Abstract
Candida albicans infections range from superficial to systemic and are one of the leading causes of fungus-associated nosocomial infections. The innate immune responses during these various infection types differ, suggesting that the host environment plays a key role in modulating the host–pathogen interaction. In addition, C. albicans is able to remodel its cell wall in response to environmental conditions to evade host clearance mechanisms and establish infection in niches, such as the oral and vaginal mucosa. Phagocytes play a key role in clearing C. albicans, which is primarily mediated by Pathogen Associated Molecular Pattern (PAMP)–Pattern Recognition Receptor (PRR) interactions. PRRs such as Dectin-1, DC-SIGN, and TLR2 and TLR4 interact with PAMPs such as β-glucans, N-mannan and O-mannan, respectively, to trigger the activation of innate immune cells. Innate immune cells exhibit distinct yet overlapping repertoires of PAMPs, resulting in the preferential recognition of particular Candida morphotypes by them. The role of phagocytes in the context of individual infection types also differs, with neutrophils playing a prominent role in kidney infections, and dendritic cells playing a prominent role in skin infections. In this review, we provide an overview of the key receptors involved in the detection of C. albicans and discuss the differential innate immune responses to C. albicans seen in different infection types such as vulvovaginal candidiasis (VVC) and oral candidiasis.
1. Introduction
Candida albicans is both a commensal and opportunistic fungal pathogen of humans, the delicate balance of which is maintained by the actions of the innate immune system and resident microbiota. C. albicans is able to colonise most areas of the body, including the nails, skin, mucosal membranes, and internal organs. The colonisation of the skin and gastrointestinal tract is thought to occur at birth when the infant passes through the birth canal, where C. albicans is a natural resident [1]. During periods of immune suppression, such as in those with HIV, neutropenia, or dysbiosis of the microbiota, C. albicans can overcome the innate immune system to establish various infections, such as superficial mucosal infections (oral and genital thrush), subcutaneous infections and systemic disease (candidemia). Although mucosal infections are not life-threatening, they pose high morbidity rates and are expensive to treat, while candidemia is associated with a poor prognosis and high mortality (40–50%) [2]. C. albicans is armed with a plethora of virulence factors including adhesins (i.e., Hwp1, Als3, and Als5), secreted hydrolytic enzymes (i.e., SAPs and phospholipases) and the recently identified cytolytic toxin candidalysin [3,4]. C. albicans can also form biofilms in nail infections; on teeth exacerbating, tooth decay; on mucosal surfaces; and on the surfaces of medical devices, including catheters, long lines and voice prostheses [5,6]. This capacity to form biofilms has made C. albicans one of the leading causes of nosocomial infections, which are difficult to treat due to increasing antifungal resistance.
Despite the causal agent being the same in each infection, the innate immune responses elicited during these infections are very different, suggesting that the host environment plays a key role in modulating the host–pathogen interaction. The majority of innate immune responses are mediated by the interaction between Pattern Recognition Receptors (PRRs) expressed on the surface of innate immune cells, and Pathogen Associated Molecular Patterns (PAMPs) on the fungal surface. The Candida cell wall consists of an inner skeletal layer of β-glucan and chitin, and an outer layer of glycosylated proteins [7,8]. These proteins play essential roles in adhesion to host cells, while carbohydrate PAMPs are recognised by PRRs on the surface of innate immune cells. The main families of receptors involved in the recognition of C. albicans are the C-type lectin receptors (CLRs), RIG I-like receptors (RLRs), NOD-like receptors (NLRs) and Toll-like receptors (TLRs), which recognise distinct PAMPs (Table 1). Each innate immune cell has a distinct, but overlapping, repertoire of receptors, giving rise to its preferential recognition of certain morphotypes. The response elicited against a microorganism is governed by the mosaic of receptor complexes engaged. In this review, we provide an overview of the key receptors involved in the detection of C. albicans, summarise the immune sensing of C. albicans occurring in different infection types, and discuss the potential role the host environment plays in modulating these responses.

Table 1.
Pattern Recognition Receptors (PRRs) involved in the recognition of C. albicans.
2. Recognition by Phagocytes
The successful clearance of C. albicans from the host tissue relies heavily upon the phagocytosis of the fungal pathogen by innate immune cells (i.e., macrophages, neutrophils and dendritic cells). Phagocytosis is a multi-step process initiated by pathogen recognition. This leads to the engulfment of the pathogen, phagosome maturation to eliminate the pathogen and then the resolution of the phagolysosome, and this has been extensively reviewed by Levin et al. [26]. Innate immune cells interact differently with the yeast and hyphal morphologies of Candida [27]. This is partly due to the differential exposure of β-glucan, with β-glucan exposure at yeast bud scars provoking Dectin-1-dependent proinflammatory innate immune responses [11]. Hyphae generally have less β-glucan exposed on their cell surface and, therefore, are less effectively recognized by Dectin-1 [11]. However, activated neutrophils can expose β-glucan in hyphal cells, leading to Dectin-1-dependent recognition and proinflammatory cytokine secretion [28]. Furthermore, the hyphal orientation during contact with phagocytic cells also impacts phagocytosis, with macrophages encountering hypha tips phagocytosing the Candida quicker [29]. Therefore, there are multiple factors that contribute to how macrophages interact with Candida. Dendritic cells (DCs) also discriminate between the different morphologies, and this differentially stimulates adaptive immunity, with yeast cells provoking a Th1 response, while hyphae initiate a Th2 response [30].
Neutrophils are the major immune cells in the control of candidiasis, and thus, neutropenic individuals and mice have an increased susceptibility to candidiasis [31,32,33,34]. The C-type lectin receptor Dectin-1 is the most important neutrophil PRR in C. albicans recognition. The engagement of Dectin-1 leads to the activation of integrin Mac-1 (CR3), and both receptors are essential for fungal engulfment and elimination [12]. Macrophages also play a key role in phagocytosis and Candida clearance in various niches, while dendritic cells specialise in processing and presenting antigenic material from Candida to lymphocytes, acting as a bridge between the innate and acquired immune systems [35]. Like neutrophils, these innate immune cells recognise C. albicans by its cell wall PAMPs; β-1,3-glucan is recognised by the C-type lectin receptor Dectin-1 and complement receptor 3 (CR3); N-mannan, by Dectin-2, DC-SIGN and MINCLE; and O-mannan, by TLR4 (Table 1 and Figure 1) [14,36,37,38,39].
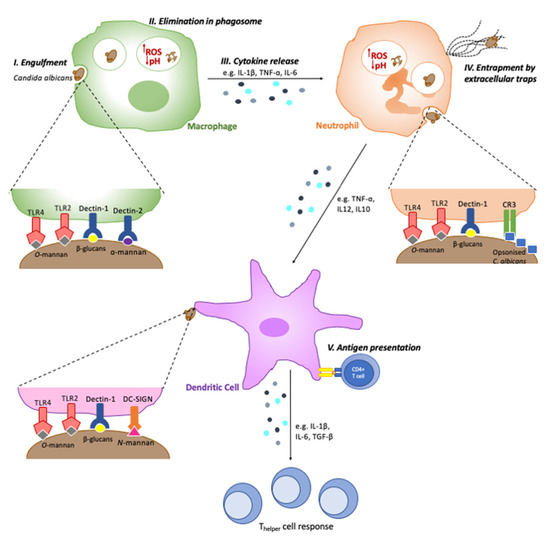
Figure 1.
Key response pathways activated by innate immune cells against Candida albicans. Following successful recognition of Candida PAMPs (such as O-mannan, α-mannan, N-mannan and β-glucans) by immune cell PRRs (such as TLR2, TLR4, Dectin-1 and DC-SIGN), immune cells use a number of response mechanisms to deal with Candida infections. These include (I) engulfment and (II) elimination of the pathogen within the phagosome, where it encounters acidic pH and increased reactive oxygen species (ROS) production; (III) cytokine release, aiding immune cell recruitment, and exerting direct antimicrobial effects; (IV) entrapment and elimination of pathogens by releasing extracellular traps comprised of DNA with antimicrobial agents; (V) antigen presentation, activating the adaptive immune system by presenting antigenic peptides to T-cells on their major histocompatibility complex (MHC) molecules.
The recognition of these PAMPs initiates phagocytosis, which involves two main stages: (i) phagosome formation and (ii) maturation. Phagosomes are formed when phagocytic receptors such as CR3, Fcγ receptors (FcγRs) and Dectin-1 bind to phagocytic targets and result in receptor clustering. Subsequent to receptor clustering, the phosphorylation of their immunoreceptor tyrosine-based activation motifs (ITAMs) and the activation of Rho-family GTPases occur. Rho-GTPases activate nucleation-promoting factors (NPFs), which cause actin-driven protrusions from the plasma membrane to advance around the target [40,41]. The activation of myosin by cytosolic Ca2+ has been suggested to be necessary in phagocytic cup formation and sealing, to form the phagosome [26,42].
Phagosome maturation is a gradual process whereby the phagosome is converted into a hostile and degradative environment to achieve pathogen elimination. The process differs between neutrophils and macrophages, which has been reviewed in detail [26,43]. Briefly, in neutrophils, phagosome maturation occurs rapidly and involves fusion with lysosomes and neutrophil granules containing antimicrobial peptides and proteolytic enzymes [43,44,45]. In macrophages, endosomal fusion begins early during phagosome maturation and is driven by the recruitment of active Rab5 and then Rab7 as they transition into late phagosomes. Eventually, lysosomes fuse with late phagosomes to form a phagolysosome, which is armed with lytic enzymes and is highly acidic (i.e., in M2 macrophages, which are involved in tissue repair and the resolution of inflammation) and oxidative, to provide an extremely degradative environment for pathogens [26,46,47,48]. Notably, neutrophils and M1 macrophages (proinflammatory macrophages)—the cells mainly responsible for pathogen elimination—maintain a slightly alkaline compartment during this transition. These phenomena are attributed to the increased proton consumption during superoxide dismutation, reduced granule fusion observed during reactive oxygen species (ROS) production, and increased leakiness for H+ caused by the oxidative effect of ROS [49,50]. The sustained alkalinization of the phagosomal compartment is also true for dendritic cells. Dendritic cells prioritise antigen presentation and, therefore, must regulate their phagosomal contents for partial degradation [51]. To achieve this, ROS production is maintained at low levels for sustained periods by the recruitment of NADPH oxidase, NOX2, to phagosomes shortly after phagocytosis [51].
ROS generation is initiated by NADPH activation in neutrophils and potentiates Candida killing within phagosomes [39,52,53]. In neutrophils, NADPH is produced by two separate, glucose-dependent pathways: glycolysis and the pentose phosphate pathway [54]. Kidney patients with reduced expression of a glucose transporter, GLUT1, and patients with chronic granulomatous disease have increased susceptibility to candidiasis, exemplifying the importance of NADPH oxidase [55]. In macrophages, the engagement of Dectin-1 causes the phosphorylation of the ITAM-like motif in its cytoplasmic tail. This leads to the subsequent recruitment of the tyrosine kinase, Syk, and the activation of NF- ϰB, which is essential for proinflammatory cytokine production, phagosome acidification and ROS generation [56,57,58,59]. Together, these stresses eliminate and dispose of C. albicans. However, achieving this end goal is made difficult by the morphological change that occurs in C. albicans as they transition from yeast to hyphae. This imposes mechanical stress on the phagosome and may lead to phagosomal rupture, although other factors such as candidalysin release have also been implicated in phagosomal rupture and escape [60]. To circumvent this problem, macrophages increase lysosome production upon the recognition and engulfment of Candida. These lysosomes are increasingly fused to the phagolysosomes to increase their surface area as the hyphae cause the distention of the phagolysosomal membrane, causing transient releases of Ca2+ into the cytosol. This ensures Candida remains confined within intact phagolysosomes long enough to effect killing [60].
The recognition of Candida by macrophages, neutrophils and dendritic cells, alike, results in the release of inflammatory and chemotactic cytokines, aiding further immune cell recruitment and infection resolution (Figure 1). Candida exposure results in the macrophage release of proinflammatory cytokines such as TNF-α, IL-1β and IL-6, enhancing Candida clearance and neutrophil recruitment [61,62,63,64]. Neutrophils play a key role in Candida clearance, by releasing proinflammatory cytokines such as TNF-α and IL-12, through the formation of neutrophil extracellular traps (NETs) and enhance dendritic cell recruitment and activation [65]. The release of IL-6 and IL-23 by dendritic cells augments the Thelper cell response to Candida infection, including the key Th17 response [66] (Figure 1).
3. Niche-Specific Immune Sensing of C. albicans
3.1. Skin and Nail Infections
Warm and moist environments promote the expansion of C. albicans on the skin, leading to superficial skin infections, such as nappy rash, athlete’s foot and nail infections. C. albicans uses hydrolytic enzymes, such as secreted aspartic proteinases, shown to be important in tissue invasion and believed to be involved in establishing nail and other superficial infections [67,68,69]. Such infections are potentiated by increased phospholipase activity, which also correlates with biofilm formation, particularly in nail infections [67,69]. Although little is known about immune sensing in the skin and nails, the presence of DCs in the skin suggests their involvement in C. albicans sensing and the immune response in this context. Skin-derived DCs from foetal mouse skin phagocytose both yeast and hyphal forms of Candida [30]. The phagocytosis of the different morphotypes induces differential cytokine profiles, influencing the T-helper activation pathways. Yeast cells induce the production of IL-12 and activation of the protective Th1 pathway, while hyphae induce IL-4 production and Th2 activation [30,35]. Furthermore, the activation of the Th17 pathway in the skin requires the secretion of IL-1β and IL-6 [70]. This illustrates the discriminatory ability of DCs in sensing and responding to the different morphotypes of Candida (Table 2).
Table 2.
Summary of immune sensing and responses at different infection sites.
3.2. Oral Candidiasis
C. albicans is a commensal of the oral mucosa, but during periods of immune suppression, the balance between commensalism and pathogenesis is lost, leading to Candida overgrowth and infection. These infections are commonly associated with biofilm formation, especially in patients with dentures, voice protheses and xerostomia (reduced or absent saliva flow). Biofilm formation makes these infections difficult to treat because they are generally polymicrobial in nature, and the production of extracellular matrix increases the resistance to antimicrobial therapy [5,6,84]. Oral candidiasis is prevalent in immunosuppressed individuals such as HIV patients, mouth and throat cancer patients, and patients taking oral steroids making them highly susceptible to infection. Although the surveillance for oral candidiasis is limited, it is estimated that 20–50% of HIV patients develop oral candidiasis [85,86]. The prevalence of oral candidiasis in HIV/AIDS patients suggests that CD4+ cells play an essential role in combating oral colonisation, which is in contrast to the case for other Candida infections.
The immune sensing of Candida at the oral mucosa is initiated by the oral epithelial cells, by a biphasic MAPK response, which has been extensively reviewed elsewhere [87,88,89,90]. Briefly, low levels of Candida colonisation on the oral mucosa activate NF-ϰB signalling, Pi3K signalling and the three major MAPK pathways (p38, JNK and ERK1/2), through the recognition of components of the fungal cell wall (i.e., β-glucan and mannans) by traditional PRRs (i.e., TLR2, TLR4, EphA2 and Dectin-1) [91,92,93] as outlined in Table 1. However, this low-level activation does not result in a significant proinflammatory innate immune response, indicating that the recognition of cell wall PAMPs is not sufficient to fully activate oral epithelial cells. Instead, the induction of mucosal immunity requires the overgrowth of Candida on the oral mucosa coupled with hyphal formation. This increase in fungal burden on the oral mucosa activates the second MAPK response involving MEK1/2-ERK1/2, leading to the activation of c-Fos and MPK1—the “danger response” (Figure 2, Table 2). This strong activation of MAPK signalling results in the production of antimicrobial peptides, the secretion of proinflammatory cytokines, and subsequent phagocyte recruitment [91]. The activation of this second MAPK response is dependent on the secretion of the peptide toxin candidalysin [3,94], suggesting that epithelial cells primarily respond to damage rather than traditional PAMP–PRR interactions.
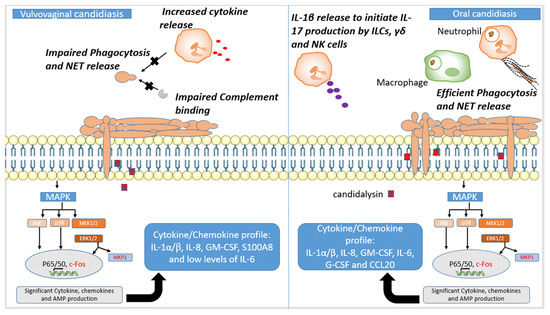
Figure 2.
Host mucosal responses to C. albicans. Both vaginal and oral epithelial cells respond to C. albicans in a biphasic way. During colonisation, C. albicans only induces a mild response through MAPK signalling, and this does not result in cytokine release. However, a high burden of C. albicans coupled with hyphal growth and candidalysin release activates the second stage of the biphasic MAPK pathway, which involves activation of transcription factor c-Fos and MAPK phosphatase (MKP1), both depicted in red in this figure. MEK1/2 activates ERK1/2, which is the predominant regulator of MKP1. The activation of c-Fos, however, is predominantly regulated by p38. The ensuing cytokine profiles are distinct between the different sites as indicated in the boxes. This leads to immune cell recruitment to the mucosa. While this leads to the effective phagocytosis of Candida yeast and NET release to contain and kill hyphae in oral candidiasis, neutrophil recruitment causes the aggravation of disease in vulvovaginal candidiasis (VVC). In VVC, neutrophil phagocytosis is impaired, and this is partly due to reduced complement binding to Candida. This culminates in increased cytokine release, causing heightened nonprotective inflammation. In contrast to in VVC, innate lymphoid cells, γδ cells and natural killer cells play a vital role in Candida control by producing IL-17, known to potentiate the proinflammatory response of epithelial cells.
The recruited macrophages and neutrophils function to clear C. albicans through PAMP–PRR-directed phagocytosis, as detailed above, with NETs killing large hyphal cells that cannot be phagocytosed [95]. The subsequent production of TNF-α by phagocytes results in increased TLR4 expression on the epithelial cell surface, which confers mucosal protection [71]. In addition to the recruitment of phagocytes, the candidalysin-dependent activation of epithelial cells also recruits lymphoid cell type 3 (ILC3), γδ T-cells and natural Th17 cells, which are activated by IL-1β secreted by neutrophils and epithelial cells [74,75,76,96,97]. The activation of these cells results in the production of IL-17A and IL-17F, which potentiates the epithelial response by further inducing the secretion of proinflammatory cytokines, histatins and β-defensins from the epithelial cells and salivary glands [72,77,78,98,99] (Table 2). As a result, individuals with defective IL-17 signalling are highly susceptible to oral candidiasis, highlighting the importance of IL-17 in oral mucosal immunity to Candida [100,101].
3.3. Vaginal Candidiasis
Vaginal candidiasis (VVC) is one of the most common Candida infections, infecting 75% of the female population, with up to 10–15% developing recurrent infections (RVVC), defined as three or more episodes in a twelve-month period [102,103]. Unlike for oral candidiasis, immune suppression is not a prerequisite for infection, and HIV-positive women are not more prone to the development of VVC. Instead, the predisposing factors include vaginal dysbiosis after antimicrobial therapy, elevated oestrogen levels (as a result of pregnancy, oral contraceptive use, or hormone-replacement therapy) and uncontrolled diabetes, and some single-nucleotide polymorphisms are enriched in women with RVVC [104,105,106,107,108,109]. Although it is a mucosal infection, the innate immune response in VVC is different to that in oral candidiasis, with IL-17 playing a minimal role [110,111,112].
Symptomatic vaginal infections are characterised by a nonprotective proinflammatory innate immune response, mediated by the influx of neutrophils, which has been reviewed in detail [113,114,115]. To summarise, the interaction of C. albicans with vaginal epithelial cells results in the production of IL-1β and S100A8 alarmin, which are potent neutrophil chemoattractants and function to recruit neutrophils to the vaginal epithelium [79,80,116] (Figure 2, Table 2). Neutrophil recruitment and, therefore, symptomatic disease are dependent on the production of candidalysin, with strains defective in candidalysin maintaining high levels of asymptomatic colonisation [94,117]. However, unlike in oral candidiasis, where neutrophils are efficient at phagocytosing yeast and trapping hyphae by NET release, these recruited neutrophils are unable to phagocytose and clear the fungal pathogen, leading to a heightened nonprotective proinflammatory response [79,118]. The neutrophil dysfunction in VVC and RVVC has been linked to the inability of complement receptor 3 (CR3) to interact with the fungal cell wall protein, Pra1 [119]. Under normal circumstances, the interaction of CR3 with Pra1 results in the formation of NETs and efficient killing of fungal hyphae [73]. However, this interaction is inhibited in vaginal fluid due to heparan sulphate, which competitively binds with CR3, preventing its interaction with Pra1, and promotes Candida survival and symptomatic infection (Figure 2) [115,120]. Heparan sulphate is induced by oestrogen, suggesting that women with high circulatory levels of oestrogen are more prone to symptomatic infection due to higher vaginal mucosal levels of heparin sulphate [121].
In addition to the role of host factors, C. albicans has been shown to remodel its cell wall in response to specific environmental cues encountered at the vaginal mucosa [122]. The growth of C. albicans in lactate—the predominant carbon source at the vaginal mucosa due to the presence of lactobacilli—promotes the concealment of β-glucans, reducing innate immune recognition and, therefore, possibly contributing to the reduced neutrophil phagocytosis rates [123,124]. On the other hand, acidic environments promote β-glucan exposure, leading to the hyperactivation of innate immune responses and significant neutrophil influx [125]. These acid-adapted fungal cells are more resistant to neutrophil killing and could also potentiate proinflammatory innate immune responses during VVC [126]. Analysis of the fungal cell wall from cells isolated from VVC patients identified β-glucan exposure on hyphae [127]. Neutrophils have been shown to expose β-glucan during NETosis, which is proposed to be the cause of glucan exposure on hyphae isolated from VVC patients, but given that neutrophils are defective in fungal killing at the vaginal mucosa, this remains to be confirmed [28,127].
3.4. Candidemia
In systemic infection, Candida enters the bloodstream and disseminates throughout the body. The presence of Candida in the blood is known as candidemia, the most common form of invasive candidiasis [128]. Candidemia can result in sepsis, infective endocarditis and vascular thrombosis, with often fatal outcomes [82]. In healthy individuals, C. albicans is rapidly removed from the bloodstream by the actions of neutrophils. Neutrophils are the most abundant leukocyte in the bloodstream, and whilst they are not essential in protection against other fungal pathogens such as Cryptococcus, neutropenic patients are more susceptible to systemic candidiasis. The chemokine receptor CXCR1 is essential in neutrophil granulogenesis, degranulation and non-oxidative killing of C. albicans, alongside the endoplasmic reticulum transmembrane protein Jagunal homolog 1 (JAGN1) [81]. The most infected organ in systemic candidiasis is the kidney, and neutrophils play a crucial role in host immunity to Candida [129]. In mice lacking the chemokine receptor CCR1, neutrophil accumulation in the infected kidney is impaired and delayed [130]. Interestingly, ccr1−/− mice display higher survival rates and lower kidney tissue damage, and ccr1+/+ mice appear to succumb to uncontrolled tissue damage due to excessive neutrophil recruitment and activity, suggesting that the neutrophil CCR1 receptor mediates neutrophil tissue injury in invasive candidiasis [130]. Conversely, the absence of the chemokine receptor CXCR1 is associated with increased mortality and higher kidney fungal burden [131]. Neutrophil recruitment was not affected in cxcr1−/− mice, but the neutrophils displayed impaired degranulation and capacity to kill Candida [131].
The role of platelets in the innate immune response to Candida is often overlooked. Although traditionally described as playing a key role in maintaining haemostasis, thrombosis and inflammation, platelets have more recently been recognised as key players of the innate immune response to infection [132]. Whilst nonphagocytic, platelets have been shown to interact with and be activated by bacteria, viruses and fungi, leading to the release of various platelet antimicrobial peptides [133,134,135,136,137]. Thrombocytopenia is a known risk factor for invasive fungal infections, including candidiasis [136]. The Candida–platelet interaction is poorly understood and controversial, with the role of thrombosis in candidiasis yet to be determined. Several studies have shown that Candida species can bind and induce the activation of platelets in vitro, although the degree of this activation is strain and species specific [82,136]. Of those Candida species that can induce platelet aggregation, only small subpopulations of platelets have been shown to directly interact with Candida [136]. Additional studies have suggested that C. albicans might inhibit platelet aggregation through the production of gliotoxin, a fungal secondary metabolite, similar to Aspergillus [138]. However, the production of gliotoxin by Candida species has been debated [139]. The associated risk of candidiasis with thrombocytopenia highlights the importance of platelets as immune regulators in bloodstream Candida infection. However, current studies suggest that the Candida–platelet interaction is complex, and likely dependent on the platelet donor and fungal strain under investigation. Understanding the underlying mechanisms of the Candida–platelet interaction and the subsequent clinical outcomes surrounding the interaction could prove fruitful for developing novel antifungal therapies based on anticoagulants.
In order to reach internal organs, Candida must cross the endothelium lining the blood vessels. To achieve this, Candida adheres to and transmigrates across endothelial cells. C. albicans express an array of adhesins that facilitate adherence to endothelial cells, such as αMβ2-like adhesin, which interacts with endothelial ICAM-1 and -2; αvβ3-like adhesin, which interacts with PECAM-1; and agglutinin-like protein 3, which interacts with N-cadherin [140]. The transmigration of C. albicans across endothelial cells is required for Candida to exit the bloodstream and enter the tissue. There are various models for how Candida transmigrates across endothelial cells. One model is that transmigration is facilitated by the endothelial endocytosis of Candida, mediated by the endothelial cell receptor N-cadherin [141]. Phan et al. showed that C. albicans hyphae bind to N-cadherin with greater affinity than yeast, and human CHO cells expressing N-cadherin endocytose significantly more C. albicans hyphae than CHO cells without cadherin expression [141]. C. albicans strains with homozygous null mutations in genes integral to filamentation regulation, such as EFG1, TUP1 and CLA4, display impaired capacity to invade and damage endothelial cells [142]. Furthermore, the ∆tup1 mutant produced pseudohyphae on endothelial cells and displayed lower endothelial cell invasion and injury compared to the ∆cla4 mutants, which were aberrantly shaped but produced hyphae, collectively suggesting that the formation of true hyphae is required for endothelial invasion and damage [142]. Conversely, Shintaku et al. [143] showed that like that of C. albicans, the internalisation of Candida parapsilosis yeast cells is facilitated by N-WASP activation. However, N-cadherin does not appear to play a role in C. parapsilosis internalisation [143]. Whilst most Candida–endothelial cell interactions have been studied in vitro, there are also a few in vivo studies investigating the mechanism(s) underlying Candida adhesion to, and transmigration across, the endothelial lining of blood vessels. However, our current understanding of Candida–endothelial cell interactions must be validated in vivo and would benefit from the inclusion of other physical parameters including blood flow, which likely has an impact on the Candida–endothelial interaction.
4. Summary
Candidiasis encompasses a range of infections, such as low-mortality-associated infections such as oral thrush, and fatal infections such as candidemia. The ability of Candida to establish disease in host niche environments and our understanding of the mechanisms underpinning this are paramount in our bid to tackle candidiasis. The ability of the host immune cells—namely, but not solely, phagocytic cells—to identify and destroy Candida is key in our understanding of how the pathogenic fungus is able to evade immune clearance. Research on the interplay between Candida and phagocytic cells, such as macrophages and neutrophils, has alluded to the various PAMPs and PRRs involved in Candida detection. CLRs, such as Dectin-1 and DC-SIGN; TLRs, such as TLR2 and TLR4; and other receptors such as Galectin-3 and CR3 have all been shown to play a role in Candida infection. Likewise, corresponding PAMPs implicated in Candida–host interaction have also been identified, such as β-glucan and O-mannan. Whilst much has been done to identify which PAMPs and PRRs are involved in host–Candida interplay, there is much research to be conducted on the roles of these, and the subsequent activation pathways implicated in Candida engulfment and destruction, under the niche host conditions where Candida establishes itself. For example, under normal conditions, neutrophils are able to recognise, engulf and destroy Candida via the receptors CR3 and FcγR, and NADPH oxidase activity [144]. In VVC, however, there is significant neutrophil recruitment, but these neutrophils are unable to control the infection, and several environmental factors, including the heparan-sulphate-dependent inhibition of CR3 recognition and reduced Dectin-1 recognition due to lactate inducing β-glucan masking, have been suggested to account for this nonprotective innate immune response. Therefore, Candida appears to have a remarkable capacity to adapt to the host environment and subsequently evade clearance by the immune system. The key to developing therapeutics against candidiasis lies in our understanding of how Candida does this, and how it affects its interaction with immune cells and subsequent immune cell activity.
Author Contributions
H.G. and E.B. wrote the majority of the review, and prepared the figures and tables; P.K. contributed a section to the review; R.A.H. oversaw the collation of the review. All authors have read and agreed to the published version of the manuscript.
Funding
H.G. is supported by an MRC doctoral training partnership: an Integrated Midlands Partnership for Biomedical Training (MR/N013913/1). E.B. is supported by the MIDAS Wellcome Trust programme, P.K. is supported by a Wellcome Trust Strategic Award for Medical Mycology and Fungal Immunology (097377/Z/11/Z), and R.A.H. is supported by the BBSRC (BB/R00966X/1). The funding bodies had no role in the collation of this work.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
We would like to acknowledge all the contributions in this field of research that could not be included here due to space limitations.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ward, T.L.; Knights, D.; Gale, C.A. Infant fungal communities: Current knowledge and research opportunities. BMC Med. 2017, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Tso, G.H.W.; Reales-Calderon, J.A.; Pavelka, N. The elusive anti-Candida vaccine: Lessons from the past and opportunities for the future. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Moyes, D.L.; Wilson, D.; Richardson, J.P.; Mogavero, S.; Tang, S.X.; Wernecke, J.; Höfs, S.; Gratacap, R.L.; Robbins, J.; Runglall, M.; et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 2016, 532, 64–68. [Google Scholar] [CrossRef]
- Zhu, W.; Filler, S.G. Interactions of Candida albicans with epithelial cells. Cell. Microbiol. 2010, 12, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Cerdeira, C.; Gregorio, M.C.; Molares-Vila, A.; López-Barcenas, A.; Fabbrocini, G.; Bardhi, B.; Sinani, A.; Sánchez-Blanco, E.; Arenas-Guzmán, R.; Hernandez-Castro, R. Biofilms and vulvovaginal candidiasis. Coll. Surf. B Biointerfaces 2019, 174, 110–125. [Google Scholar] [CrossRef]
- Du, Q.; Ren, B.; He, J.; Peng, X.; Guo, Q.; Zheng, L.; Li, J.; Dai, H.; Chen, V.; Zhang, L.; et al. Candida albicans promotes tooth decay by inducing oral microbial dysbiosis. ISME J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.A. Dressed to impress: Impact of environmental adaptation on the Candida albicans cell wall. Mol. Microbiol. 2015, 97, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Gow, N.A.R.; Latge, J.-P.; Munro, C.A. The Fungal cell wall: Structure, biosynthesis, and function. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef]
- Chen, S.M.; Shen, H.; Zhang, T.; Huang, X.; Liu, X.Q.; Guo, S.Y.; Zhao, J.J.; Wang, C.F.; Yan, L.; Xu, G.T.; et al. Dectin-1 plays an important role in host defense against systemic Candida glabrata infection. Virulence 2017, 8, 1643–1656. [Google Scholar] [CrossRef]
- Esteban, A.; Popp, M.W.; Vyas, V.K.; Strijbis, K.; Ploegh, H.L.; Fink, G.R. Fungal recognition is mediated by the association of dectin-1 and galectin-3 in macrophages. Proc. Nat. Acad. Sci. USA 2011, 108, 14270–14275. [Google Scholar] [CrossRef] [PubMed]
- Gantner, B.N.; Simmons, R.M.; Underhill, D.M. Dectin-1 mediates macrophage recognition of Candida albicans yeast but not filaments. EMBO J. 2005, 24, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Utomo, A.; Cullere, X.; Choi, M.M.; Milner, D.A., Jr.; Venkatesh, D.; Yun, S.H.; Mayadas, T.N. The β-glucan receptor Dectin-1 activates the integrin Mac-1 in neutrophils via Vav protein signaling to promote Candida albicans clearance. Cell Host Microbe 2011, 10, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Ifrim, D.C.; Quintin, J.; Courjol, F.; Verschueren, I.; van Krieken, J.H.; Koentgen, F.; Fradin, C.; Gow, N.A.; Joosten, L.A.; van der Meer, J.W.; et al. The role of dectin-2 for host defense against disseminated Candidiasis. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2016, 36, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Saijo, S.; Ikeda, S.; Yamabe, K.; Kakuta, S.; Ishigame, H.; Akitsu, A.; Fujikado, N.; Kusaka, T.; Kubo, S.; Chung, S.H.; et al. Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity 2010, 32, 681–691. [Google Scholar] [CrossRef]
- Zhu, L.L.; Zhao, X.Q.; Jiang, C.; You, Y.; Chen, X.P.; Jiang, Y.Y.; Jia, X.M.; Lin, X. C-type lectin receptors Dectin-3 and Dectin-2 form a heterodimeric pattern-recognition receptor for host defense against fungal infection. Immunity 2013, 39, 324–334. [Google Scholar] [CrossRef]
- Cambi, A.; Gijzen, K.; de Vries l, J.; Torensma, R.; Joosten, B.; Adema, G.J.; Netea, M.G.; Kullberg, B.J.; Romani, L.; Figdor, C.G. The C-type lectin DC-SIGN (CD209) is an antigen-uptake receptor for Candida albicans on dendritic cells. Eur. J. Immunol. 2003, 33, 532–538. [Google Scholar] [CrossRef]
- Cambi, A.; Netea, M.G.; Mora-Montes, H.M.; Gow, N.A.; Hato, S.V.; Lowman, D.W.; Kullberg, B.J.; Torensma, R.; Williams, D.L.; Figdor, C.G. Dendritic cell interaction with Candida albicans critically depends on N-linked mannan. J. Biol. Chem. 2008, 283, 20590–20599. [Google Scholar] [CrossRef]
- Wells, C.A.; Salvage-Jones, J.A.; Li, X.; Hitchens, K.; Butcher, S.; Murray, R.Z.; Beckhouse, A.G.; Lo, Y.L.; Manzanero, S.; Cobbold, C.; et al. The macrophage-inducible C-type lectin, mincle, is an essential component of the innate immune response to Candida albicans. J. immunol. 2008, 180, 7404–7413. [Google Scholar] [CrossRef]
- Netea, M.G.; Gow, N.A.; Munro, C.A.; Bates, S.; Collins, C.; Ferwerda, G.; Hobson, R.P.; Bertram, G.; Hughes, H.B.; Jansen, T.; et al. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J. Clin. Investig. 2006, 116, 1642–1650. [Google Scholar] [CrossRef] [PubMed]
- van de Veerdonk, F.L.; Marijnissen, R.J.; Kullberg, B.J.; Koenen, H.J.; Cheng, S.C.; Joosten, I.; van den Berg, W.B.; Williams, D.L.; van der Meer, J.W.; Joosten, L.A.; et al. The macrophage mannose receptor induces IL-17 in response to Candida albicans. Cell Host Microbe 2009, 5, 329–340. [Google Scholar] [CrossRef]
- Blasi, E.; Mucci, A.; Neglia, R.; Pezzini, F.; Colombari, B.; Radzioch, D.; Cossarizza, A.; Lugli, E.; Volpini, G.; Del Giudice, G.; et al. Biological importance of the two Toll-like receptors, TLR2 and TLR4, in macrophage response to infection with Candida albicans. FEMS Immunol. Med. Microbiol. 2005, 44, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Tessarolli, V.; Gasparoto, T.H.; Lima, H.R.; Figueira, E.A.; Garlet, T.P.; Torres, S.A.; Garlet, G.P.; da Silva, J.S.; Campanelli, A.P. Absence of TLR2 influences survival of neutrophils after infection with Candida albicans. Med. Mycol. 2010, 48, 129–140. [Google Scholar] [CrossRef]
- Jouault, T.; Ibata-Ombetta, S.; Takeuchi, O.; Trinel, P.A.; Sacchetti, P.; Lefebvre, P.; Akira, S.; Poulain, D. Candida albicans phospholipomannan is sensed through toll-like receptors. J. Infect. Dis. 2003, 188, 165–172. [Google Scholar] [CrossRef]
- Netea, M.G.; van de Veerdonk, F.; Verschueren, I.; van der Meer, J.W.; Kullberg, B.J. Role of TLR1 and TLR6 in the host defense against disseminated candidiasis. FEMS Immunol. Med. Microbiol. 2008, 52, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Halder, L.D.; Jo, E.A.H.; Hasan, M.Z.; Ferreira-Gomes, M.; Krüger, T.; Westermann, M.; Palme, D.I.; Rambach, G.; Beyersdorf, N.; Speth, C.; et al. Immune modulation by complement receptor 3-dependent human monocyte TGF-β1-transporting vesicles. Nat. Commun. 2020, 11, 2331. [Google Scholar] [CrossRef] [PubMed]
- Levin, R.; Grinstein, S.; Canton, J. The life cycle of phagosomes: Formation, maturation, and resolution. Immunol. Rev. 2016, 273, 156–179. [Google Scholar] [CrossRef] [PubMed]
- Mukaremera, L.; Lee, K.K.; Mora-Montes, H.M.; Gow, N.A.R. Candida albicans yeast, pseudohyphal, and hyphal morphogenesis differentially affects immune recognition. Front. Immunol. 2017, 8, 629. [Google Scholar] [CrossRef] [PubMed]
- Hopke, A.; Nicke, N.; Hidu, E.E.; Degani, G.; Popolo, L.; Wheeler, R.T. Neutrophil Attack triggers extracellular trap-dependent Candida cell wall remodeling and altered immune recognition. PLoS Pathog. 2016, 12, e1005644. [Google Scholar] [CrossRef]
- Lewis, L.E.; Bain, J.M.; Lowes, C.; Gillespie, C.; Rudkin, F.M.; Gow, N.A.R.; Erwig, L.-P. Stage specific assessment of Candida albicans phagocytosis by macrophages identifies cell wall composition and morphogenesis as key determinants. PLoS Pathog. 2012, 8, e1002578. [Google Scholar] [CrossRef]
- D’Ostiani, C.F.; Del Sero, G.; Bacci, A.; Montagnoli, C.; Spreca, A.; Mencacci, A.; Ricciardi-Castagnoli, P.; Romani, L. Dendritic cells discriminate between yeasts and hyphae of the fungus Candida albicans. Implications for initiation of T helper cell immunity in vitro and in vivo. J. Exp. Med. 2000, 191, 1661–1674. [Google Scholar] [CrossRef] [PubMed]
- Hope, W.W.; Drusano, G.L.; Moore, C.B.; Sharp, A.; Louie, A.; Walsh, T.J.; Denning, D.W.; Warn, P.A. Effect of neutropenia and treatment delay on the response to antifungal agents in experimental disseminated candidiasis. Antimicrob. Agents Chemother. 2007, 51, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Ramy, N.; Hashim, M.; Abou Hussein, H.; Sawires, H.; Gaafar, M.; El Maghraby, A. Role of early onset neutropenia in development of Candidemia in premature infants. J. Trop. Pediatr. 2017, 64, 51–59. [Google Scholar] [CrossRef]
- Son, H.-J.; Kim, M.J.; Lee, S.; Choi, S.; Jung, K.H.; Jung, J.; Chong, Y.P.; Kim, S.-H.; Choi, S.-H.; Kim, Y.S.; et al. Risk factors and outcomes of patients with ocular involvement of candidemia. PLoS ONE 2019, 14, e0222356. [Google Scholar] [CrossRef]
- Fradin, C.; De Groot, P.; MacCallum, D.; Schaller, M.; Klis, F.; Odds, F.C.; Hube, B. Granulocytes govern the transcriptional response, morphology and proliferation of Candida albicans in human blood. Mol. Microbiol. 2005, 56, 397–415. [Google Scholar] [CrossRef]
- Newman, S.L.; Holly, A. Candida albicans is phagocytosed, killed, and processed for antigen presentation by human dendritic cells. Infect. Immun. 2001, 69, 6813–6822. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Gordon, S. A new receptor for β-glucans. Nature 2001, 413, 36–37. [Google Scholar] [CrossRef] [PubMed]
- Erwig, L.P.; Gow, N.A.R. Interactions of fungal pathogens with phagocytes. Nat. Rev. Microbiol. 2016, 14, 163–176. [Google Scholar] [CrossRef]
- Thornton, B.P.; Vĕtvicka, V.; Pitman, M.; Goldman, R.C.; Ross, G.D. Analysis of the sugar specificity and molecular location of the beta-glucan-binding lectin site of complement receptor type 3 (CD11b/CD18). J. Immunol. 1996, 156, 1235–1246. [Google Scholar] [PubMed]
- Netea, M.G.; Brown, G.D.; Kullberg, B.J.; Gow, N.A. An integrated model of the recognition of Candida albicans by the innate immune system. Nat. Rev. Microbiol. 2008, 6, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Marie-Anaïs, F.; Mazzolini, J.; Herit, F.; Niedergang, F. Dynamin-actin cross talk contributes to phagosome formation and closure. Traffic 2016, 17, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Gold, E.S.; Underhill, D.M.; Morrissette, N.S.; Guo, J.; McNiven, M.A.; Aderem, A. Dynamin 2 is required for phagocytosis in macrophages. J. Exp. Med. 1999, 190, 1849–1856. [Google Scholar] [CrossRef] [PubMed]
- Campellone, K.G.; Welch, M.D. A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 2010, 11, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.L.; Harrison, R.E.; Grinstein, S. Phagocytosis by neutrophils. Microb. Infect. 2003, 5, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Nauclér, C.; Grinstein, S.; Sundler, R.; Tapper, H. Signaling to localized degranulation in neutrophils adherent to immune complexes. J. Leukoc. Biol. 2002, 71, 701–710. [Google Scholar] [CrossRef]
- Nordenfelt, P.; Tapper, H. Phagosome dynamics during phagocytosis by neutrophils. J. Leukoc. Biol. 2011, 90, 271–284. [Google Scholar] [CrossRef]
- Roberts, R.L.; Barbieri, M.A.; Ullrich, J.; Stahl, P.D. Dynamics of Rab5 activation in endocytosis and phagocytosis. J. Leukoc. Biol. 2000, 68, 627–632. [Google Scholar] [PubMed]
- Vieira, O.V.; Bucci, C.; Harrison, R.E.; Trimble, W.S.; Lanzetti, L.; Gruenberg, J.; Schreiber, A.D.; Stahl, P.D.; Grinstein, S. Modulation of Rab5 and Rab7 recruitment to phagosomes by phosphatidylinositol 3-kinase. Mol. Cell. Biol. 2003, 23, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Vylkova, S.; Lorenz, M.C. Modulation of phagosomal pH by Candida albicans promotes hyphal morphogenesis and requires Stp2p, a regulator of amino acid transport. PLoS Pathog. 2014, 10, e1003995. [Google Scholar] [CrossRef] [PubMed]
- Canton, J.; Khezri, R.; Glogauer, M.; Grinstein, S. Contrasting phagosome pH regulation and maturation in human M1 and M2 macrophages. Mol. Biol. Cell 2014, 25, 3330–3341. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, A.; Scott, C.C.; Grinstein, S. Determinants of the phagosomal pH in neutrophils. J. Biol. Chem. 2002, 277, 6059–6066. [Google Scholar] [CrossRef]
- Savina, A.; Jancic, C.; Hugues, S.; Guermonprez, P.; Vargas, P.; Moura, I.C.; Lennon-Duménil, A.M.; Seabra, M.C.; Raposo, G.; Amigorena, S. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 2006, 126, 205–218. [Google Scholar] [CrossRef]
- Chauhan, N.; Latge, J.P.; Calderone, R. Signalling and oxidant adaptation in Candida albicans and Aspergillus fumigatus. Nat. Rev. Microbiol. 2006, 4, 435–444. [Google Scholar] [CrossRef]
- Thompson, H.L.; Wilton, J.M. Interaction and intracellular killing of Candida albicans blastospores by human polymorphonuclear leucocytes, monocytes and monocyte-derived macrophages in aerobic and anaerobic conditions. Clin. Exp. Immunol. 1992, 87, 316–321. [Google Scholar] [CrossRef]
- Azevedo, E.P.; Rochael, N.C.; Guimarães-Costa, A.B.; de Souza-Vieira, T.S.; Ganilho, J.; Saraiva, E.M.; Palhano, F.L.; Foguel, D. A metabolic shift toward pentose phosphate pathway is necessary for amyloid fibril- and phorbol 12-myristate 13-acetate-induced Neutrophil Extracellular Trap (NET) Formation. J. Biol. Chem. 2015, 290, 22174–22183. [Google Scholar] [CrossRef] [PubMed]
- Jawale, C.V.; Ramani, K.; Li, D.-d.; Coleman, B.M.; Oberoi, R.S.; Kupul, S.; Lin, L.; Desai, J.V.; Delgoffe, G.M.; Lionakis, M.S.; et al. Restoring glucose uptake rescues neutrophil dysfunction and protects against systemic fungal infection in mouse models of kidney disease. Sci. Transl. Med. 2020, 12, eaay5691. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Ishihara, C.; Takeuchi, A.; Imanishi, T.; Xue, L.; Morris, S.W.; Inui, M.; Takai, T.; Shibuya, A.; Saijo, S.; et al. The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat. Immunol. 2007, 8, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Rogers, N.C.; Slack, E.C.; Edwards, A.D.; Nolte, M.A.; Schulz, O.; Schweighoffer, E.; Williams, D.L.; Gordon, S.; Tybulewicz, V.L.; Brown, G.D.; et al. Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity 2005, 22, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Pearlman, E. Immune interactions with pathogenic and commensal fungi: A two-way street. Immunity 2015, 43, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Hernanz-Falcón, P.; Joffre, O.; Williams, D.L.; Reis e Sousa, C. Internalization of dectin-1 terminates induction of inflammatory responses. Eur. J. Immunol. 2009, 39, 507–513. [Google Scholar] [CrossRef]
- Westman, J.; Walpole, G.F.W.; Kasper, L.; Xue, B.Y.; Elshafee, O.; Hube, B.; Grinstein, S. Lysosome fusion maintains phagosome integrity during fungal infection. Cell Host Microbe 2020. [Google Scholar] [CrossRef]
- Aybay, C.; Imir, T. Tumor necrosis factor (TNF) induction from monocyte/macrophages by Candida species. Immunobiology 1996, 196, 363–374. [Google Scholar] [CrossRef]
- Ganesan, S.; Rathinam, V.A.K.; Bossaller, L.; Army, K.; Kaiser, W.J.; Mocarski, E.S.; Dillon, C.P.; Green, D.R.; Mayadas, T.N.; Levitz, S.M.; et al. Caspase-8 modulates dectin-1 and complement receptor 3-driven IL-1β production in response to β-glucans and the fungal pathogen, Candida albicans. J. Immunol. 2014, 193, 2519–2530. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Klein, T.W.; Friedman, H. Involvement of mannose receptor in cytokine interleukin-1beta (IL-1beta), IL-6, and granulocyte-macrophage colony-stimulating factor responses, but not in chemokine macrophage inflammatory protein 1beta (MIP-1beta), MIP-2, and KC responses, caused by attachment of Candida albicans to macrophages. Infect. Immun. 1997, 65, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Chin, V.K.; Foong, K.J.; Maha, A.; Rusliza, B.; Norhafizah, M.; Chong, P.P. Early expression of local cytokines during systemic Candida albicans infection in a murine intravenous challenge model. Biomed. Rep. 2014, 2, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Romani, L.; Bistoni, F.; Puccetti, P. Initiation of T-helper cell immunity to Candida albicans by IL-12: The role of neutrophils. Chem. Immunol. 1997, 68, 110–135. [Google Scholar] [CrossRef] [PubMed]
- Maher, C.O.; Dunne, K.; Comerford, R.; O’Dea, S.; Loy, A.; Woo, J.; Rogers, T.R.; Mulcahy, F.; Dunne, P.J.; Doherty, D.G. Candida albicans stimulates IL-23 release by human dendritic cells and downstream IL-17 secretion by Vδ1 T cells. J. Immunol. 2015, 194, 5953–5960. [Google Scholar] [CrossRef]
- Mohammadi, F.; Ghasemi, Z.; Familsatarian, B.; Salehi, E.; Sharifynia, S.; Barikani, A.; mirzadeh, M.; Hosseini, M.A. Relationship between antifungal susceptibility profile and virulence factors in Candida albicans isolated from nail specimens. Rev. Soc. Bras. Med. Trop. 2020, 53, doi. [Google Scholar] [CrossRef] [PubMed]
- Naglik, J.; Albrecht, A.; Bader, O.; Hube, B. Candida albicans proteinases and host/pathogen interactions. Cell. Microbiol. 2004, 6, 915–926. [Google Scholar] [CrossRef]
- Schaller, M.; Borelli, C.; Korting, H.C.; Hube, B. Hydrolytic enzymes as virulence factors of Candida albicans. Mycoses 2005, 48, 365–377. [Google Scholar] [CrossRef]
- Hu, W.; Troutman, T.D.; Edukulla, R.; Pasare, C. Priming microenvironments dictate cytokine requirements for T helper 17 cell lineage commitment. Immunity 2011, 35, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Weindl, G.; Naglik, J.R.; Kaesler, S.; Biedermann, T.; Hube, B.; Korting, H.C.; Schaller, M. Human epithelial cells establish direct antifungal defense through TLR4-mediated signaling. J. Clin. Investig. 2007, 117, 3664–3672. [Google Scholar] [CrossRef] [PubMed]
- Tomalka, J.; Azodi, E.; Narra, H.P.; Patel, K.; O’Neill, S.; Cardwell, C.; Hall, B.A.; Wilson, J.M.; Hise, A.G. β-Defensin 1 plays a role in acute mucosal defense against Candida albicans. J. Immunol. 2015, 194, 1788–1795. [Google Scholar] [CrossRef]
- Byrd, A.S.; O’Brien, X.M.; Johnson, C.M.; Lavigne, L.M.; Reichner, J.S. An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to Candida albicans. J. Immunol. 2013, 190, 4136–4148. [Google Scholar] [CrossRef] [PubMed]
- Gladiator, A.; Wangler, N.; Trautwein-Weidner, K.; LeibundGut-Landmann, S. Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J. Immunol. 2013, 190, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Peterson, A.C.; Brane, L.; Huppler, A.R.; Hernández-Santos, N.; Whibley, N.; Garg, A.V.; Simpson-Abelson, M.R.; Gibson, G.A.; Mamo, A.J.; et al. Oral-resident natural Th17 cells and γδ T cells control opportunistic Candida albicans infections. J. Exp. Med. 2014, 211, 2075–2084. [Google Scholar] [CrossRef] [PubMed]
- Bishu, S.; Su, E.W.; Wilkerson, E.R.; Reckley, K.A.; Jones, D.M.; McGeachy, M.J.; Gaffen, S.L.; Levesque, M.C. Rheumatoid arthritis patients exhibit impaired Candida albicans-specific Th17 responses. Arthr. Res. Ther. 2014, 16, R50. [Google Scholar] [CrossRef]
- Conti, H.R.; Bruno, V.M.; Childs, E.E.; Daugherty, S.; Hunter, J.P.; Mengesha, B.G.; Saevig, D.L.; Hendricks, M.R.; Coleman, B.M.; Brane, L.; et al. IL-17 receptor signaling in oral epithelial cells is critical for protection against oropharyngeal candidiasis. Cell Host Microbe 2016, 20, 606–617. [Google Scholar] [CrossRef]
- Conti, H.R.; Shen, F.; Nayyar, N.; Stocum, E.; Sun, J.N.; Lindemann, M.J.; Ho, A.W.; Hai, J.H.; Yu, J.J.; Jung, J.W.; et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 2009, 206, 299–311. [Google Scholar] [CrossRef]
- Yano, J.; Lilly, E.; Barousse, M.; Fidel, P.L., Jr. Epithelial cell-derived S100 calcium-binding proteins as key mediators in the hallmark acute neutrophil response during Candida vaginitis. Infect. Immun. 2010, 78, 5126–5137. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.; Palmer, G.E.; Eberle, K.E.; Peters, B.M.; Vogl, T.; McKenzie, A.N.; Fidel, P.L., Jr. Vaginal epithelial cell-derived S100 alarmins induced by Candida albicans via pattern recognition receptor interactions are sufficient but not necessary for the acute neutrophil response during experimental vaginal candidiasis. Infect. Immun. 2014, 82, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Wirnsberger, G.; Zwolanek, F.; Stadlmann, J.; Tortola, L.; Liu, S.W.; Perlot, T.; Järvinen, P.; Dürnberger, G.; Kozieradzki, I.; Sarao, R.; et al. Jagunal homolog 1 is a critical regulator of neutrophil function in fungal host defense. Nat. Genet. 2014, 46, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- Willcox, M.D.P.; Webb, B.C.; Thakur, A.; Harty, D.W.S. Interactions between Candida species and platelets. J. Med. Microbiol. 1998, 47, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.R.; Soldan, S.S.; Ghannoum, M.A.; Edwards, J.E., Jr.; Filler, S.G.; Bayer, A.S. Resistance to platelet microbicidal protein results in increased severity of experimental Candida albicans endocarditis. Infect. Immun. 1996, 64, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Alam, F.; Catlow, D.; Di Maio, A.; Blair, J.M.A.; Hall, R.A. Candida albicans enhances meropenem tolerance of Pseudomonas aeruginosa in a dual-species biofilm. J. Antimicrob. Chemother. 2020, 75, 925–935. [Google Scholar] [CrossRef]
- Ambe, N.F.; Longdoh, N.A.; Tebid, P.; Bobga, T.P.; Nkfusai, C.N.; Ngwa, S.B.; Nsai, F.S.; Cumber, S.N. The prevalence, risk factors and antifungal sensitivity pattern of oral candidiasis in HIV/AIDS patients in Kumba District Hospital, South West Region, Cameroon. Pan Afr. Med. J. 2020, 36, 23. [Google Scholar] [CrossRef] [PubMed]
- Kirti, Y.K. Prevalence of oral Candidiasis in Indian HIV sero-positive patients with CD4(+) cell count correlation. Indian J. Otolaryngol. Head Neck Surg. Off. Publ. Assoc. Otolaryngol. India 2019, 71, 124–127. [Google Scholar] [CrossRef]
- Naglik, J.R.; König, A.; Hube, B.; Gaffen, S.L. Candida albicans-epithelial interactions and induction of mucosal innate immunity. Curr. Opin. Microbiol. 2017, 40, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Nikou, S.A.; Kichik, N.; Brown, R.; Ponde, N.O.; Ho, J.; Naglik, J.R.; Richardson, J.P. Candida albicans interactions with mucosal surfaces during health and disease. Pathogens 2019, 8, 53. [Google Scholar] [CrossRef]
- Pavlova, A.; Sharafutdinov, I. Recognition of Candida albicans and role of innate type 17 immunity in oral candidiasis. Microorganisms 2020, 8, 1340. [Google Scholar] [CrossRef]
- Richardson, J.P.; Moyes, D.L.; Ho, J.; Naglik, J.R. Candida innate immunity at the mucosa. Semin. Cell Dev. Biol. 2019, 89, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Moyes, D.L.; Runglall, M.; Murciano, C.; Shen, C.; Nayar, D.; Thavaraj, S.; Kohli, A.; Islam, A.; Mora-Montes, H.; Challacombe, S.J.; et al. A biphasic innate immune MAPK response discriminates between the yeast and hyphal forms of Candida albicans in epithelial cells. Cell Host Microbe 2010, 8, 225–235. [Google Scholar] [CrossRef]
- Moyes, D.L.; Shen, C.; Murciano, C.; Runglall, M.; Richardson, J.P.; Arno, M.; Aldecoa-Otalora, E.; Naglik, J.R. Protection against epithelial damage during Candida albicans infection is mediated by PI3K/Akt and mammalian target of rapamycin signaling. J. Infect. Dis. 2014, 209, 1816–1826. [Google Scholar] [CrossRef] [PubMed]
- Swidergall, M.; Solis, N.V.; Lionakis, M.S.; Filler, S.G. EphA2 is an epithelial cell pattern recognition receptor for fungal β-glucans. Nat. Microbiol. 2018, 3, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.P.; Mogavero, S.; Moyes, D.L.; Blagojevic, M.; Krüger, T.; Verma, A.H.; Coleman, B.M.; De La Cruz Diaz, J.; Schulz, D.; Ponde, N.O.; et al. Processing of Candida albicans Ece1p Is critical for Candidalysin maturation and fungal virulence. mBio 2018, 9, e02178-17. [Google Scholar] [CrossRef]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell. Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef]
- Altmeier, S.; Toska, A.; Sparber, F.; Teijeira, A.; Halin, C.; LeibundGut-Landmann, S. IL-1 coordinates the neutrophil response to C. albicans in the oral mucosa. PLoS Pathog. 2016, 12, e1005882. [Google Scholar] [CrossRef] [PubMed]
- Hise, A.G.; Tomalka, J.; Ganesan, S.; Patel, K.; Hall, B.A.; Brown, G.D.; Fitzgerald, K.A. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 2009, 5, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Baker, O.; Freeman, A.F.; Jang, W.S.; Holland, S.M.; Li, R.A.; Edgerton, M.; Gaffen, S.L. New mechanism of oral immunity to mucosal candidiasis in hyper-IgE syndrome. Mucosal Immunol. 2011, 4, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Huppler, A.R.; Verma, A.H.; Conti, H.R.; Gaffen, S.L. Neutrophils do not express IL-17A in the context of acute oropharyngeal Candidiasis. Pathogens 2015, 4, 559–572. [Google Scholar] [CrossRef]
- Liu, L.; Okada, S.; Kong, X.-F.; Kreins, A.Y.; Cypowyj, S.; Abhyankar, A.; Toubiana, J.; Itan, Y.; Audry, M.; Nitschke, P.; et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med. 2011, 208, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Puel, A.; Cypowyj, S.; Bustamante, J.; Wright, J.F.; Liu, L.; Lim, H.K.; Migaud, M.; Israel, L.; Chrabieh, M.; Audry, M.; et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 2011, 332, 65–68. [Google Scholar] [CrossRef]
- Sobel, J.D. Pathogenesis and treatment of recurrent vulvovaginal candidiasis. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 1992, 14, S148–S153. [Google Scholar] [CrossRef]
- Sobel, J.D. Vaginitis. N. Eng. J. Med. 1997, 337, 1896–1903. [Google Scholar] [CrossRef]
- Gonçalves, B.; Ferreira, C.; Alves, C.T.; Henriques, M.; Azeredo, J.; Silva, S. Vulvovaginal candidiasis: Epidemiology, microbiology and risk factors. Crit. Rev. Microbiol. 2016, 42, 905–927. [Google Scholar] [CrossRef] [PubMed]
- Sobel, J.D. Vulvovaginal candidosis. Lancet 2007, 369, 1961–1971. [Google Scholar] [CrossRef]
- Giraldo, P.C.; Babula, O.; Gonçalves, A.K.; Linhares, I.M.; Amaral, R.L.; Ledger, W.J.; Witkin, S.S. Mannose-binding lectin gene polymorphism, vulvovaginal candidiasis, and bacterial vaginosis. Obstet. Gynecol. 2007, 109, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Hammad, N.M.; El Badawy, N.E.; Nasr, A.M.; Ghramh, H.A.; Al Kady, L.M. Mannose-binding lectin gene polymorphism and its association with susceptibility to recurrent vulvovaginal candidiasis. BioMed. Res. Int. 2018, 2018, 7648152. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, M.; Carvalho, A.; Cunha, C.; Plantinga, T.S.; van de Veerdonk, F.; Puccetti, M.; Galosi, C.; Joosten, L.A.; Dupont, B.; Kullberg, B.J.; et al. Association of a variable number tandem repeat in the NLRP3 gene in women with susceptibility to RVVC. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. European Soc. Clin. Microbiol. 2016, 35, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Rosentul, D.C.; Delsing, C.E.; Jaeger, M.; Plantinga, T.S.; Oosting, M.; Costantini, I.; Venselaar, H.; Joosten, L.A.; van der Meer, J.W.; Dupont, B.; et al. Gene polymorphisms in pattern recognition receptors and susceptibility to idiopathic recurrent vulvovaginal candidiasis. Front. Microbiol. 2014, 5, 483. [Google Scholar] [CrossRef] [PubMed]
- Fidel, P.L., Jr.; Lynch, M.E.; Sobel, J.D. Circulating CD4 and CD8 T cells have little impact on host defense against experimental vaginal candidiasis. Infect. Immun. 1995, 63, 2403–2408. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.E.; Barousse, M.; Swoboda, R.K.; Myers, T.; Hager, S.; Wolf, N.A.; Cutright, J.L.; Thompson, J.; Sobel, J.D.; Fidel, P.L., Jr. Candida-specific systemic cell-mediated immune reactivities in human immunodeficiency virus-positive persons with mucosal candidiasis. J. Infect. Dis. 2001, 183, 277–285. [Google Scholar] [CrossRef] [PubMed]
- White, M.H. Is vulvovaginal candidiasis an AIDS-related illness? Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 1996, 22, S124–S127. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Bernardis, F.; Graziani, S.; Tirelli, F.; Antonopoulou, S. Candida vaginitis: Virulence, host response and vaccine prospects. Med. Mycol. 2018, 56, 26–31. [Google Scholar] [CrossRef]
- Rosati, D.; Bruno, M.; Jaeger, M.; Ten Oever, J.; Netea, M.G. Recurrent vulvovaginal Candidiasis: An immunological perspective. Microorganisms 2020, 8, 144. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.; Peters, B.M.; Noverr, M.C.; Fidel, P.L., Jr. Novel mechanism behind the immunopathogenesis of vulvovaginal Candidiasis: “Neutrophil Anergy”. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.M.; Yano, J.; Noverr, M.C.; Fidel, P.L., Jr. Candida vaginitis: When opportunism knocks, the host responds. PLoS Pathog. 2014, 10, e1003965. [Google Scholar] [CrossRef]
- Fidel, P.L., Jr.; Barousse, M.; Espinosa, T.; Ficarra, M.; Sturtevant, J.; Martin, D.H.; Quayle, A.J.; Dunlap, K. An intravaginal live Candida challenge in humans leads to new hypotheses for the immunopathogenesis of vulvovaginal candidiasis. Infect. Immun. 2004, 72, 2939–2946. [Google Scholar] [CrossRef] [PubMed]
- Nomanbhoy, F.; Steele, C.; Yano, J.; Fidel, P.L., Jr. Vaginal and oral epithelial cell anti-Candida activity. Infect. Immun. 2002, 70, 7081–7088. [Google Scholar] [CrossRef] [PubMed]
- Soloviev, D.A.; Jawhara, S.; Fonzi, W.A. Regulation of innate immune response to Candida albicans infections by αMβ2-Pra1p interaction. Infect. Immun. 2011, 79, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.; Noverr, M.C.; Fidel, P.L., Jr. Vaginal heparan sulfate linked to neutrophil dysfunction in the acute inflammatory response associated with experimental vulvovaginal Candidiasis. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Hayashi, M.; Boutin, E.; Cunha, G.R.; Bernfield, M.; Trelstad, R.L. Hormonal modification of epithelial differentiation and expression of cell surface heparan sulfate proteoglycan in the mouse vaginal epithelium. An immunohistochemical and electron microscopic study. Lab. Investig. J. Tech. Methods Pathol. 1988, 58, 68–76. [Google Scholar]
- Hopke, A.; Brown, A.J.P.; Hall, R.A.; Wheeler, R.T. Dynamic fungal cell wall architecture in stress adaptation and immune evasion. Trends Microbiol. 2018, 26, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Ballou, E.R.; Avelar, G.M.; Childers, D.S.; Mackie, J.; Bain, J.M.; Wagener, J.; Kastora, S.L.; Panea, M.D.; Hardison, S.E.; Walker, L.A.; et al. Lactate signalling regulates fungal β-glucan masking and immune evasion. Nat. Microbiol. 2016, 2, 16238. [Google Scholar] [CrossRef]
- Ene, I.V.; Cheng, S.C.; Netea, M.G.; Brown, A.J. Growth of Candida albicans cells on the physiologically relevant carbon source lactate affects their recognition and phagocytosis by immune cells. Infect. Immun. 2013, 81, 238–248. [Google Scholar] [CrossRef]
- Sherrington, S.L.; Sorsby, E.; Mahtey, N.; Kumwenda, P.; Lenardon, M.D.; Brown, I.; Ballou, E.R.; MacCallum, D.M.; Hall, R.A. Adaptation of Candida albicans to environmental pH induces cell wall remodelling and enhances innate immune recognition. PLoS Pathog. 2017, 13, e1006403. [Google Scholar] [CrossRef] [PubMed]
- Cottier, F.; Sherrington, S.; Cockerill, S.; Del Olmo Toledo, V.; Kissane, S.; Tournu, H.; Orsini, L.; Palmer, G.E.; Pérez, J.C.; Hall, R.A. Remasking of Candida albicans β-glucan in response to environmental pH is regulated by quorum sensing. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Pericolini, E.; Perito, S.; Castagnoli, A.; Gabrielli, E.; Mencacci, A.; Blasi, E.; Vecchiarelli, A.; Wheeler, R.T. Epitope unmasking in vulvovaginal candidiasis is associated with hyphal growth and neutrophilic infiltration. PLoS ONE 2018, 13, e0201436. [Google Scholar] [CrossRef] [PubMed]
- Wisplinghoff, H.; Bischoff, T.; Tallent, S.M.; Seifert, H.; Wenzel, R.P.; Edmond, M.B. Nosocomial bloodstream infections in US hospitals: Analysis of 24,179 cases from a prospective nationwide surveillance study. Clin. Infect. Dis. 2004, 39, 309–317. [Google Scholar] [CrossRef]
- Spellberg, B.; Ibrahim, A.S.; Edwards, J.E., Jr.; Filler, S.G. Mice with disseminated candidiasis die of progressive sepsis. J. Infect. Dis. 2005, 192, 336–343. [Google Scholar] [CrossRef]
- Lionakis, M.S.; Fischer, B.G.; Lim, J.K.; Swamydas, M.; Wan, W.; Richard Lee, C.C.; Cohen, J.I.; Scheinberg, P.; Gao, J.L.; Murphy, P.M. Chemokine receptor Ccr1 drives neutrophil-mediated kidney immunopathology and mortality in invasive candidiasis. PLoS Pathog. 2012, 8, e1002865. [Google Scholar] [CrossRef] [PubMed]
- Swamydas, M.; Gao, J.L.; Break, T.J.; Johnson, M.D.; Jaeger, M.; Rodriguez, C.A.; Lim, J.K.; Green, N.M.; Collar, A.L.; Fischer, B.G.; et al. CXCR1-mediated neutrophil degranulation and fungal killing promote Candida clearance and host survival. Sci. Transl. Med. 2016, 8, 322ra310. [Google Scholar] [CrossRef] [PubMed]
- Jenne, C.N.; Kubes, P. Platelets in inflammation and infection. Platelets 2015, 26, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Arman, M.; Krauel, K.; Tilley, D.O.; Weber, C.; Cox, D.; Greinacher, A.; Kerrigan, S.W.; Watson, S.P. Amplification of bacteria-induced platelet activation is triggered by FcγRIIA, integrin αIIbβ3, and platelet factor 4. Blood 2014, 123, 3166–3174. [Google Scholar] [CrossRef]
- Seyoum, M.; Enawgaw, B.; Melku, M. Human blood platelets and viruses: Defense mechanism and role in the removal of viral pathogens. Thromb. J. 2018, 16, 16. [Google Scholar] [CrossRef] [PubMed]
- Ghuman, H.; Shepherd-Roberts, A.; Watson, S.; Zuidscherwoude, M.; Watson, S.P.; Voelz, K. Mucor circinelloides induces platelet aggregation through integrin αIIbβ3 and FcγRIIA. Platelets 2019, 30, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Eberl, C.; Speth, C.; Jacobsen, I.D.; Hermann, M.; Hagleitner, M.; Deshmukh, H.; Ammann, C.G.; Lass-Flörl, C.; Rambach, G. Candida: Platelet interaction and platelet activity in vitro. J. Innate Immun. 2019, 11, 52–62. [Google Scholar] [CrossRef]
- Watson, C.N.; Kerrigan, S.W.; Cox, D.; Henderson, I.R.; Watson, S.P.; Arman, M. Human platelet activation by Escherichia coli: Roles for FcγRIIA and integrin αIIbβ3. Platelets 2016, 27, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Bertling, A.; Niemann, S.; Uekötter, A.; Fegeler, W.; Lass-Flörl, C.; von Eiff, C.; Kehrel, B.E. Candida albicans and its metabolite gliotoxin inhibit platelet function via interaction with thiols. Thromb. Haemost. 2010, 104, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Kupfahl, C.; Ruppert, T.; Dietz, A.; Geginat, G.; Hof, H. Candida species fail to produce the immunosuppressive secondary metabolite gliotoxin in vitro. FEMS Yeast Res. 2007, 7, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Grubb, S.E.; Murdoch, C.; Sudbery, P.E.; Saville, S.P.; Lopez-Ribot, J.L.; Thornhill, M.H. Candida albicans-endothelial cell interactions: A key step in the pathogenesis of systemic candidiasis. Infect. Immun. 2008, 76, 4370–4377. [Google Scholar] [CrossRef]
- Phan, Q.T.; Fratti, R.A.; Prasadarao, N.V.; Edwards, J.E., Jr.; Filler, S.G. N-cadherin mediates endocytosis of Candida albicans by endothelial cells. J. Biol. Chem. 2005, 280, 10455–10461. [Google Scholar] [CrossRef] [PubMed]
- Phan, Q.T.; Belanger, P.H.; Filler, S.G. Role of hyphal formation in interactions of Candida albicans with endothelial cells. Infect. Immun. 2000, 68, 3485–3490. [Google Scholar] [CrossRef] [PubMed]
- Shintaku, T.; Glass, K.A.; Hirakawa, M.P.; Longley, S.J.; Bennett, R.J.; Bliss, J.M.; Shaw, S.K. Human endothelial cells internalize Candida parapsilosis via N-WASP-mediated endocytosis. Infect. Immun. 2013, 81, 2777–2787. [Google Scholar] [CrossRef] [PubMed]
- Gazendam, R.P.; van Hamme, J.L.; Tool, A.T.; van Houdt, M.; Verkuijlen, P.J.; Herbst, M.; Liese, J.G.; van de Veerdonk, F.L.; Roos, D.; van den Berg, T.K.; et al. Two independent killing mechanisms of Candida albicans by human neutrophils: Evidence from innate immunity defects. Blood 2014, 124, 590–597. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).