
The Lysine Demethylases KdmA and KdmB Differently Regulate Asexual Development, Stress Response, and Virulence in Aspergillus fumigatus



Abstract
1. Introduction
2. Materials and Methods
2.1. Bioinformatic Analysis of KdmA and KdmB
2.2. Strains and Culture Conditions
2.3. Construction of Mutant A. fumigatus Strains
2.4. Nucleic Acid Isolation and Manipulation
2.5. Physiological Experiments
2.6. Enzyme Assay and Western Blotting
2.7. Transcriptome Analysis
2.8. Murine Virulence Assay
2.9. Statistical Analysis
3. Results
3.1. Structure of KdmA and KdmB of A. fumigatus
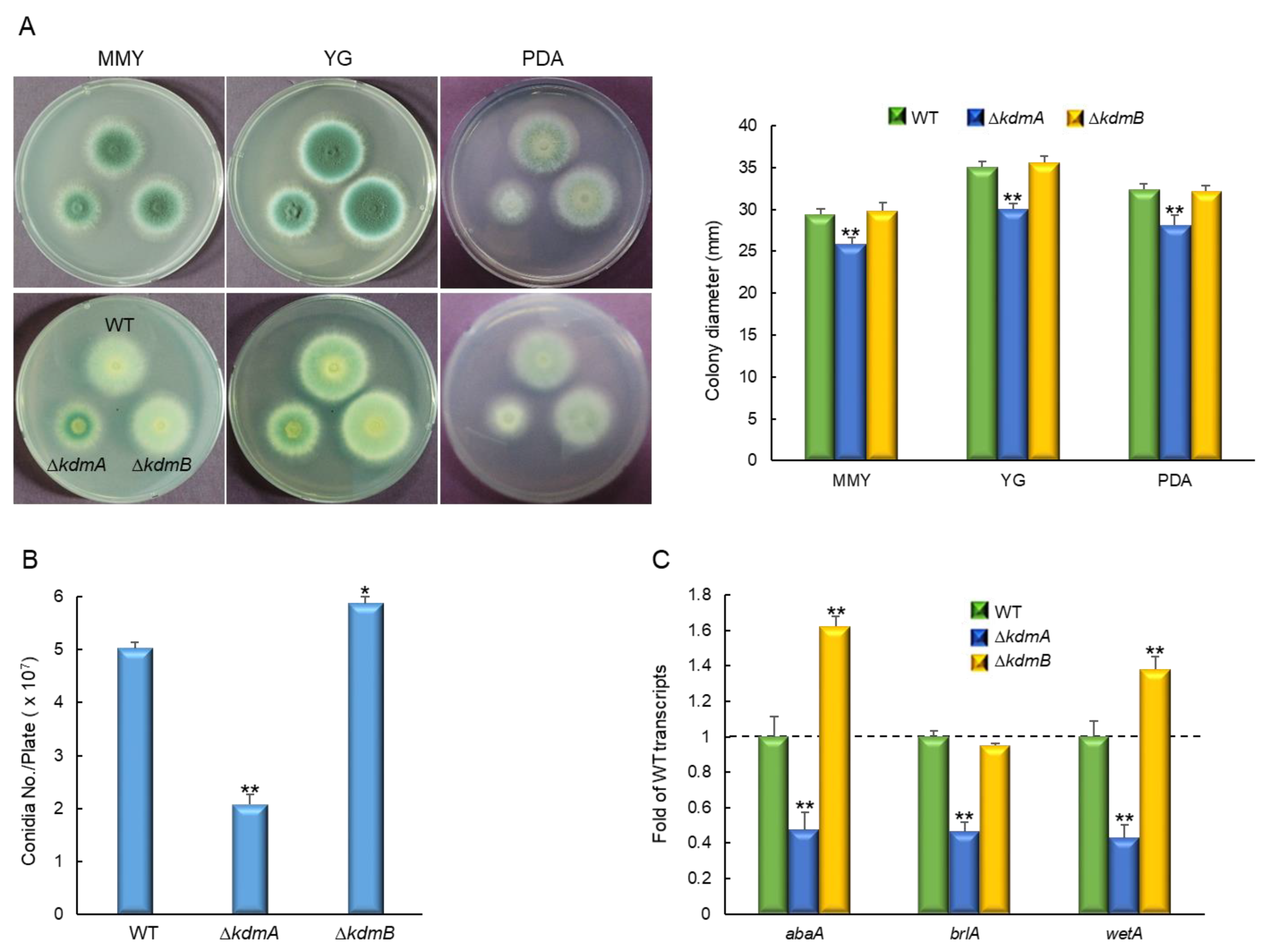
3.2. KdmA and KdmB Influence Vegetative Growth and Asexual Sporulation
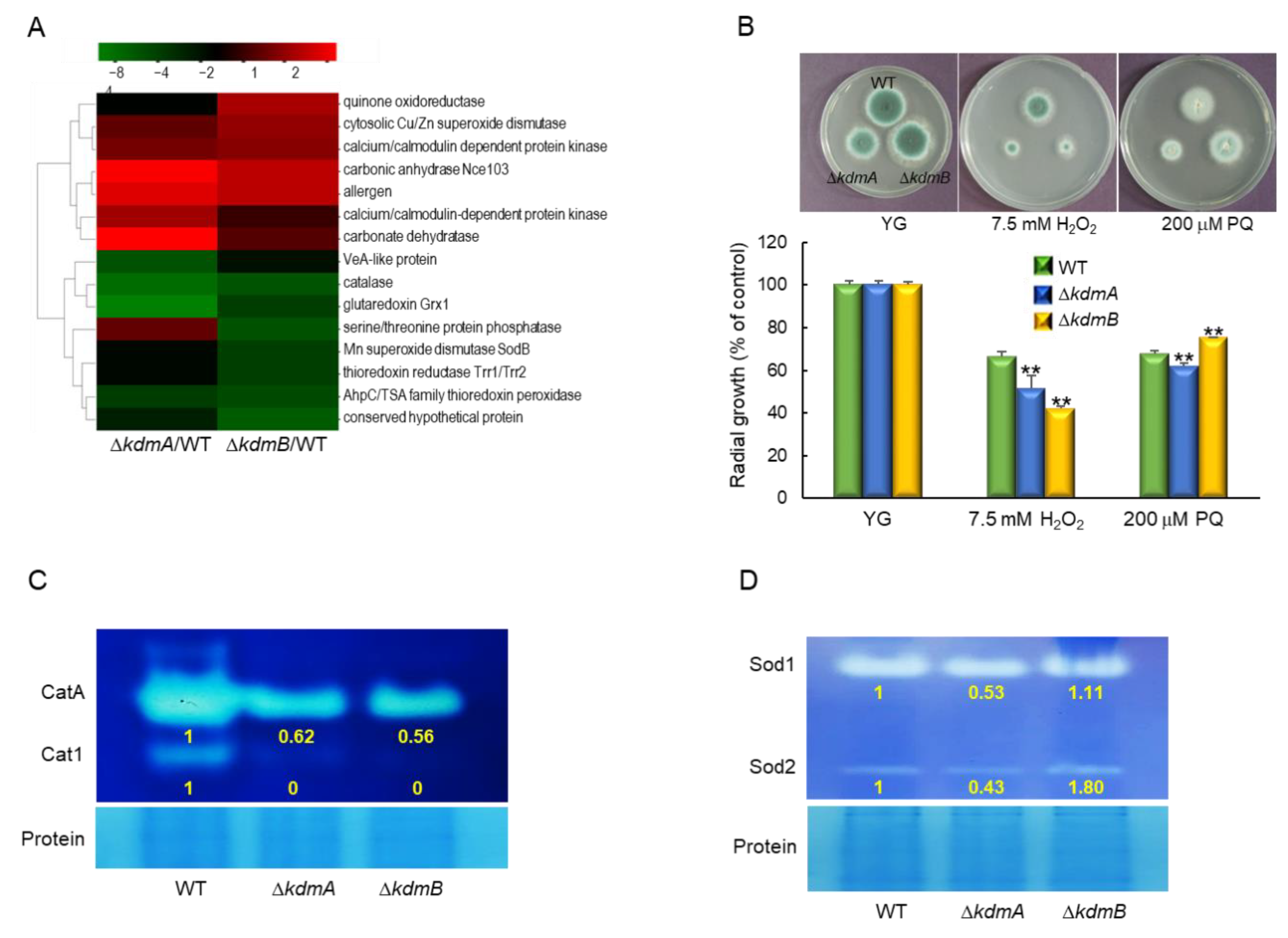
3.3. KdmA and KdmB Affect Response to Oxidative Stress
3.4. KdmA and KdmB Regulate Sensitivity to 6-Azauracil
3.5. KdmA and KdmB Oppositely Affect Gliotoxin Synthesis
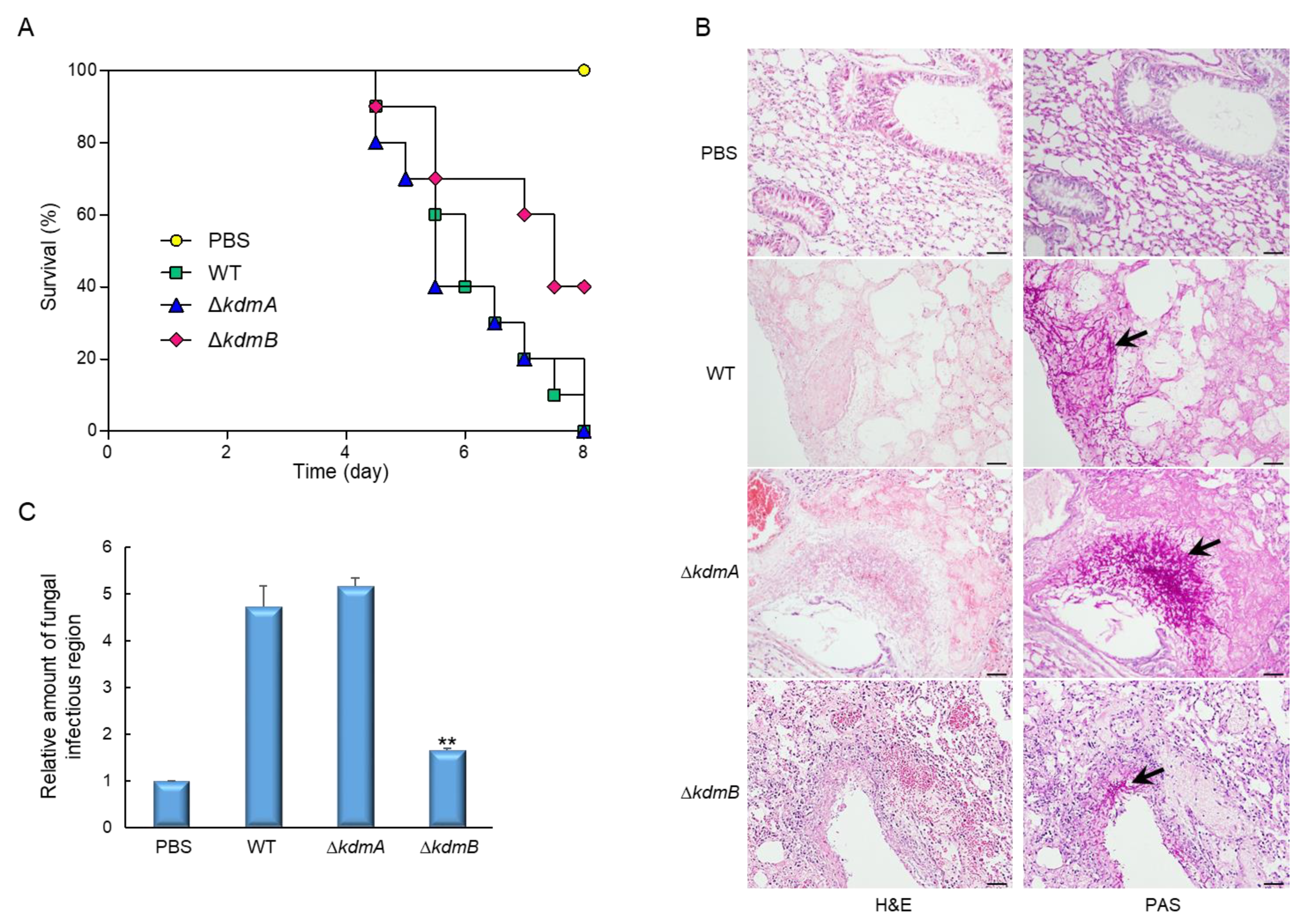
3.6. Virulence of the ΔkdmA and ΔkdmB Strains in Immunocompromised Mice
3.7. Transcriptome Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mosammaparast, N.; Shi, Y. Reversal of histone methylation: Biochemical and molecular mechanisms of histone demethylases. Annu. Rev. Biochem. 2010, 79, 155–179. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Gardner, K.E.; Allis, C.D.; Strahl, B.D. Operating on chromatin, a colorful language where context matters. J. Mol. Biol. 2011, 409, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Shwab, E.K.; Bok, J.W.; Tribus, M.; Galehr, J.; Graessle, S.; Keller, N.P. Histone deacetylase activity regulates chemical diversity in Aspergillus. Eukaryot. Cell 2007, 6, 1656–1664. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.; Reyes-Dominguez, Y. Regulation of secondary metabolism by chromatin structure and epigenetic codes. Fungal Genet. Biol. 2011, 48, 62–69. [Google Scholar] [CrossRef]
- Gacek-Matthews, A.; Noble, L.M.; Gruber, C.; Berger, H.; Sulyok, M.; Marcos, A.T.; Strauss, J.; Andrianopoulos, A. KdmA, a histone H3 demethylase with bipartite function, differentially regulates primary and secondary metabolism in Aspergillus nidulans. Mol. Microbiol. 2015, 96, 839–860. [Google Scholar] [CrossRef]
- Gacek, A.; Strauss, J. The chromatin code of fungal secondary metabolite gene clusters. Appl. Microbiol. Biotechnol. 2012, 95, 1389–1404. [Google Scholar] [CrossRef]
- Klose, R.J.; Kallin, E.M.; Zhang, Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006, 7, 715–727. [Google Scholar] [CrossRef]
- Dimitrova, E.; Turberfield, A.H.; Klose, R.J. Histone demethylases in chromatin biology and beyond. EMBO Rep. 2015, 16, 1620–1639. [Google Scholar] [CrossRef]
- Shi, Y.G.; Tsukada, Y. The discovery of histone demethylases. Cold Spring Harb. Perspect. Biol. 2013, 5, a017947. [Google Scholar] [CrossRef]
- Gacek-Matthews, A.; Berger, H.; Sasaki, T.; Wittstein, K.; Gruber, C.; Lewis, Z.A.; Strauss, J. KdmB, a Jumonji Histone H3 Demethylase, Regulates Genome-Wide H3K4 Trimethylation and Is Required for Normal Induction of Secondary Metabolism in Aspergillus nidulans. PLoS Genet. 2016, 12, e1006222. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Quan, Z.; Oliver, S.G.; Zhang, N. JmjN interacts with JmjC to ensure selective proteolysis of Gis1 by the proteasome. Microbiology 2011, 157, 2694–2701. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Chandrasekharan, M.B.; Chen, Y.C.; Bhaskara, S.; Hiebert, S.W.; Sun, Z.W. The JmjN domain of Jhd2 is important for its protein stability, and the plant homeodomain (PHD) finger mediates its chromatin association independent of H3K4 methylation. J. Biol. Chem. 2010, 285, 24548–24561. [Google Scholar] [CrossRef]
- Brookman, J.L.; Denning, D.W. Molecular genetics in Aspergillus fumigatus. Curr. Opin. Microbiol. 2000, 3, 468–474. [Google Scholar] [CrossRef]
- Kafer, E. Meiotic and mitotic recombination in Aspergillus and its chromosomal aberrations. Adv. Genet. 1977, 19, 33–131. [Google Scholar]
- Palmer, J.M.; Perrin, R.M.; Dagenais, T.R.; Keller, N.P. H3K9 methylation regulates growth and development in Aspergillus fumigatus. Eukaryot. Cell 2008, 7, 2052–2060. [Google Scholar] [CrossRef]
- Xue, T.; Nguyen, C.K.; Romans, A.; Kontoyiannis, D.P.; May, G.S. Isogenic auxotrophic mutant strains in the Aspergillus fumigatus genome reference strain AF293. Arch. Microbiol. 2004, 182, 346–353. [Google Scholar] [CrossRef]
- Yu, J.H.; Hamari, Z.; Han, K.H.; Seo, J.A.; Reyes-Dominguez, Y.; Scazzocchio, C. Double-joint PCR: A PCR-based molecular tool for gene manipulations in filamentous fungi. Fungal Genet. Biol. 2004, 41, 973–981. [Google Scholar] [CrossRef]
- Szewczyk, E.; Nayak, T.; Oakley, C.E.; Edgerton, H.; Xiong, Y.; Taheri-Talesh, N.; Osmani, S.A.; Oakley, B.R. Fusion PCR and gene targeting in Aspergillus nidulans. Nat. Protoc. 2006, 1, 3111–3120. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.S.; Kim, Y.H.; Yu, J.H. Proteomic analyses reveal the key roles of BrlA and AbaA in biogenesis of gliotoxin in Aspergillus fumigatus. Biochem. Biophys. Res. Commun. 2015, 463, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Mah, J.H.; Yu, J.H. Upstream and downstream regulation of asexual development in Aspergillus fumigatus. Eukaryot. Cell 2006, 5, 1585–1595. [Google Scholar] [CrossRef]
- Han, K.H.; Seo, J.A.; Yu, J.H. A putative G protein-coupled receptor negatively controls sexual development in Aspergillus nidulans. Mol. Microbiol. 2004, 51, 1333–1345. [Google Scholar] [CrossRef]
- Song, H.; Dang, X.; He, Y.Q.; Zhang, T.; Wang, H.Y. Selection of housekeeping genes as internal controls for quantitative RT-PCR analysis of the veined rapa whelk (Rapana venosa). PeerJ 2017, 5, e3398. [Google Scholar] [CrossRef]
- Huan, P.; Wang, H.; Liu, B. Assessment of housekeeping genes as internal references in quantitative expression analysis during early development of oyster. Genes Genet. Syst. 2017, 91, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−DDCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Choi, Y.H.; Jun, S.C.; Lee, M.W.; Yu, J.H.; Shin, K.S. Characterization of the mbsA Gene Encoding a Putative APSES Transcription Factor in Aspergillus fumigatus. Int. J. Mol. Sci. 2021, 22, 3777. [Google Scholar] [CrossRef]
- Bok, J.W.; Keller, N.P. LaeA, a regulator of secondary metabolism in Aspergillus spp. Eukaryot. Cell 2004, 3, 527–535. [Google Scholar] [CrossRef]
- Wayne, L.G.; Diaz, G.A. A double staining method for differentiating between two classes of mycobacterial catalase in polyacrylamide electrophoresis gels. Anal. Biochem. 1986, 157, 89–92. [Google Scholar] [CrossRef]
- Beauchamp, C.; Fridovich, I. Superoxide dismutase: Improved assays and an assay applicable to acrylamide gels. Anal. Biochem. 1971, 44, 276–287. [Google Scholar] [CrossRef]
- Palmer, J.M.; Keller, N.P. Secondary metabolism in fungi: Does chromosomal location matter? Curr. Opin. Microbiol. 2010, 13, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef]
- Jun, S.C.; Choi, Y.H.; Lee, M.W.; Yu, J.H.; Shin, K.S. The Putative APSES Transcription Factor RgdA Governs Growth, Development, Toxigenesis, and Virulence in Aspergillus fumigatus. Msphere 2020, 5, e00998-20. [Google Scholar] [CrossRef]
- Stolz, D.J.; Sands, E.M.; Amarsaikhan, N.; Tsoggerel, A.; Templeton, S.P. Histological Quantification to Determine Lung Fungal Burden in Experimental Aspergillosis. J. Vis. Exp. 2018, 133, e57155. [Google Scholar] [CrossRef]
- Sun, Q.; Zhou, D.X. Rice jmjC domain-containing gene JMJ706 encodes H3K9 demethylase required for floral organ development. Proc. Natl. Acad. Sci. USA 2008, 105, 13679–13684. [Google Scholar] [CrossRef]
- Ponnaluri, V.K.; Vavilala, D.T.; Putty, S.; Gutheil, W.G.; Mukherji, M. Identification of non-histone substrates for JMJD2A-C histone demethylases. Biochem. Biophys. Res. Commun. 2009, 390, 280–284. [Google Scholar] [CrossRef]
- Klose, R.J.; Gardner, K.E.; Liang, G.; Erdjument-Bromage, H.; Tempst, P.; Zhang, Y. Demethylation of histone H3K36 and H3K9 by Rph1: A vestige of an H3K9 methylation system in Saccharomyces cerevisiae? Mol. Cell Biol. 2007, 27, 3951–3961. [Google Scholar] [CrossRef]
- Kim, T.; Buratowski, S. Two Saccharomyces cerevisiae JmjC domain proteins demethylate histone H3 Lys36 in transcribed regions to promote elongation. J. Biol. Chem. 2007, 282, 20827–20835. [Google Scholar] [CrossRef] [PubMed]
- Cloos, P.A.; Christensen, J.; Agger, K.; Maiolica, A.; Rappsilber, J.; Antal, T.; Hansen, K.H.; Helin, K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 2006, 442, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Wu, J.; Tong, X.J.; Zhou, J.Q.; Ding, J. Crystal structure of the catalytic core of Saccharomyces cerevesiae histone demethylase Rph1: Insights into the substrate specificity and catalytic mechanism. Biochem. J. 2011, 433, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Lambou, K.; Lamarre, C.; Beau, R.; Dufour, N.; Latge, J.P. Functional analysis of the superoxide dismutase family in Aspergillus fumigatus. Mol. Microbiol. 2010, 75, 910–923. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Sekimizu, K. SDT1/SSM1, a multicopy suppressor of S-II null mutant, encodes a novel pyrimidine 5′-nucleotidase. J. Biol. Chem. 2002, 277, 22103–22106. [Google Scholar] [CrossRef]
- Escobar-Henriques, M.; Daignan-Fornier, B. Transcriptional regulation of the yeast GMP synthesis pathway by its end products. J. Biol. Chem. 2001, 276, 1523–1530. [Google Scholar] [CrossRef]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef]
- South, P.F.; Harmeyer, K.M.; Serratore, N.D.; Briggs, S.D. H3K4 methyltransferase Set1 is involved in maintenance of ergosterol homeostasis and resistance to Brefeldin A. Proc. Natl. Acad. Sci. USA 2013, 110, E1016–E1025. [Google Scholar] [CrossRef]
- Serratore, N.D.; Baker, K.M.; Macadlo, L.A.; Gress, A.R.; Powers, B.L.; Atallah, N.; Westerhouse, K.M.; Hall, M.C.; Weake, V.M.; Briggs, S.D. A Novel Sterol-Signaling Pathway Governs Azole Antifungal Drug Resistance and Hypoxic Gene Repression in Saccharomyces cerevisiae. Genetics 2018, 208, 1037–1055. [Google Scholar] [CrossRef]
- Verweij, P.E.; Te Dorsthorst, D.T.; Janssen, W.H.; Meis, J.F.; Mouton, J.W. In vitro activities at pH 5.0 and pH 7.0 and in vivo efficacy of flucytosine against Aspergillus fumigatus. Antimicrob. Agents Chemother. 2008, 52, 4483–4485. [Google Scholar] [CrossRef]
- Shaw, R.J.; Reines, D. Saccharomyces cerevisiae transcription elongation mutants are defective in PUR5 induction in response to nucleotide depletion. Mol. Cell Biol. 2000, 20, 7427–7437. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Nakano, A.; Nomura, K.; Sekimizu, K.; Natori, S. Purification, gene cloning, and gene disruption of the transcription elongation factor S-II in Saccharomyces cerevisiae. J. Biol. Chem. 1992, 267, 13200–13204. [Google Scholar] [CrossRef]
- Exinger, F.; Lacroute, F. 6-Azauracil inhibition of GTP biosynthesis in Saccharomyces cerevisiae. Curr. Genet. 1992, 22, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Howe, L.; Anderson, S.; Yates, J.R., 3rd; Workman, J.L. The Set2 histone methyltransferase functions through the phosphorylated carboxyl-terminal domain of RNA polymerase II. J. Biol. Chem. 2003, 278, 8897–8903. [Google Scholar] [CrossRef] [PubMed]
- Krogan, N.J.; Kim, M.; Tong, A.; Golshani, A.; Cagney, G.; Canadien, V.; Richards, D.P.; Beattie, B.K.; Emili, A.; Boone, C.; et al. Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol. Cell Biol. 2003, 23, 4207–4218. [Google Scholar] [CrossRef]
- De Castro, P.A.; Colabardini, A.C.; Moraes, M.; Horta, M.A.C.; Knowles, S.L.; Raja, H.A.; Oberlies, N.H.; Koyama, Y.; Ogawa, M.; Gomi, K.; et al. Regulation of gliotoxin biosynthesis and protection in Aspergillus species. PLoS Genet. 2022, 18, e1009965. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Genotype | Reference |
|---|---|---|
| A. nidulans FGSC4 | veA+ (Wild type) | FGSC a |
| Af293 | Wild type | [16] |
| Af293.1 | pyrG1 | [19] |
| ΔkdmA | ΔkdmA::AnipyrG; AfupyrG1 | This study |
| ΔkdmB | ΔkdmB::AnipyrG; AfupyrG1 | This study |
| C’ kdmA | ΔkdmA::AnipyrG; AfupyrG1; kdmA::hygB | This study |
| C’ kdmB | ΔkdmB::AnipyrG; AfupyrG1; kdmB::hygB | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, Y.-H.; Lee, M.-W.; Shin, K.-S. The Lysine Demethylases KdmA and KdmB Differently Regulate Asexual Development, Stress Response, and Virulence in Aspergillus fumigatus. J. Fungi 2022, 8, 590. https://doi.org/10.3390/jof8060590
Choi Y-H, Lee M-W, Shin K-S. The Lysine Demethylases KdmA and KdmB Differently Regulate Asexual Development, Stress Response, and Virulence in Aspergillus fumigatus. Journal of Fungi. 2022; 8(6):590. https://doi.org/10.3390/jof8060590
Chicago/Turabian StyleChoi, Yong-Ho, Min-Woo Lee, and Kwang-Soo Shin. 2022. "The Lysine Demethylases KdmA and KdmB Differently Regulate Asexual Development, Stress Response, and Virulence in Aspergillus fumigatus" Journal of Fungi 8, no. 6: 590. https://doi.org/10.3390/jof8060590
APA StyleChoi, Y.-H., Lee, M.-W., & Shin, K.-S. (2022). The Lysine Demethylases KdmA and KdmB Differently Regulate Asexual Development, Stress Response, and Virulence in Aspergillus fumigatus. Journal of Fungi, 8(6), 590. https://doi.org/10.3390/jof8060590