrid DFT to model electronic properties in two-dimensional MOFs for thermoelectric applications. J. Chem. Theory Comput. 2024, 20, 3976–3992. [Google Scholar] [CrossRef] [PubMed]
- Timsorn, K.; Wongchoosuk, C. Inkjet printing of room-temperature gas sensors for identification of formalin contamination in squids. J. Mater. Sci. Mater. Electron. 2019, 30, 4782–4791. [Google Scholar] [CrossRef]
- Marutaphan, A.; Seekaew, Y.; Wongchoosuk, C. Self-consistent charge density functional tight-binding study of poly(3,4-ethylenedioxythiphene): Poly(styrenesulfonate) ammonia gas sensor. Nanoscale Res. Lett. 2017, 12, 90. [Google Scholar] [CrossRef]
- Arunragsa, S.; Seekaew, Y.; Pon-on, W.; Wongchoosuk, C. Hydroxyl edge-functionalized graphene quantum dots for gas-sensing applications. Diamond Relat. Mater. 2020, 105, 107790. [Google Scholar] [CrossRef]
- Novotný, M.; Domίnguez-Gutiérrez, F.J.; Krstić, P. A computational study of hydrogen detection by borophene. J. Mater. Chem. C 2017, 5, 5426–5433. [Google Scholar] [CrossRef]
- Lukose, B.; Kuc, A.; Frenzel, J.; Heine, T. On the reticular construction concept of covalent organic frameworks. Beilstein J. Nanotechnol. 2010, 1, 60–70. [Google Scholar] [CrossRef]
- Gemming, S.; Enyashin, A.; Andrey, N.; Frenzel, J.; Seifert, G. Adsorption of nucleotides on the rutile (110) surface. Int. J. Mater. Res. 2010, 101, 758–764. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, R.; Zhang, D. Adsorption of formaldehyde molecule on the pristine and silicon-doped boron nitride nanotubes. Chem. Phys. Lett. 2008, 467, 131–135. [Google Scholar] [CrossRef]
- Zhai, H.J.; Kiran, B.; Li, J.; Wang, L.S. Hydrocarbon analogues of boron clusters-planarity aromaticity and antiaromaticity. Nat. Mater. 2003, 2, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, A.N.; Boldyrev, A.I.; Zhai, H.J.; Wang, L.S. All-boron aromatic clusters as potential new inorganic ligands and building blocks in chemistry. Coord. Chem. Rev. 2006, 250, 2811–2866. [Google Scholar] [CrossRef]
- Qin, X.; Yan, W.; Li, D.; Zhang, Z.; Chen, S. A first-principles study of gas molecule adsorption on carbon-, nitrogen-, and oxygen-doped two-dimensional borophene. Adv. Cond. Matter Phys. 2021, 2021, 3760631. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, Z.; Yan, W.; Qin, X. Effect of doping on the photoelectric properties of borophene. Adv. Cond. Matter Phys. 2021, 3718040, 7. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, Z. Effect of Co-doping on the photoelectric properties of the novel two-dimensional material borophene. Int. J. Opt. 2023, 1603014, 9. [Google Scholar] [CrossRef]
- Kistanov, A.A.; Cai, Y.; Zhou, K.; Srikanth, N.; Dmitriev, S.V.; Zhang, Y.W. Exploring the charge localization and band gap opening of borophene: A first-principles study. Nanoscale 2018, 10, 1403. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).