
Immune Response Analysis of Head Kidney in Large Yellow Croaker (Larimichthys crocea) Following Nocardia seriolae Infection



Abstract
1. Introduction
2. Materials and Methods
2.1. L. crocea and N. seriolae
2.2. Infection Experiment
2.3. Sample Collection
2.4. Total RNA Extraction, Library Construction, and Sequencing
2.5. RNA Sequencing Data Processing and Analysis
2.6. Identification and Analysis of Differentially Expressed Genes (DEGs)
2.7. Quantitative Real-Time PCR (qPCR) Analysis
3. Results
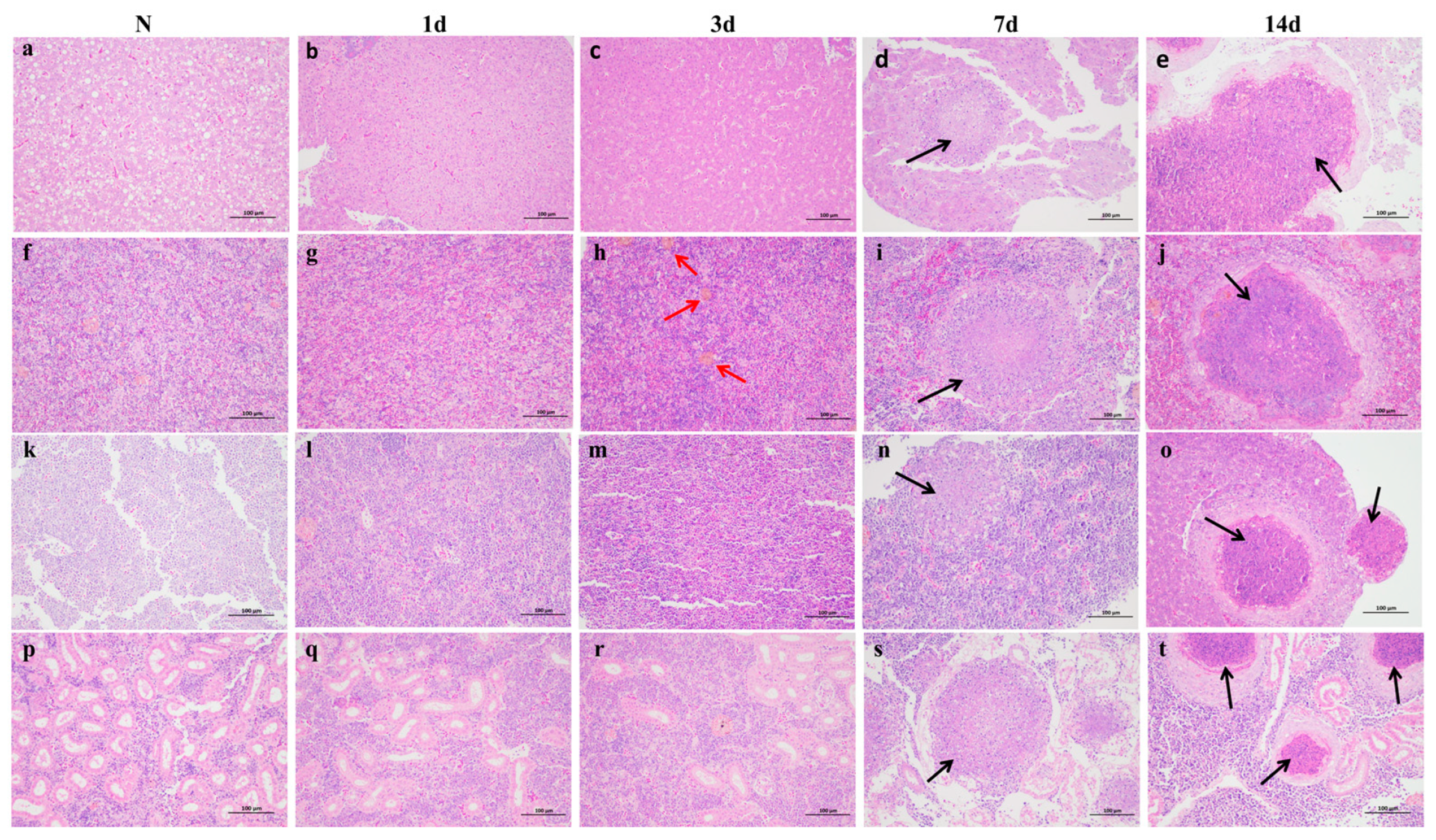
3.1. Clinical Symptoms and Histopathological Observations
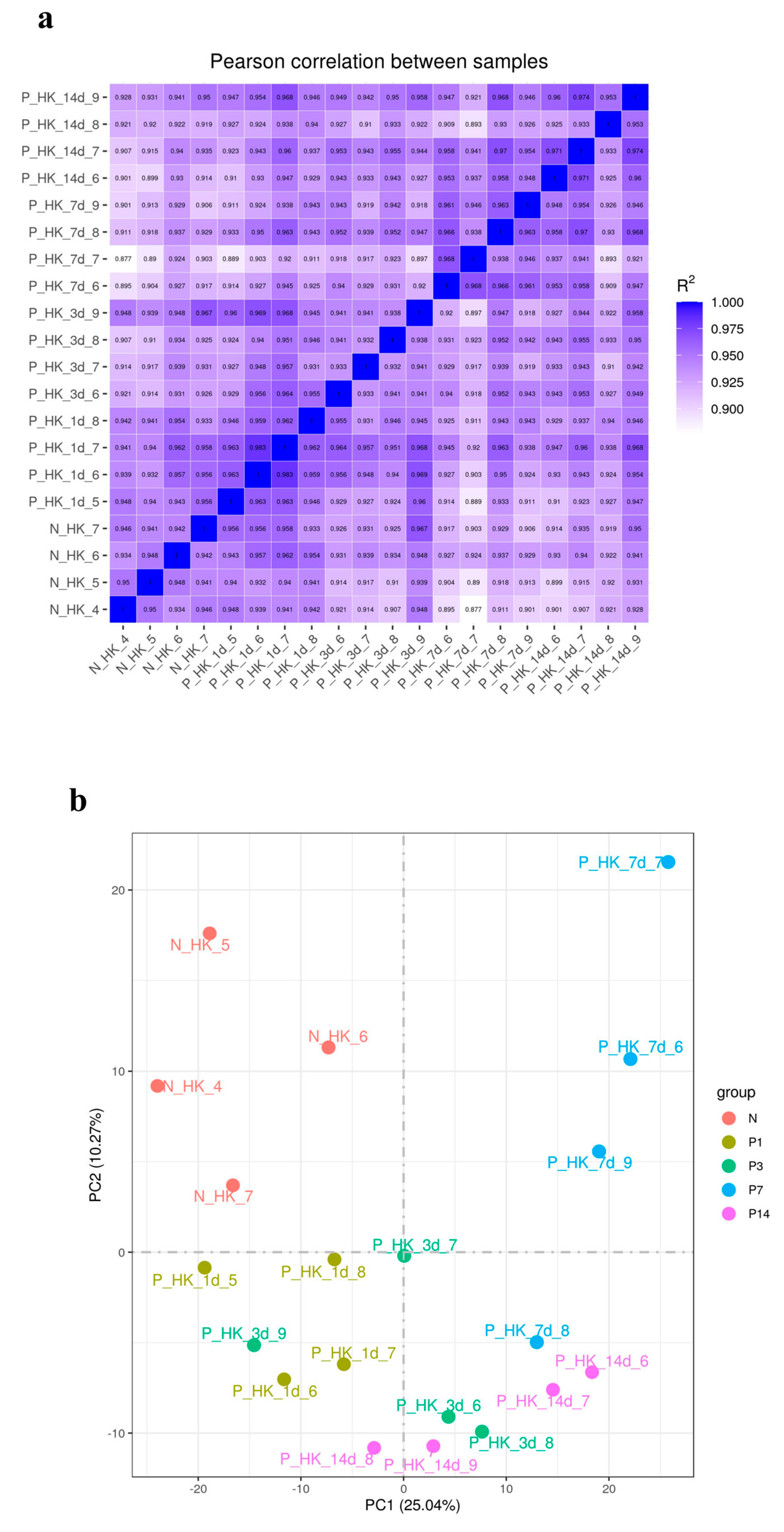
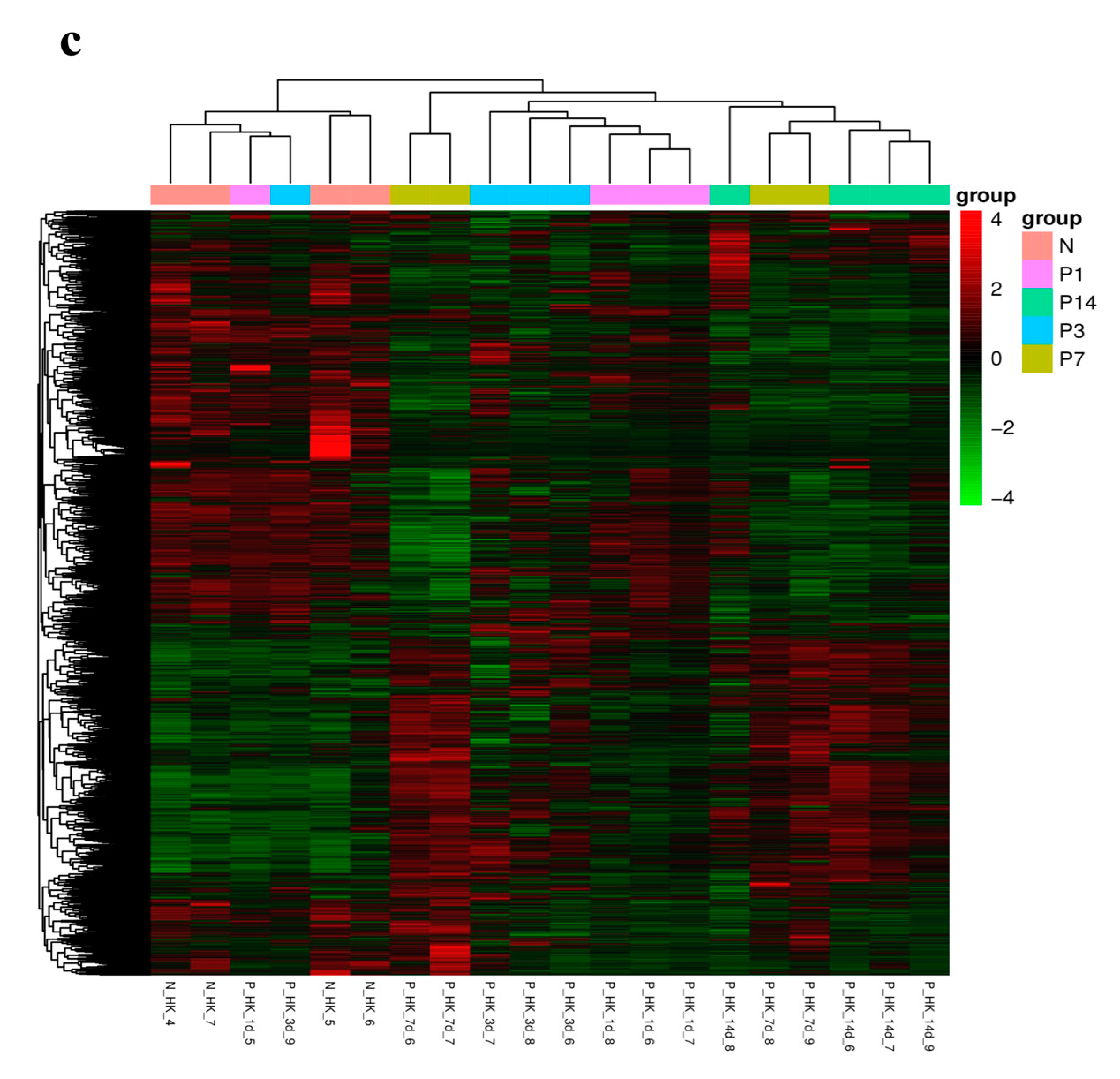
3.2. RNA Sequencing and Transcriptome Assembly
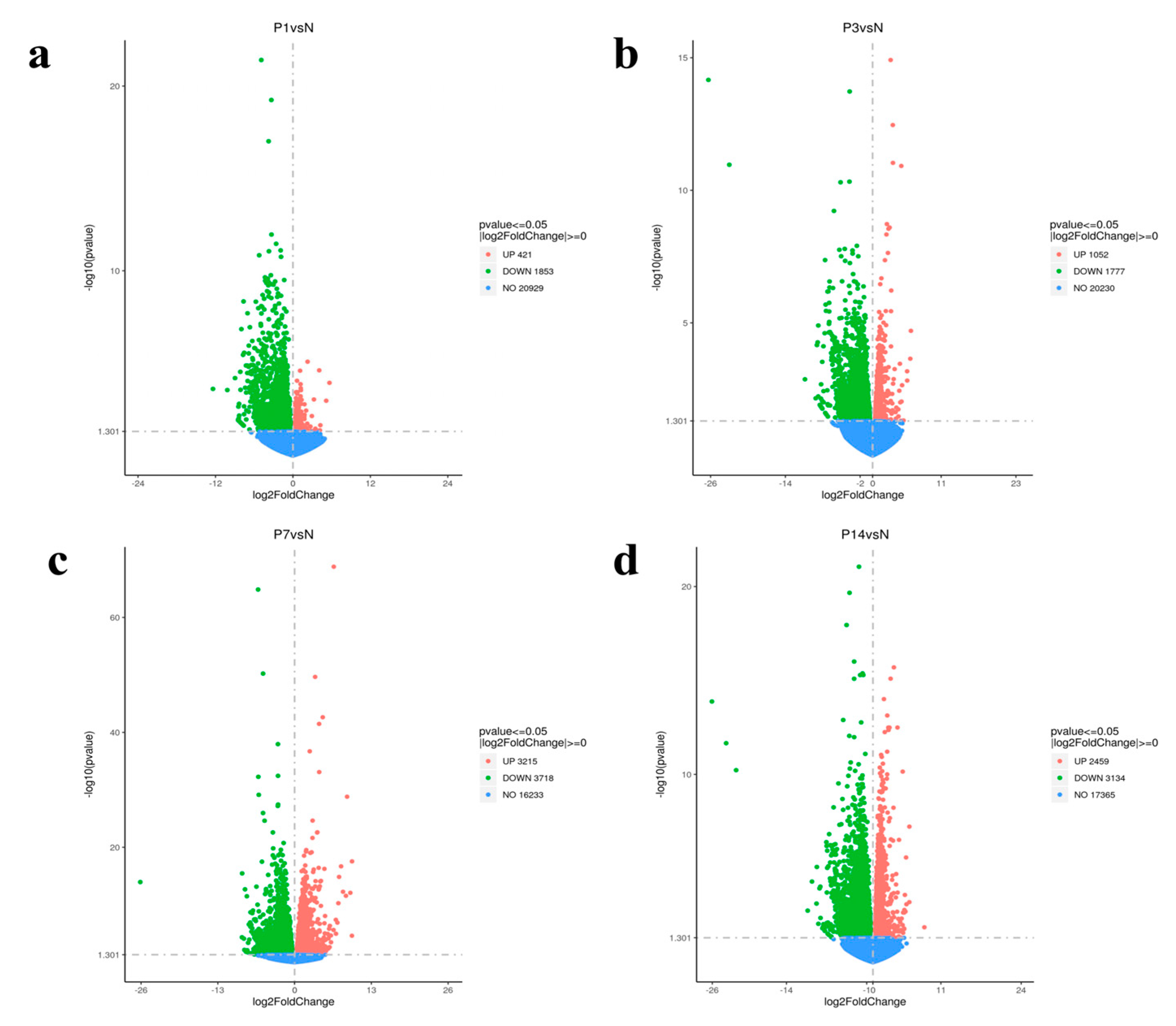
3.3. Identification of Differential Expression Gene
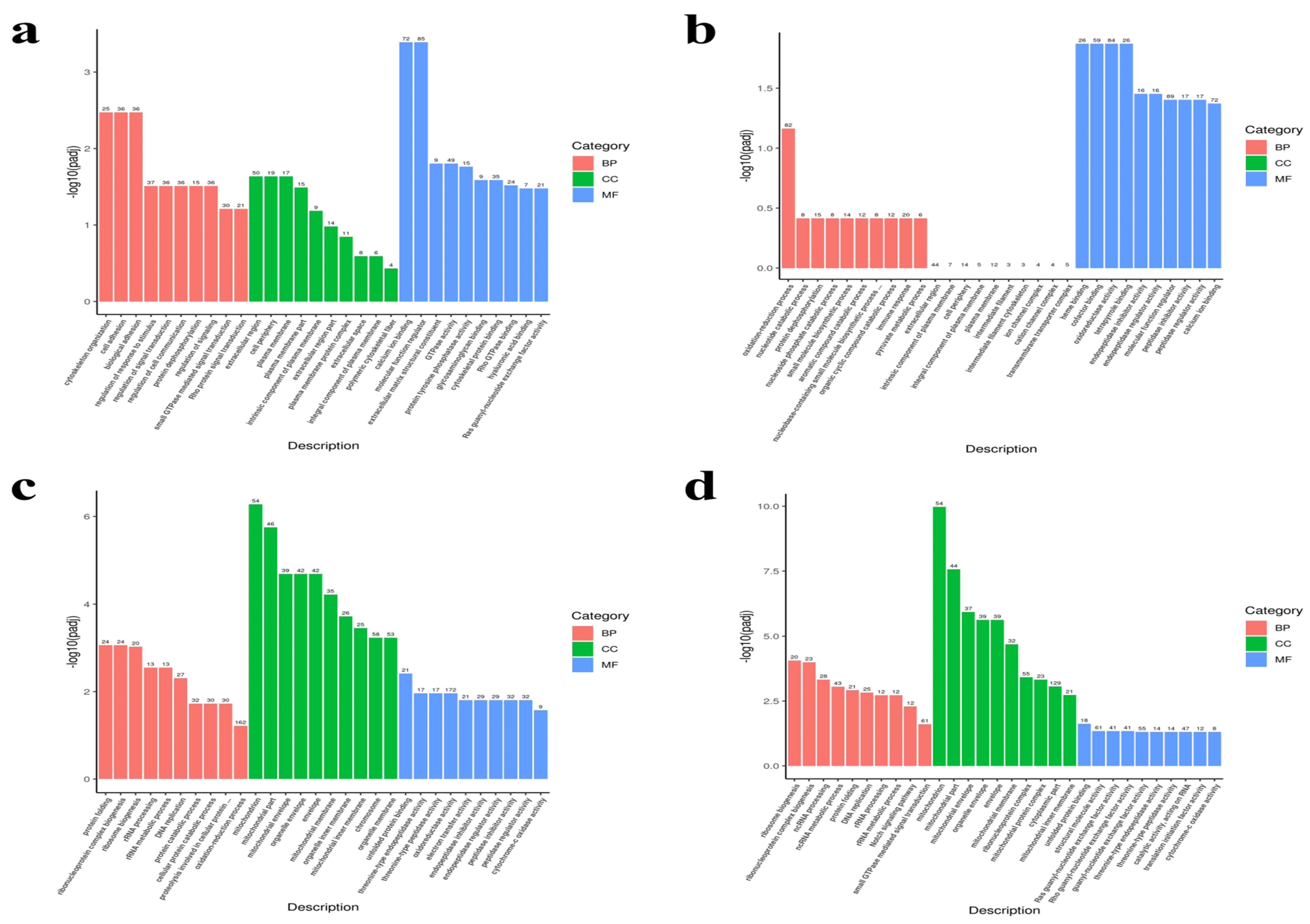
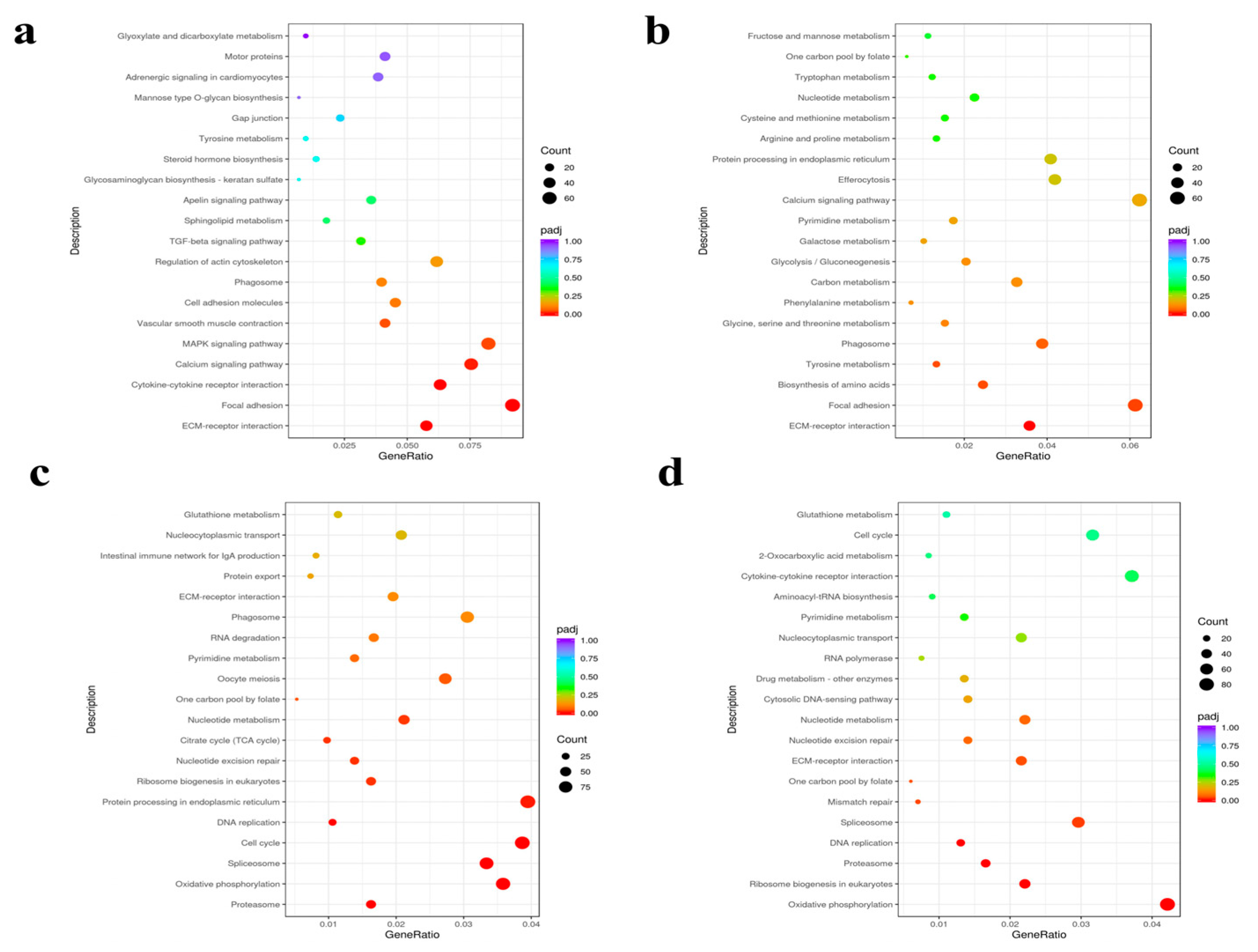
3.4. Functional Annotation and Enrichment Analysis
3.5. qPCR Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Forward Primer | Reverse Primer |
|---|---|---|
| β-actin | GTGATGGTTGGTATGGGCCA | TCGATGGGGTACTTCAGGGT |
| CDHE | GTGTGGATAAGGAGACGGGC | ACGTTCCTCAATGGTGGGTG |
| CDH5 | TGCAAACCTGCGTTTTCCTG | GCTGTGACCGCTCCTATGTT |
| TRX1 | ACTGTATGCCCACGTTCCAG | CTATGACGTGCCAGCTTTTCTT |
| LAMP | ACGATCAACACCACCTACCG | TGGAGTTACGGTTGTAGCGG |
| CXCL12 | GTCAACAACATCCCACGAAGC | GCTTGGCAATCACTTGGAAGG |
| IL7R | TGGCAACACACAGGACAACT | TAGGATGAGGGATGCTTGGC |
| CXCR3 | TCTCTTGGGCACCGTACAAC | GGTTCAGAGCACAATGCGAC |
| CARD9 | GGACGGAGTGTGTGAGGATG | TGTCGGAGGTATGGGGTGAT |
| MKK4 | GATAAGCCTGTCTGGAGTGTCC | GACGACTCGATGCTGTGTGT |
| Samples | Raw Reads | Raw Bases | Clean Reads | Clean Bases | Error Rate | Q20 | Q30 | GC |
|---|---|---|---|---|---|---|---|---|
| N_HK_4 | 45,270,980 | 6.79G | 43,075,476 | 6.46G | 0.01 | 98.09 | 95.44 | 47.84 |
| N_HK_5 | 46,974,800 | 7.05G | 44,636,102 | 6.7G | 0.01 | 98.14 | 95.57 | 47.98 |
| N_HK_6 | 40,982,186 | 6.15G | 38,848,096 | 5.83G | 0.01 | 98.03 | 95.37 | 47.55 |
| N_HK_7 | 40,274,004 | 6.04G | 38,171,012 | 5.73G | 0.01 | 98.02 | 95.28 | 48.5 |
| P_HK_1d_5 | 47,345,312 | 7.1G | 44,994,378 | 6.75G | 0.01 | 98.08 | 95.48 | 48.22 |
| P_HK_1d_6 | 45,743,588 | 6.86G | 43,437,776 | 6.52G | 0.01 | 98.02 | 95.3 | 47.81 |
| P_HK_1d_7 | 47,512,608 | 7.13G | 45,120,636 | 6.77G | 0.01 | 98.03 | 95.33 | 47.91 |
| P_HK_1d_8 | 56,976,132 | 8.55G | 54,292,304 | 8.14G | 0.01 | 97.99 | 95.32 | 46.91 |
| P_HK_3d_6 | 48,648,122 | 7.3G | 46,120,906 | 6.92G | 0.01 | 97.96 | 95.2 | 47.53 |
| P_HK_3d_7 | 46,225,332 | 6.93G | 44,490,220 | 6.67G | 0.01 | 98.07 | 95.47 | 45.84 |
| P_HK_3d_8 | 50,153,988 | 7.52G | 47,511,608 | 7.13G | 0.01 | 97.98 | 95.27 | 45.81 |
| P_HK_3d_9 | 44,439,958 | 6.67G | 42,185,068 | 6.33G | 0.01 | 98.12 | 95.55 | 48.21 |
| P_HK_7d_6 | 46,642,454 | 7G | 44,231,964 | 6.63G | 0.01 | 98.01 | 95.22 | 48.68 |
| P_HK_7d_7 | 44,570,078 | 6.69G | 42,994,850 | 6.45G | 0.01 | 97.78 | 94.6 | 47.08 |
| P_HK_7d_8 | 43,698,142 | 6.55G | 42,023,454 | 6.3G | 0.01 | 97.87 | 94.78 | 47.93 |
| P_HK_7d_9 | 48,596,398 | 7.29G | 47,487,732 | 7.12G | 0.01 | 97.51 | 93.16 | 46.78 |
| P_HK_14d_6 | 41,531,408 | 6.23G | 39,872,020 | 5.98G | 0.01 | 97.69 | 94.5 | 47.71 |
| P_HK_14d_7 | 45,741,040 | 6.86G | 44,079,956 | 6.61G | 0.01 | 97.86 | 94.78 | 48.03 |
| P_HK_14d_8 | 48,603,658 | 7.29G | 46,527,064 | 6.98G | 0.01 | 97.71 | 94.53 | 46.74 |
| P_HK_14d_9 | 50,941,480 | 7.64G | 48,972,860 | 7.35G | 0.01 | 97.88 | 94.86 | 48.18 |
References
- Chen, Y.-J.; Lin, L.-S.; Li, Y.; Zhang, J.; Song, P.-Q.; Zhang, R.; Zhong, Z.-H. Research review of fish species diversity in taiwan strait. Acta Hydrobiol. Sin. 2016, 40, 157–164. [Google Scholar]
- Yan, L.-T.; Jiang, Y.; Xu, Q.; Ding, G.-M.; Chen, X.-Y.; Liu, M. Reproductive dynamics of the large yellow croaker Larimichthys crocea (Sciaenidae), A commercially important fishery species in China. Front. Mar. Sci. 2022, 9, 154–196. [Google Scholar] [CrossRef]
- Hu, F.; Zhong, H.; Wu, C.; Wang, S.; Guo, Z.; Tao, M.; Zhang, C.; Gong, D.; Gao, X.; Tang, C.; et al. Development of fisheries in China. Reprod. Breed. 2021, 1, 64–79. [Google Scholar] [CrossRef]
- Zeng, J.; Long, F.; Wang, J.; Zhao, J.; Ke, Q.; Gong, J.; Bai, Y.; Deng, Y.; Jiang, P.; Qu, A.; et al. GWAS reveals heritable individual variations in the inherent swimming performance of juvenile large yellow croaker. Aquaculture. 2022, 559, 738419. [Google Scholar] [CrossRef]
- Beaman, B.L.; Beaman, L. Nocardia species: Host-parasite relationships. Clin. Microbiol. Rev. 1994, 7, 213–264. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, S.; Yoshida, T.; Wang, P.C.; Chen, S.C. Current knowledge of nocardiosis in teleost fish. J. Fish. Dis. 2018, 41, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Itano, T.; Kawakami, H.; Kono, T.; Sakai, M. Experimental induction of nocardiosis in yellowtail, Seriola quinqueradiata Temminck and Schlegel by artificial challenge. J. Fish Dis. 2006, 29, 529–534. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Z.; Wang, W.; Hou, S.; Cai, J.; Xia, L.; Lu, Y. Development of DNA vaccines encoding ribosomal proteins (RplL and RpsA) against Nocardia seriolae infection in fish. Fish Shellfish Immunol. 2020, 96, 201–212. [Google Scholar] [CrossRef]
- Xi, Q. RNA-Seq technology and its application in fish transcriptomics. Omics A J. Integr. Biol. 2014, 18, 98–110. [Google Scholar]
- Li, Q.; Ai, Q.; Mai, K.; Xu, W.; Zheng, Y. In vitro effects of arachidonic acid on immune functions of head kidney macrophages isolated from large yellow croaker (Larmichthys crocea). Aquaculture 2012, 330, 47–53. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, G.; Xia, L.; Lu, Y. A review on the pathogenic bacterium Nocardia seriolae: Aetiology, pathogenesis, diagnosis and vaccine development. Rev. Aquac. 2022, 15, 14–34. [Google Scholar] [CrossRef]
- Casadevall, A.; Pirofski, L.A. Host-pathogen interactions: Basic concepts of microbial commensalism, colonization, infection, and disease. Infect. Immun. 2000, 68, 6511–6518. [Google Scholar]
- Huang, X.; Liu, S.; Chen, X.; Zhang, H.; Yao, J.; Geng, Y.; Ou, Y.; Chen, D.; Yin, L.; Li, L.; et al. Comparative pathological description of nocardiosis in largemouth bass (Micropterus salmoides) and other Perciformes. Aquaculture 2021, 534, 736193. [Google Scholar] [CrossRef]
- Bransden, M.P.; Carson, J.; Munday, B.L.; Handlinger, J.H.; Carter, C.G.; Nowak, B.F. Nocardiosis in tank-reared Atlantic salmon, Salmo salar L. J. Fish Dis. 2000, 23, 83–85. [Google Scholar]
- Zhang, Y.; Hu, J.; Li, Y.; Yuan, F.; Yan, K.; Gu, W.; Zhang, M.; Li, Y.; Huang, X.; Zhang, C.; et al. Immune strategies of silver pomfret (Pampus argenteus) infected with Nocardia seriolae at different infection stages. Aquaculture 2024, 582, 740449. [Google Scholar]
- Zhou, T.; Cai, P.; Li, J.; Dan, X.; Li, Z. Pathological variations and immune response in Channa argus infected with pathogenic Nocardia seriolae strain. Fish Shellfish Immunol. 2024, 150, 109554. [Google Scholar] [PubMed]
- Itano, T.; Nakaoka, N.; Kawakami, H.; Kono, T.; Sakai, M. Comparison of Sensitivity between Yellowtail Seriola quinqueradiata and Red Sea Bream Pagrus major to Nocardia seriolae. Fish Pathol. 2006, 41, 135–139. [Google Scholar]
- Medzhitov, R.; Janeway, C., Jr. Innate immunity. N. Engl. J. Med. 2000, 343, 338–344. [Google Scholar]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar]
- Dong, Y.; Wen, H.; Zhang, Y.; Qi, X.; Wang, L.; Li, H.; Zhang, K.; Li, Y. Unveiling the molecular regulatory mechanisms of immune responses in the spleen of spotted sea bass (Lateolabrax maculatus) against Nocardia seriolae infection. Aquaculture 2024, 592, 741178. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar]
- Dinarello, C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011, 117, 3720–3732. [Google Scholar] [CrossRef]
- Rauta, P.R.; Samanta, M.; Dash, H.R.; Nayak, B.; Das, S. Toll-like receptors (TLRs) in aquatic animals: Signaling pathways, expressions and immune responses. Immunol. Lett. 2014, 158, 14–24. [Google Scholar] [PubMed]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A.E. Toll-like receptors activation, signaling, and targeting: An overview. Bull. Natl. Res. Cent. 2019, 43, 1–12. [Google Scholar]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar]
- Celhar, T.; Magalhães, R.; Fairhurst, A.M. TLR7 and TLR9 in SLE: When sensing self goes wrong. Immunol. Res. 2012, 53, 58–77. [Google Scholar] [PubMed]
- Uematsu, S.; Fujimoto, K.; Jang, M.H.; Yang, B.G.; Jung, Y.J.; Nishiyama, M.; Sato, S.; Tsujimura, T.; Yamamoto, M.; Yokota, Y.; et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat. Immunol. 2008, 9, 769–776. [Google Scholar]
- Ribeiro, C.M.; Hermsen, T.; Taverne-Thiele, A.J.; Savelkoul, H.F.; Wiegertjes, G.F. Evolution of recognition of ligands from Gram-positive bacteria: Similarities and differences in the TLR2-mediated response between mammalian vertebrates and teleost fish. J. Immunol. 2010, 184, 2355–2368. [Google Scholar]
- Tomlin, H.; Piccinini, A.M. A complex interplay between the extracellular matrix and the innate immune response to microbial pathogens. Immunology 2018, 155, 186–201. [Google Scholar]
- Bhattacharjee, O.; Ayyangar, U.; Kurbet, A.S.; Ashok, D.; Raghavan, S. Unraveling the ECM-Immune Cell Crosstalk in Skin Diseases. Front. Cell Dev. Biol. 2019, 7, 68. [Google Scholar]
- Gulati, K.; Guhathakurta, S.; Joshi, J.; Rai, N.; Ray, A.J. Cytokines and their role in health and disease: A brief overview. MOJ Immunol. 2016, 4, 00121. [Google Scholar]
- Bonecchi, R.; Galliera, E.; Borroni, E.M.; Corsi, M.M.; Locati, M.; Mantovani, A. Chemokines and chemokine receptors: An overview. Front. Biosci. 2009, 14, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kang, L.; Liu, W.; Lou, B.; Wu, C.; Jiang, L. Molecular characterization and expression analysis of the large yellow croaker (Larimichthys crocea) chemokine receptors CXCR2, CXCR3, and CXCR4 after bacterial and poly I:C challenge. Fish Shellfish Immunol. 2017, 70, 228–239. [Google Scholar] [CrossRef]
- Takamura, K.; Fukuyama, S.; Nagatake, T.; Kim, D.Y.; Kawamura, A.; Kawauchi, H.; Kiyono, H. Regulatory role of lymphoid chemokine CCL19 and CCL21 in the control of allergic rhinitis. J. Immunol. 2007, 179, 5897–5906. [Google Scholar] [CrossRef]
- Nawaz, M.; Gouife, M.; Zhu, S.; Yue, X.; Huang, K.; Ma, R.; Jiang, J.; Jin, S.; Zhu, J.; Xie, J. Transcriptome profiling and differential expression analysis of altered immune-related genes in goldfish (Carassius auratus) infected with Aeromonas hydrophila. Fish Shellfish Immunol. 2023, 137, 108789. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yu, J.; Chu, Q. Comparative transcriptome analysis reveals potential regulatory mechanisms of genes and immune pathways following Vibrio harveyi infection in red drum (Sciaenops ocellatus). Fish Shellfish Immunol. 2024, 146, 109386. [Google Scholar] [CrossRef]
- Stansberg, C.; Subramaniam, S.; Collet, B.; Secombes, C.J.; Cunningham, C. Cloning of the Atlantic salmon (Salmo salar) IL-1 receptor associated protein. Fish Shellfish Immunol. 2005, 19, 53–65. [Google Scholar] [CrossRef]
- Li, X.; chen, f.; Kong, J.; Shang, B.; li, z.; Du, Q.; Zhang, X.; Shen, X. Transcriptome analysis of Aeromonas hydrophila infected Channel catfish (Ictalurus punctatus). Res. Sq. 2024, 8, 17925. [Google Scholar]
- Brown, N.D.; Vomhof-DeKrey, E.E. Focal Adhesion Kinase and Colony Stimulating Factors: Intestinal Homeostasis and Innate Immunity Crosstalk. Cells 2024, 13, 1178. [Google Scholar] [CrossRef]
- Liu, H.; Hu, X.; Lian, Z.; Luo, Z.; Lv, A.; Tan, J. Focal adhesion signaling pathway involved in skin immune response of tongue sole Cynoglossus semilaevis to Vibrio vulnificus infection. Fish Shellfish Immunol. 2023, 135, 108651. [Google Scholar] [CrossRef]
- Esteban, M.Á. An Overview of the Immunological Defenses in Fish Skin. Int. Sch. Res. Not. 2012, 2012, 1–29. [Google Scholar]
- Ganeshan, K.; Chawla, A. Metabolic regulation of immune responses. Annu. Rev. Immunol. 2014, 32, 609–634. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Gong, H.; Ke, Q.; Li, A. Stress, antioxidant defence and mucosal immune responses of the large yellow croaker Pseudosciaena crocea challenged with Cryptocaryon irritans. Fish Shellfish Immunol. 2015, 47, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, L.; Li, C.; Shao, G.; Chen, X. Transcriptome analysis revealed multiple immune processes and energy metabolism pathways involved in the defense response of the large yellow croaker Larimichthys crocea against Pseudomonas plecoglossicida. Comp. Biochem. Physiol. Part. D Genom. Proteom. 2021, 40, 100886. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, L.; Ban, Z.; Huang, K.; Ma, R.; Zhou, S.; Jiang, J.; Fei, C.; Xie, J. Immune Response Analysis of Head Kidney in Large Yellow Croaker (Larimichthys crocea) Following Nocardia seriolae Infection. Fishes 2025, 10, 167. https://doi.org/10.3390/fishes10040167
Yuan L, Ban Z, Huang K, Ma R, Zhou S, Jiang J, Fei C, Xie J. Immune Response Analysis of Head Kidney in Large Yellow Croaker (Larimichthys crocea) Following Nocardia seriolae Infection. Fishes. 2025; 10(4):167. https://doi.org/10.3390/fishes10040167
Chicago/Turabian StyleYuan, Lu, Ziqi Ban, Kejing Huang, Rongrong Ma, Suming Zhou, Jianhu Jiang, Chenjie Fei, and Jiasong Xie. 2025. "Immune Response Analysis of Head Kidney in Large Yellow Croaker (Larimichthys crocea) Following Nocardia seriolae Infection" Fishes 10, no. 4: 167. https://doi.org/10.3390/fishes10040167
APA StyleYuan, L., Ban, Z., Huang, K., Ma, R., Zhou, S., Jiang, J., Fei, C., & Xie, J. (2025). Immune Response Analysis of Head Kidney in Large Yellow Croaker (Larimichthys crocea) Following Nocardia seriolae Infection. Fishes, 10(4), 167. https://doi.org/10.3390/fishes10040167