Considerations on the Development of Therapeutics in Vascular Calcification




Abstract

{kind=link}
{kind=link}
{kind=link}
1. Introduction to the Development of Therapies in Vascular Calcification
1.1. Establishing a Need for Vascular Calcification Therapies
1.2. Active Mechanisms in the Development of Vascular Calcification
1.3. Master Regulators of Vascular Smooth Muscle Cell Osteogenic Differentiation
1.3.1. Wnt Signaling
1.3.2. Bone Morphogenic Proteins
1.4. Comorbidities and the Pathophysiology of Vascular Calcification
1.4.1. Intimal Versus Medial Calcification
1.4.2. Medial Calcification
1.4.3. Intimal Calcification
1.4.4. Genetic Predisposition to Ectopic Calcification
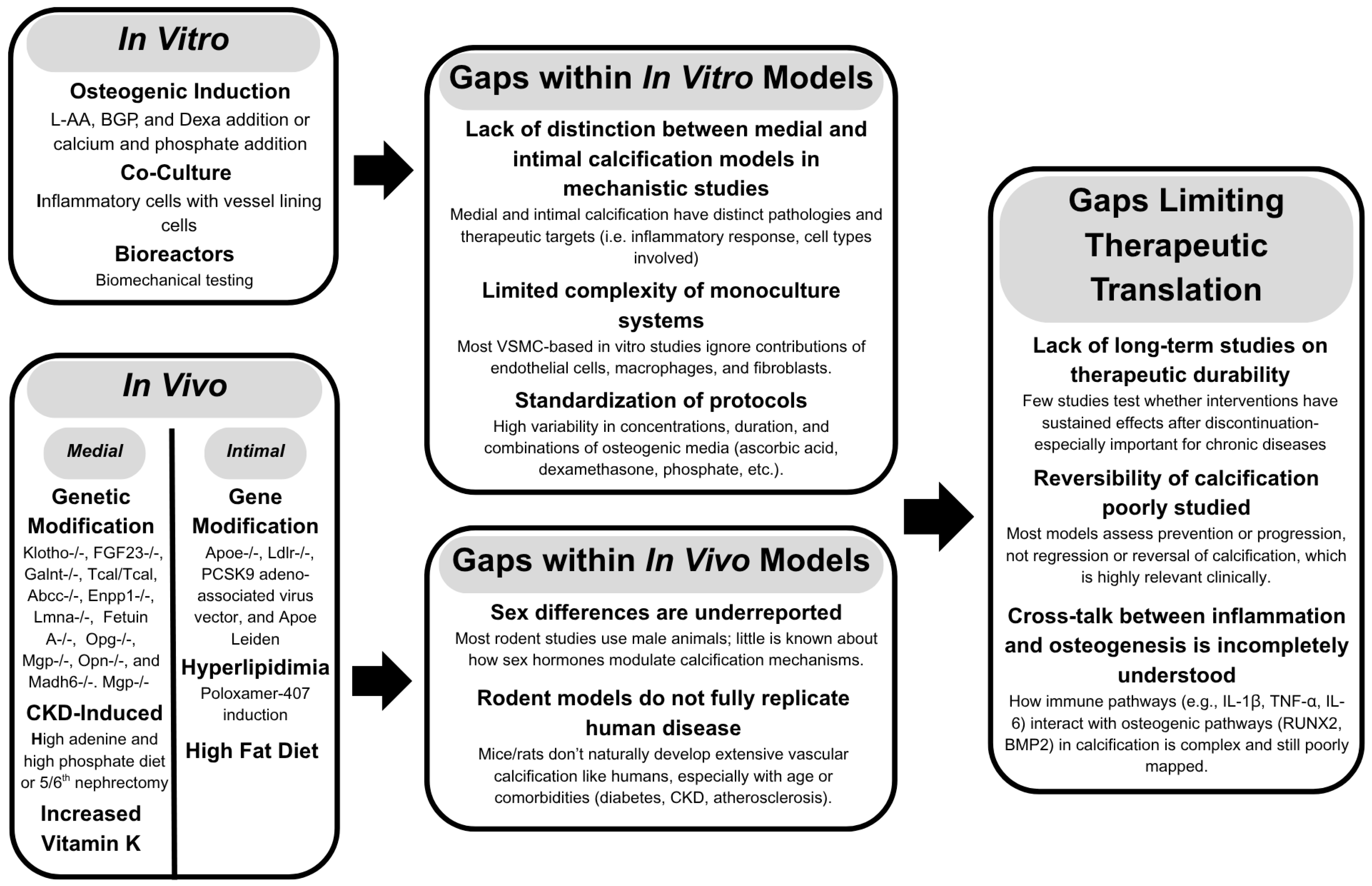
2. Common Models for Target Discovery
2.1. In Vitro Studies
2.2. In Vivo Studies
2.2.1. Medial Calcification
2.2.2. Intimal Calcification
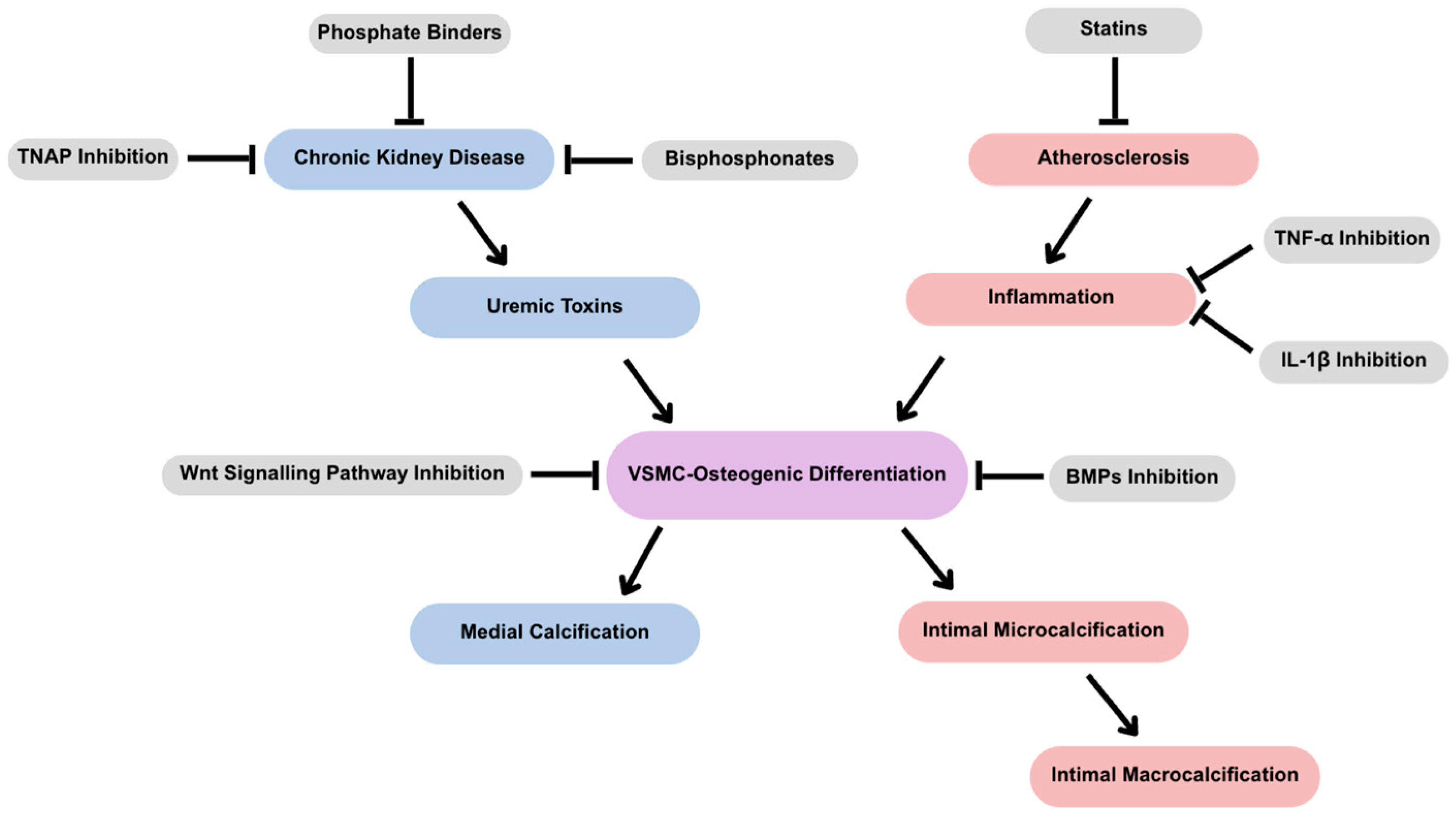
3. Considerations for Therapeutic Development
3.1. Modulating Mineral Formation
3.1.1. Bisphosphonates
3.1.2. Phosphate Binders
3.1.3. Tissue Non-Specific Alkaline Phosphatase
3.1.4. Vitamin K
3.1.5. Receptor for Advanced Glycation End Products
3.2. Modulating Inflammation
3.2.1. Tumor Necrosis Factor-α
3.2.2. IL-1β
3.3. Calcification Paradox
3.4. Micro- vs. Macrocalcifications
3.4.1. Macrocalcifications
3.4.2. Microcalcifications
3.5. Timing of Treatment
4. Conclusions and Future Directions
4.1. Summary of Key Findings
4.2. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CVD | Cardiovascular Disease |
| CKD | Chronic Kidney Disease |
| VSMC | Vascular Smooth Muscle Cells |
| EV | Extracellular Vesicles |
| LRP5/6 | Low Density Lipoprotein Receptor-Related Protein |
| DVL | Disheveled Protein |
| GSK-3 | Glycogen Synthase 3 |
| APC | Adenomatous Polyposis Coli |
| RANKL | Receptor Activator Nuclear Factor- κB Ligand |
| OPG | Osteoprotegerin |
| VCAN | Versican |
| F-CPE | Full-Length Carboxypeptidase E |
| SOST | Sclorostin |
| BMP | Bone Morphogenic Protein |
| TGF-β | Transforming Growth Factor Beta |
| MGP | Matrix Gla Protein |
| LDL | Low-Density Lipoprotein |
| IL-1β | Interleukin β |
| ACDC | Arterial Calcification due to Deficiency of CD73 |
| PPi | Pyrophosphate |
| Pi | Inorganic Phosphate |
| TNAP | Tissue Nonspecific Alkaline Phosphate |
| mTOR | Mammalian Target of Rapamycin |
| CT | Computed Tomography |
| P-407 | Poloxamer 407 |
| LPL | Lipoprotein Lipase |
| AGEs | Advanced Glycation End Products |
| RAGEs | Receptor for Advanced Glycation End Products |
| TNFα | Tumor Necrosis Factor-α |
| PPARγ | Peroxisome Proliferator-Activated Receptor Gamma |
| CANTOS | Canakinumab Anti-inflammatory Thrombosis Outcome Study |
| MRI | Magnetic Resonance Imaging |
| PET | Positron Emission Tomography |
| 18F-NaF | 18F-sodium fluoride |
| 18F-DG | 18F-fluorodeoxyglucose |
| OCT | Optical Coherence Tomography |
References
- Leopold, J.A. Vascular calcification: An age-old problem of old age. Circulation 2013, 127, 2380–2382. [Google Scholar] [CrossRef] [PubMed]
- Liberman, M.; Pesaro, A.E.; Carmo, L.S.; Serrano, C.V., Jr. Vascular calcification: Pathophysiology and clinical implications. Einstein (Sao Paulo) 2013, 11, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Rifkin, D.E.; Ix, J.H.; Wassel, C.L.; Criqui, M.H.; Allison, M.A. Renal artery calcification and mortality among clinically asymptomatic adults. J. Am. Coll. Cardiol. 2012, 60, 1079–1085. [Google Scholar] [CrossRef]
- Rocha-Singh, K.J.; Zeller, T.; Jaff, M.R. Peripheral arterial calcification: Prevalence, mechanism, detection, and clinical implications. Catheter. Cardiovasc. Interv. 2014, 83, E212–E220. [Google Scholar] [CrossRef]
- Wang, X.; Chen, X.; Chen, Z.; Zhang, M. Arterial Calcification and Its Association With Stroke: Implication of Risk, Prognosis, Treatment Response, and Prevention. Front. Cell Neurosci. 2022, 16, 845215. [Google Scholar] [CrossRef]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. Suppl. 2013, 3, 1–150. [Google Scholar] [CrossRef]
- Chen, N.X.; Moe, S.M. Vascular calcification: Pathophysiology and risk factors. Curr. Hypertens. Rep. 2012, 14, 228–237. [Google Scholar] [CrossRef]
- London, G.M.; Guerin, A.P.; Marchais, S.J.; Metivier, F.; Pannier, B.; Adda, H. Arterial media calcification in end-stage renal disease: Impact on all-cause and cardiovascular mortality. Nephrol. Dial. Transplant. 2003, 18, 1731–1740. [Google Scholar] [CrossRef]
- Mehrotra, R.; Budoff, M.; Hokanson, J.E.; Ipp, E.; Takasu, J.; Adler, S. Progression of coronary artery calcification in diabetics with and without chronic kidney disease. Kidney Int. 2005, 68, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Moe, S.M.; O’Neill, K.D.; Reslerova, M.; Fineberg, N.; Persohn, S.; Meyer, C.A. Natural history of vascular calcification in dialysis and transplant patients. Nephrol. Dial. Transplant. 2004, 19, 2387–2393. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, X.; Wu, H. Arterial Stiffness. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1078–1093. [Google Scholar] [CrossRef] [PubMed]
- Tyson, J.; Bundy, K.; Roach, C.; Douglas, H.; Ventura, V.; Segars, M.F.; Schwartz, O.; Simpson, C.L. Mechanisms of the Osteogenic Switch of Smooth Muscle Cells in Vascular Calcification: WNT Signaling, BMPs, Mechanotransduction, and EndMT. Bioengineering 2020, 7, 88. [Google Scholar] [CrossRef]
- Owens, G.K. Regulation of differentiation of vascular smooth muscle cells. Physiol. Rev. 1995, 75, 487–517. [Google Scholar] [CrossRef]
- Bakhshian Nik, A.; Hutcheson, J.D.; Aikawa, E. Extracellular Vesicles As Mediators of Cardiovascular Calcification. Front. Cardiovasc. Med. 2017, 4, 78. [Google Scholar] [CrossRef]
- Davidson, S.M.; Boulanger, C.M.; Aikawa, E.; Badimon, L.; Barile, L.; Binder, C.J.; Brisson, A.; Buzas, E.; Emanueli, C.; Jansen, F.; et al. Methods for the identification and characterization of extracellular vesicles in cardiovascular studies: From exosomes to microvesicles. Cardiovasc. Res. 2022, 119, 45–63. [Google Scholar] [CrossRef] [PubMed]
- Chiva-Blanch, G.; Davidson, S.M. Extracellular vesicles signal from bones to vessels: An answer to the ‘calcification paradox’? Cardiovasc. Res. 2022, 118, e75–e77. [Google Scholar] [CrossRef]
- Loyer, X.; Vion, A.-C.; Tedgui, A.; Boulanger, C.M. Microvesicles as Cell–Cell Messengers in Cardiovascular Diseases. Circ. Res. 2014, 114, 345–353. [Google Scholar] [CrossRef]
- Hutcheson, J.D.; Goettsch, C.; Bertazzo, S.; Maldonado, N.; Ruiz, J.L.; Goh, W.; Yabusaki, K.; Faits, T.; Bouten, C.; Franck, G.; et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat. Mater. 2016, 15, 335–343. [Google Scholar] [CrossRef]
- Chaudhary, S.C.; Khalid, S.; Smethurst, V.; Monier, D.; Mobley, J.; Huet, A.; Conway, J.F.; Napierala, D. Proteomic profiling of extracellular vesicles released from vascular smooth muscle cells during initiation of phosphate-induced mineralization. Connect. Tissue Res. 2018, 59, 55–61. [Google Scholar] [CrossRef]
- Lv, Y.; Tan, J.; Miao, Y.; Zhang, Q. The role of microvesicles and its active molecules in regulating cellular biology. J. Cell. Mol. Med. 2019, 23, 7894–7904. [Google Scholar] [CrossRef]
- Baba, I.; Matoba, T.; Katsuki, S.; Koga, J.I.; Kawahara, T.; Kimura, M.; Akita, H.; Tsutsui, H. EVs-miR-17-5p attenuates the osteogenic differentiation of vascular smooth muscle cells potentially via inhibition of TGF-beta signaling under high glucose conditions. Sci. Rep. 2024, 14, 16323. [Google Scholar] [CrossRef]
- Krohn, J.B.; Hutcheson, J.D.; Martinez-Martinez, E.; Aikawa, E. Extracellular vesicles in cardiovascular calcification: Expanding current paradigms. J. Physiol. 2016, 594, 2895–2903. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Chatrou, M.L.; Drozdov, I.; Zheng, Y.; Davidson, S.M.; Soong, D.; Furmanik, M.; Sanchis, P.; De Rosales, R.T.; Alvarez-Hernandez, D.; et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ. Res. 2015, 116, 1312–1323. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef]
- Bundy, K.; Boone, J.; Simpson, C.L. Wnt Signaling in Vascular Calcification. Front. Cardiovasc. Med. 2021, 8, 708470. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wang, S.; Li, L. New insights into the regulation of Axin function in canonical Wnt signaling pathway. Protein Cell 2014, 5, 186–193. [Google Scholar] [CrossRef]
- Khan, K.; Yu, B.; Tardif, J.C.; Rheaume, E.; Al-Kindi, H.; Filimon, S.; Pop, C.; Genest, J.; Cecere, R.; Schwertani, A. Significance of the Wnt signaling pathway in coronary artery atherosclerosis. Front. Cardiovasc. Med. 2024, 11, 1360380. [Google Scholar] [CrossRef]
- Lin, M.E.; Chen, T.; Leaf, E.M.; Speer, M.Y.; Giachelli, C.M. Runx2 Expression in Smooth Muscle Cells Is Required for Arterial Medial Calcification in Mice. Am. J. Pathol. 2015, 185, 1958–1969. [Google Scholar] [CrossRef]
- Skalka, N.; Caspi, M.; Caspi, E.; Loh, Y.P.; Rosin-Arbesfeld, R. Carboxypeptidase E: A negative regulator of the canonical Wnt signaling pathway. Oncogene 2013, 32, 2836–2847. [Google Scholar] [CrossRef]
- McArthur, K.; Kay, A.M.; Mosier, J.A.; Grant, J.; Stewart, J.A.; Simpson, C.L. Manipulating the Plasticity of Smooth Muscle Cells to Regulate Vascular Calcification. AIMS Cell Tissue Eng. 2017, 1, 165–179. [Google Scholar] [CrossRef]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Hruska, K.A.; Mathew, S.; Saab, G. Bone morphogenetic proteins in vascular calcification. Circ. Res. 2005, 97, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Lowery, J.W.; de Caestecker, M.P. BMP signaling in vascular development and disease. Cytokine Growth Factor. Rev. 2010, 21, 287–298. [Google Scholar] [CrossRef]
- Zebboudj, A.F.; Shin, V.; Bostrom, K. Matrix GLA protein and BMP-2 regulate osteoinduction in calcifying vascular cells. J. Cell Biochem. 2003, 90, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Derwall, M.; Malhotra, R.; Lai, C.S.; Beppu, Y.; Aikawa, E.; Seehra, J.S.; Zapol, W.M.; Bloch, K.D.; Yu, P.B. Inhibition of bone morphogenetic protein signaling reduces vascular calcification and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 613–622. [Google Scholar] [CrossRef]
- Braam, L.A.; Hoeks, A.P.; Brouns, F.; Hamulyak, K.; Gerichhausen, M.J.; Vermeer, C. Beneficial effects of vitamins D and K on the elastic properties of the vessel wall in postmenopausal women: A follow-up study. Thromb. Haemost. 2004, 91, 373–380. [Google Scholar] [CrossRef]
- Galvin, K.M.; Donovan, M.J.; Lynch, C.A.; Meyer, R.I.; Paul, R.J.; Lorenz, J.N.; Fairchild-Huntress, V.; Dixon, K.L.; Dunmore, J.H.; Gimbrone, M.A., Jr.; et al. A role for smad6 in development and homeostasis of the cardiovascular system. Nat. Genet. 2000, 24, 171–174. [Google Scholar] [CrossRef]
- Demetriou, M.; Binkert, C.; Sukhu, B.; Tenenbaum, H.C.; Dennis, J.W. Fetuin/alpha2-HS glycoprotein is a transforming growth factor-beta type II receptor mimic and cytokine antagonist. J. Biol. Chem. 1996, 271, 12755–12761. [Google Scholar] [CrossRef]
- Zhu, W.; Kim, J.; Cheng, C.; Rawlins, B.A.; Boachie-Adjei, O.; Crystal, R.G.; Hidaka, C. Noggin regulation of bone morphogenetic protein (BMP) 2/7 heterodimer activity in vitro. Bone 2006, 39, 61–71. [Google Scholar] [CrossRef]
- Peeters, T.; Monteagudo, S.; Tylzanowski, P.; Luyten, F.P.; Lories, R.; Cailotto, F. SMOC2 inhibits calcification of osteoprogenitor and endothelial cells. PLoS ONE 2018, 13, e0198104. [Google Scholar] [CrossRef]
- Majesky, M.W.; Dong, X.R.; Hoglund, V.; Mahoney, W.M., Jr.; Daum, G. The adventitia: A dynamic interface containing resident progenitor cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1530–1539. [Google Scholar] [CrossRef] [PubMed]
- Lanzer, P.; Boehm, M.; Sorribas, V.; Thiriet, M.; Janzen, J.; Zeller, T.; St Hilaire, C.; Shanahan, C. Medial vascular calcification revisited: Review and perspectives. Eur. Heart J. 2014, 35, 1515–1525. [Google Scholar] [CrossRef]
- Lanzer, P.; Hannan, F.M.; Lanzer, J.D.; Janzen, J.; Raggi, P.; Furniss, D.; Schuchardt, M.; Thakker, R.; Fok, P.W.; Saez-Rodriguez, J.; et al. Medial Arterial Calcification: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 78, 1145–1165. [Google Scholar] [CrossRef]
- Amann, K. Media calcification and intima calcification are distinct entities in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1599–1605. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Mackenzie, N.C.; Farquharson, C.; Macrae, V.E. Mechanisms and clinical consequences of vascular calcification. Front Endocrinol 2012, 3, 95. [Google Scholar] [CrossRef]
- Wu, M.; Rementer, C.; Giachelli, C.M. Vascular calcification: An update on mechanisms and challenges in treatment. Calcif. Tissue Int. 2013, 93, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N.; Bobryshev, Y.V. Mechanisms of medial arterial calcification in diabetes. Curr. Pharm. Des. 2014, 20, 5870–5883. [Google Scholar] [CrossRef]
- Libby, P. Vascular biology of atherosclerosis: Overview and state of the art. Am. J. Cardiol. 2003, 91, 3A–6A. [Google Scholar] [CrossRef]
- Markin, A.M.; Sobenin, I.A.; Grechko, A.V.; Zhang, D.; Orekhov, A.N. Cellular Mechanisms of Human Atherogenesis: Focus on Chronification of Inflammation and Mitochondrial Mutations. Front. Pharmacol. 2020, 11, 642. [Google Scholar] [CrossRef]
- Kobiyama, K.; Ley, K. Atherosclerosis. Circ. Res. 2018, 123, 1118–1120. [Google Scholar] [CrossRef]
- Lundberg, A.M.; Hansson, G.K. Innate immune signals in atherosclerosis. Clin. Immunol. 2010, 134, 5–24. [Google Scholar] [CrossRef]
- Fernandez, D.M.; Rahman, A.H.; Fernandez, N.F.; Chudnovskiy, A.; Amir, E.D.; Amadori, L.; Khan, N.S.; Wong, C.K.; Shamailova, R.; Hill, C.A.; et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 2019, 25, 1576–1588. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, E.; Nahrendorf, M.; Figueiredo, J.L.; Swirski, F.K.; Shtatland, T.; Kohler, R.H.; Jaffer, F.A.; Aikawa, M.; Weissleder, R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation 2007, 116, 2841–2850. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.Y.; Chen, A.Q.; Zhang, H.; Gao, X.F.; Kong, X.Q.; Zhang, J.J. Vascular Smooth Muscle Cells Phenotypic Switching in Cardiovascular Diseases. Cells 2022, 11, 4060. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.H.; Huang, X.Q.; Tan, H.M. Vascular fibrosis in atherosclerosis. Cardiovasc. Pathol. 2013, 22, 401–407. [Google Scholar] [CrossRef]
- Puylaert, P.; Zurek, M.; Rayner, K.J.; De Meyer, G.R.Y.; Martinet, W. Regulated Necrosis in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 1283–1306. [Google Scholar] [CrossRef]
- Li, M.; Wang, Z.W.; Fang, L.J.; Cheng, S.Q.; Wang, X.; Liu, N.F. Programmed cell death in atherosclerosis and vascular calcification. Cell Death Dis. 2022, 13, 467. [Google Scholar] [CrossRef]
- Jin, H.; St Hilaire, C.; Huang, Y.; Yang, D.; Dmitrieva, N.I.; Negro, A.; Schwartzbeck, R.; Liu, Y.; Yu, Z.; Walts, A.; et al. Increased activity of TNAP compensates for reduced adenosine production and promotes ectopic calcification in the genetic disease ACDC. Sci. Signal 2016, 9, ra121. [Google Scholar] [CrossRef]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef]
- Ferrante, E.A.; Cudrici, C.D.; Rashidi, M.; Fu, Y.P.; Huffstutler, R.; Carney, K.; Chen, M.Y.; St Hilaire, C.; Smith, K.; Bagheri, H.; et al. Pilot study to evaluate the safety and effectiveness of etidronate treatment for arterial calcification due to deficiency of CD73 (ACDC). Vasc. Med. 2024, 29, 245–255. [Google Scholar] [CrossRef]
- Bauer, C.; le Saux, O.; Pomozi, V.; Aherrahrou, R.; Kriesen, R.; Stolting, S.; Liebers, A.; Kessler, T.; Schunkert, H.; Erdmann, J.; et al. Etidronate prevents dystrophic cardiac calcification by inhibiting macrophage aggregation. Sci. Rep. 2018, 8, 5812. [Google Scholar] [CrossRef] [PubMed]
- Jono, S.; McKee, M.D.; Murry, C.E.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 2000, 87, E10–E17. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.; Babic, M.; Tolle, M.; van der Giet, M.; Schuchardt, M. Research Models for Studying Vascular Calcification. Int. J. Mol. Sci. 2020, 21, 2204. [Google Scholar] [CrossRef] [PubMed]
- Langenbach, F.; Handschel, J. Effects of dexamethasone, ascorbic acid and beta-glycerophosphate on the osteogenic differentiation of stem cells in vitro. Stem Cell Res. Ther. 2013, 4, 117. [Google Scholar] [CrossRef]
- Arakawa, E.; Hasegawa, K.; Irie, J.; Ide, S.; Ushiki, J.; Yamaguchi, K.; Oda, S.; Matsuda, Y. L-ascorbic acid stimulates expression of smooth muscle-specific markers in smooth muscle cells both in vitro and in vivo. J. Cardiovasc. Pharmacol. 2003, 42, 745–751. [Google Scholar] [CrossRef]
- Pinnell, S.R. Regulation of collagen biosynthesis by ascorbic acid: A review. Yale J. Biol. Med. 1985, 58, 553–559. [Google Scholar]
- Byon, C.H.; Javed, A.; Dai, Q.; Kappes, J.C.; Clemens, T.L.; Darley-Usmar, V.M.; McDonald, J.M.; Chen, Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J. Biol. Chem. 2008, 283, 15319–15327. [Google Scholar] [CrossRef]
- Lian, J.B.; Javed, A.; Zaidi, S.K.; Lengner, C.; Montecino, M.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S. Regulatory controls for osteoblast growth and differentiation: Role of Runx/Cbfa/AML factors. Crit. Rev. Eukaryot. Gene Expr. 2004, 14, 1–41. [Google Scholar] [CrossRef]
- Shioi, A.; Nishizawa, Y.; Jono, S.; Koyama, H.; Hosoi, M.; Morii, H. Beta-glycerophosphate accelerates calcification in cultured bovine vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 2003–2009. [Google Scholar] [CrossRef]
- Mori, K.; Shioi, A.; Jono, S.; Nishizawa, Y.; Morii, H. Dexamethasone enhances In vitro vascular calcification by promoting osteoblastic differentiation of vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2112–2118. [Google Scholar] [CrossRef]
- Lin, X.; Shan, S.K.; Xu, F.; Zhong, J.Y.; Wu, F.; Duan, J.Y.; Guo, B.; Li, F.X.; Wang, Y.; Zheng, M.H.; et al. The crosstalk between endothelial cells and vascular smooth muscle cells aggravates high phosphorus-induced arterial calcification. Cell Death Dis. 2022, 13, 650. [Google Scholar] [CrossRef] [PubMed]
- Persiani, E.; Ceccherini, E.; Gisone, I.; Cecchettini, A.; Vozzi, F. Protocol to generate an in vitro model to study vascular calcification using human endothelial and smooth muscle cells. STAR Protoc. 2023, 4, 102328. [Google Scholar] [CrossRef] [PubMed]
- Shaver, M.; Gomez, K.; Kaiser, K.; Hutcheson, J.D. Mechanical stretch leads to increased caveolin-1 content and mineralization potential in extracellular vesicles from vascular smooth muscle cells. BMC Mol. Cell Biol. 2024, 25, 8. [Google Scholar] [CrossRef]
- Herrmann, J.; Gummi, M.R.; Xia, M.; van der Giet, M.; Tolle, M.; Schuchardt, M. Vascular Calcification in Rodent Models-Keeping Track with an Extented Method Assortment. Biology 2021, 10, 459. [Google Scholar] [CrossRef] [PubMed]
- Shobeiri, N.; Adams, M.A.; Holden, R.M. Vascular calcification in animal models of CKD: A review. Am. J. Nephrol. 2010, 31, 471–481. [Google Scholar] [CrossRef]
- Tolle, M.; Henkel, C.; Herrmann, J.; Daniel, C.; Babic, M.; Xia, M.; Schulz, A.M.; Amann, K.; van der Giet, M.; Schuchardt, M. Uremic mouse model to study vascular calcification and “inflamm-aging”. J. Mol. Med. 2022, 100, 1321–1330. [Google Scholar] [CrossRef]
- Bas, A.; Lopez, I.; Perez, J.; Rodriguez, M.; Aguilera-Tejero, E. Reversibility of calcitriol-induced medial artery calcification in rats with intact renal function. J. Bone Miner. Res. 2006, 21, 484–490. [Google Scholar] [CrossRef]
- Bouderlique, E.; Tang, E.; Zaworski, J.; Coudert, A.; Bazin, D.; Borondics, F.; Haymann, J.P.; Leftheriotis, G.; Martin, L.; Daudon, M.; et al. Vitamin D and Calcium Supplementation Accelerate Vascular Calcification in a Model of Pseudoxanthoma Elasticum. Int. J. Mol. Sci. 2022, 23, 2302. [Google Scholar] [CrossRef]
- Jono, S.; Nishizawa, Y.; Shioi, A.; Morii, H. 1,25-Dihydroxyvitamin D3 increases in vitro vascular calcification by modulating secretion of endogenous parathyroid hormone-related peptide. Circulation 1998, 98, 1302–1306. [Google Scholar] [CrossRef]
- Leon, C.; Wasan, K.M.; Sachs-Barrable, K.; Johnston, T.P. Acute P-407 administration to mice causes hypercholesterolemia by inducing cholesterolgenesis and down-regulating low-density lipoprotein receptor expression. Pharm. Res. 2006, 23, 1597–1607. [Google Scholar] [CrossRef]
- Palmer, W.K.; Emeson, E.E.; Johnston, T.P. Poloxamer 407-induced atherogenesis in the C57BL/6 mouse. Atherosclerosis 1998, 136, 115–123. [Google Scholar] [CrossRef]
- Johnston, T.P. Poloxamer 407 as a general lipase inhibitor: Its implications in lipid metabolism and atheroma formation in C57BL/6 mice. J. Pharm. Pharmacol. 2010, 62, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Goettsch, C.; Hutcheson, J.D.; Hagita, S.; Rogers, M.A.; Creager, M.D.; Pham, T.; Choi, J.; Mlynarchik, A.K.; Pieper, B.; Kjolby, M.; et al. A single injection of gain-of-function mutant PCSK9 adeno-associated virus vector induces cardiovascular calcification in mice with no genetic modification. Atherosclerosis 2016, 251, 109–118. [Google Scholar] [CrossRef]
- Lomashvili, K.A.; Monier-Faugere, M.C.; Wang, X.; Malluche, H.H.; O’Neill, W.C. Effect of bisphosphonates on vascular calcification and bone metabolism in experimental renal failure. Kidney Int. 2009, 75, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Bakhshian Nik, A.; Ng, H.H.; Garcia Russo, M.; Iacoviello, F.; Shearing, P.R.; Bertazzo, S.; Hutcheson, J.D. The Time-Dependent Role of Bisphosphonates on Atherosclerotic Plaque Calcification. J. Cardiovasc. Dev. Dis. 2022, 9, 168. [Google Scholar] [CrossRef] [PubMed]
- Price, P.A.; Faus, S.A.; Williamson, M.K. Bisphosphonates alendronate and ibandronate inhibit artery calcification at doses comparable to those that inhibit bone resorption. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 817–824. [Google Scholar] [CrossRef]
- Kawakami, K.; Ohya, M.; Yashiro, M.; Sonou, T.; Yamamoto, S.; Nakashima, Y.; Yano, T.; Tanaka, Y.; Ishida, K.; Kobashi, S.; et al. Bisphosphonate FYB-931 Prevents High Phosphate-Induced Vascular Calcification in Rat Aortic Rings by Altering the Dynamics of the Transformation of Calciprotein Particles. Calcif. Tissue Int. 2023, 113, 216–228. [Google Scholar] [CrossRef]
- Shimshi, M.; Abe, E.; Fisher, E.A.; Zaidi, M.; Fallon, J.T. Bisphosphonates induce inflammation and rupture of atherosclerotic plaques in apolipoprotein-E null mice. Biochem. Biophys. Res. Commun. 2005, 328, 790–793. [Google Scholar] [CrossRef]
- Toussaint, N.D.; Lau, K.K.; Strauss, B.J.; Polkinghorne, K.R.; Kerr, P.G. Effect of alendronate on vascular calcification in CKD stages 3 and 4: A pilot randomized controlled trial. Am. J. Kidney Dis. 2010, 56, 57–68. [Google Scholar] [CrossRef]
- Nitta, K.; Akiba, T.; Suzuki, K.; Uchida, K.; Watanabe, R.; Majima, K.; Aoki, T.; Nihei, H. Effects of cyclic intermittent etidronate therapy on coronary artery calcification in patients receiving long-term hemodialysis. Am. J. Kidney Dis. 2004, 44, 680–688. [Google Scholar] [CrossRef]
- Iseri, K.; Watanabe, M.; Yoshikawa, H.; Mitsui, H.; Endo, T.; Yamamoto, Y.; Iyoda, M.; Ryu, K.; Inaba, T.; Shibata, T. Effects of Denosumab and Alendronate on Bone Health and Vascular Function in Hemodialysis Patients: A Randomized, Controlled Trial. J. Bone Miner. Res. 2019, 34, 1014–1024. [Google Scholar] [CrossRef]
- Chen, C.L.; Chen, N.C.; Wu, F.Z.; Wu, M.T. Impact of denosumab on cardiovascular calcification in patients with secondary hyperparathyroidism undergoing dialysis: A pilot study. Osteoporos. Int. 2020, 31, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Samelson, E.J.; Miller, P.D.; Christiansen, C.; Daizadeh, N.S.; Grazette, L.; Anthony, M.S.; Egbuna, O.; Wang, A.; Siddhanti, S.R.; Cheung, A.M.; et al. RANKL inhibition with denosumab does not influence 3-year progression of aortic calcification or incidence of adverse cardiovascular events in postmenopausal women with osteoporosis and high cardiovascular risk. J. Bone Miner. Res. 2014, 29, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Block, G.A.; Klassen, P.S.; Lazarus, J.M.; Ofsthun, N.; Lowrie, E.G.; Chertow, G.M. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J. Am. Soc. Nephrol. 2004, 15, 2208–2218. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Martin, J.L.; Martinez-Camblor, P.; Dionisi, M.P.; Floege, J.; Ketteler, M.; London, G.; Locatelli, F.; Gorriz, J.L.; Rutkowski, B.; Ferreira, A.; et al. Improvement of mineral and bone metabolism markers is associated with better survival in haemodialysis patients: The COSMOS study. Nephrol. Dial. Transplant. 2015, 30, 1542–1551. [Google Scholar] [CrossRef]
- Sakaguchi, Y.; Fujii, N.; Shoji, T.; Hayashi, T.; Rakugi, H.; Isaka, Y. Hypomagnesemia is a significant predictor of cardiovascular and non-cardiovascular mortality in patients undergoing hemodialysis. Kidney Int. 2014, 85, 174–181. [Google Scholar] [CrossRef]
- Mohamed, O.N.; Mohamed, M.R.M.; Hassan, I.G.; Alakkad, A.F.; Othman, A.; Setouhi, A.; Issa, A.S. The Relationship of Fetuin-A with Coronary Calcification, Carotid Atherosclerosis, and Mortality Risk in Non-Dialysis Chronic Kidney Disease. J. Lipid Atheroscler. 2024, 13, 194–211. [Google Scholar] [CrossRef]
- Bommer, J.; Locatelli, F.; Satayathum, S.; Keen, M.L.; Goodkin, D.A.; Saito, A.; Akiba, T.; Port, F.K.; Young, E.W. Association of predialysis serum bicarbonate levels with risk of mortality and hospitalization in the Dialysis Outcomes and Practice Patterns Study (DOPPS). Am. J. Kidney Dis. 2004, 44, 661–671. [Google Scholar] [CrossRef]
- Palit, S.; Kendrick, J. Vascular calcification in chronic kidney disease: Role of disordered mineral metabolism. Curr. Pharm. Des. 2014, 20, 5829–5833. [Google Scholar] [CrossRef]
- St Peter, W.L.; Wazny, L.D.; Weinhandl, E.D. Phosphate-Binder Use in US Dialysis Patients: Prevalence, Costs, Evidence, and Policies. Am. J. Kidney Dis. 2018, 71, 246–253. [Google Scholar] [CrossRef]
- Madaj, R.; Gostynski, B.; Pawlowska, R.; Chworos, A. Tissue-Nonspecific Alkaline Phosphatase (TNAP) as the Enzyme Involved in the Degradation of Nucleotide Analogues in the Ligand Docking and Molecular Dynamics Approaches. Biomolecules 2021, 11, 1104. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, D.; Hofmann, C.; Jakob, F.; Klopocki, E.; Graser, S. Tissue-Nonspecific Alkaline Phosphatase-A Gatekeeper of Physiological Conditions in Health and a Modulator of Biological Environments in Disease. Biomolecules 2020, 10, 1648. [Google Scholar] [CrossRef] [PubMed]
- Hamczyk, M.R.; Villa-Bellosta, R. Pyrophosphate metabolism and calcification. Aging 2018, 10, 3652–3653. [Google Scholar] [CrossRef]
- Sheen, C.R.; Kuss, P.; Narisawa, S.; Yadav, M.C.; Nigro, J.; Wang, W.; Chhea, T.N.; Sergienko, E.A.; Kapoor, K.; Jackson, M.R.; et al. Pathophysiological role of vascular smooth muscle alkaline phosphatase in medial artery calcification. J. Bone Miner. Res. 2015, 30, 824–836. [Google Scholar] [CrossRef] [PubMed]
- Savinov, A.Y.; Salehi, M.; Yadav, M.C.; Radichev, I.; Millan, J.L.; Savinova, O.V. Transgenic Overexpression of Tissue-Nonspecific Alkaline Phosphatase (TNAP) in Vascular Endothelium Results in Generalized Arterial Calcification. J. Am. Heart Assoc. 2015, 4, 12. [Google Scholar] [CrossRef]
- Liu, K.; Yu, Y.; Yuan, Y.; Xu, X.; Lei, W.; Niu, R.; Shen, M.; Zhou, L.; Peng, R.; Wang, Q.; et al. Elevated Levels of Serum Alkaline Phosphatase are Associated with Increased Risk of Cardiovascular Disease: A Prospective Cohort Study. J. Atheroscler. Thromb. 2023, 30, 795–819. [Google Scholar] [CrossRef]
- Opdebeeck, B.; Neven, E.; Millan, J.L.; Pinkerton, A.B.; D’Haese, P.C.; Verhulst, A. Pharmacological TNAP inhibition efficiently inhibits arterial media calcification in a warfarin rat model but deserves careful consideration of potential physiological bone formation/mineralization impairment. Bone 2020, 137, 115392. [Google Scholar] [CrossRef]
- Bessueille, L.; Kawtharany, L.; Quillard, T.; Goettsch, C.; Briolay, A.; Taraconat, N.; Balayssac, S.; Gilard, V.; Mebarek, S.; Peyruchaud, O.; et al. Inhibition of alkaline phosphatase impairs dyslipidemia and protects mice from atherosclerosis. Transl. Res. 2023, 251, 2–13. [Google Scholar] [CrossRef]
- Andleeb, H.; Hussain, M.; Abida Ejaz, S.; Sevigny, J.; Farman, M.; Yasinzai, M.; Zhang, J.; Iqbal, J.; Hameed, S. Synthesis and computational studies of highly selective inhibitors of human recombinant tissue non-specific alkaline phosphatase (h-TNAP): A therapeutic target against vascular calcification. Bioorg. Chem. 2020, 101, 103999. [Google Scholar] [CrossRef]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997, 386, 78–81. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Spronk, H.M.; Soute, B.A.; Schiffers, P.M.; DeMey, J.G.; Vermeer, C. Regression of warfarin-induced medial elastocalcinosis by high intake of vitamin K in rats. Blood 2007, 109, 2823–2831. [Google Scholar] [CrossRef] [PubMed]
- Shea, M.K.; Holden, R.M. Vitamin K status and vascular calcification: Evidence from observational and clinical studies. Adv. Nutr. 2012, 3, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Lehto, S.; Niskanen, L.; Suhonen, M.; Ronnemaa, T.; Laakso, M. Medial artery calcification. A neglected harbinger of cardiovascular complications in non-insulin-dependent diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 978–983. [Google Scholar] [CrossRef]
- Johnson, R.C.; Leopold, J.A.; Loscalzo, J. Vascular calcification: Pathobiological mechanisms and clinical implications. Circ. Res. 2006, 99, 1044–1059. [Google Scholar] [CrossRef]
- Kay, A.M.; Simpson, C.L.; Stewart, J.A., Jr. The Role of AGE/RAGE Signaling in Diabetes-Mediated Vascular Calcification. J. Diabetes Res. 2016, 2016, 6809703. [Google Scholar] [CrossRef]
- Rhee, S.Y.; Kim, Y.S. The Role of Advanced Glycation End Products in Diabetic Vascular Complications. Diabetes Metab. J. 2018, 42, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Harja, E.; Bu, D.X.; Hudson, B.I.; Chang, J.S.; Shen, X.; Hallam, K.; Kalea, A.Z.; Lu, Y.; Rosario, R.H.; Oruganti, S.; et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE−/− mice. J. Clin. Investig. 2008, 118, 183–194. [Google Scholar] [CrossRef]
- Hofmann Bowman, M.A.; Gawdzik, J.; Bukhari, U.; Husain, A.N.; Toth, P.T.; Kim, G.; Earley, J.; McNally, E.M. S100A12 in vascular smooth muscle accelerates vascular calcification in apolipoprotein E-null mice by activating an osteogenic gene regulatory program. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 337–344. [Google Scholar] [CrossRef]
- Kawakami, R.; Katsuki, S.; Travers, R.; Romero, D.C.; Becker-Greene, D.; Passos, L.S.A.; Higashi, H.; Blaser, M.C.; Sukhova, G.K.; Buttigieg, J.; et al. S100A9-RAGE Axis Accelerates Formation of Macrophage-Mediated Extracellular Vesicle Microcalcification in Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1838–1853. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Jones, A.M.; Sidgwick, G.P.; Arafat, A.M.; Alexander, Y.M.; Wilkinson, F.L. Small Molecule Glycomimetics Inhibit Vascular Calcification via c-Met/Notch3/HES1 Signalling. Cell Physiol. Biochem. 2019, 53, 323–336. [Google Scholar] [CrossRef]
- Sidgwick, G.P.; Weston, R.; Mahmoud, A.M.; Schiro, A.; Serracino-Inglott, F.; Tandel, S.M.; Skeoch, S.; Bruce, I.N.; Jones, A.M.; Alexander, M.Y.; et al. Novel Glycomimetics Protect against Glycated Low-Density Lipoprotein-Induced Vascular Calcification In Vitro via Attenuation of the RAGE/ERK/CREB Pathway. Cells 2024, 13, 312. [Google Scholar] [CrossRef] [PubMed]
- van Loo, G.; Bertrand, M.J.M. Death by TNF: A road to inflammation. Nat. Rev. Immunol. 2023, 23, 289–303. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rifai, N.; Pfeffer, M.; Sacks, F.; Lepage, S.; Braunwald, E. Elevation of tumor necrosis factor-alpha and increased risk of recurrent coronary events after myocardial infarction. Circulation 2000, 101, 2149–2153. [Google Scholar] [CrossRef] [PubMed]
- Branen, L.; Hovgaard, L.; Nitulescu, M.; Bengtsson, E.; Nilsson, J.; Jovinge, S. Inhibition of tumor necrosis factor-alpha reduces atherosclerosis in apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2137–2142. [Google Scholar] [CrossRef]
- Zhang, L.; Peppel, K.; Sivashanmugam, P.; Orman, E.S.; Brian, L.; Exum, S.T.; Freedman, N.J. Expression of Tumor Necrosis Factor Receptor-1 in Arterial Wall Cells Promotes Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, X.; Bian, F.; Wu, P.; Xing, S.; Xu, G.; Li, W.; Chi, J.; Ouyang, C.; Zheng, T.; et al. TNF-alpha promotes early atherosclerosis by increasing transcytosis of LDL across endothelial cells: Crosstalk between NF-kappaB and PPAR-gamma. J. Mol. Cell Cardiol. 2014, 72, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Tintut, Y.; Patel, J.; Parhami, F.; Demer, L.L. Tumor necrosis factor-alpha promotes in vitro calcification of vascular cells via the cAMP pathway. Circulation 2000, 102, 2636–2642. [Google Scholar] [CrossRef]
- Song, X.; Song, Y.; Ma, Q.; Fang, K.; Chang, X. M1-Type Macrophages Secrete TNF-alpha to Stimulate Vascular Calcification by Upregulating CA1 and CA2 Expression in VSMCs. J. Inflamm. Res. 2023, 16, 3019–3032. [Google Scholar] [CrossRef]
- Oberoi, R.; Vlacil, A.K.; Schuett, J.; Schosser, F.; Schuett, H.; Tietge, U.J.F.; Schieffer, B.; Grote, K. Anti-tumor necrosis factor-alpha therapy increases plaque burden in a mouse model of experimental atherosclerosis. Atherosclerosis 2018, 277, 80–89. [Google Scholar] [CrossRef]
- Shobeiri, N.; Bendeck, M.P. Interleukin-1beta Is a Key Biomarker and Mediator of Inflammatory Vascular Calcification. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 179–180. [Google Scholar] [CrossRef]
- Shen, J.; Zhao, M.; Zhang, C.; Sun, X. IL-1beta in atherosclerotic vascular calcification: From bench to bedside. Int. J. Biol. Sci. 2021, 17, 4353–4364. [Google Scholar] [CrossRef] [PubMed]
- Ceneri, N.; Zhao, L.; Young, B.D.; Healy, A.; Coskun, S.; Vasavada, H.; Yarovinsky, T.O.; Ike, K.; Pardi, R.; Qin, L.; et al. Rac2 Modulates Atherosclerotic Calcification by Regulating Macrophage Interleukin-1beta Production. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.S.; Tong, Y.; Ling, L.; Chen, D.; Sun, H.J.; Zhou, H.; Qi, X.H.; Chen, Q.; Li, Y.H.; Kang, Y.M.; et al. NLRP3 Gene Deletion Attenuates Angiotensin II-Induced Phenotypic Transformation of Vascular Smooth Muscle Cells and Vascular Remodeling. Cell Physiol. Biochem. 2017, 44, 2269–2280. [Google Scholar] [CrossRef]
- Rothman, A.M.; MacFadyen, J.; Thuren, T.; Webb, A.; Harrison, D.G.; Guzik, T.J.; Libby, P.; Glynn, R.J.; Ridker, P.M. Effects of Interleukin-1beta Inhibition on Blood Pressure, Incident Hypertension, and Residual Inflammatory Risk: A Secondary Analysis of CANTOS. Hypertension 2020, 75, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Haybar, H.; Shokuhian, M.; Bagheri, M.; Davari, N.; Saki, N. Involvement of circulating inflammatory factors in prognosis and risk of cardiovascular disease. J. Mol. Cell Cardiol. 2019, 132, 110–119. [Google Scholar] [CrossRef]
- Awan, Z.; Denis, M.; Roubtsova, A.; Essalmani, R.; Marcinkiewicz, J.; Awan, A.; Gram, H.; Seidah, N.G.; Genest, J. Reducing Vascular Calcification by Anti-IL-1beta Monoclonal Antibody in a Mouse Model of Familial Hypercholesterolemia. Angiology 2016, 67, 157–167. [Google Scholar] [CrossRef]
- Hassan, M. CANTOS: A breakthrough that proves the inflammatory hypothesis of atherosclerosis. Glob. Cardiol. Sci. Pract. 2018, 2018, 2. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-Inflammatory Therapy With Canakinumab for the Prevention of Hospitalization for Heart Failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, B. Systematic review and meta-analysis for the association of bone mineral density and osteoporosis/osteopenia with vascular calcification in women. Int. J. Rheum. Dis. 2017, 20, 154–160. [Google Scholar] [CrossRef]
- Persy, V.; D’Haese, P. Vascular calcification and bone disease: The calcification paradox. Trends Mol. Med. 2009, 15, 405–416. [Google Scholar] [CrossRef]
- Shapiro, I.M.; Landis, W.J.; Risbud, M.V. Matrix vesicles: Are they anchored exosomes? Bone 2015, 79, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Houston, D.A.; Farquharson, C.; MacRae, V.E. Characterisation of matrix vesicles in skeletal and soft tissue mineralisation. Bone 2016, 87, 147–158. [Google Scholar] [CrossRef]
- New, S.E.; Aikawa, E. Role of extracellular vesicles in de novo mineralization: An additional novel mechanism of cardiovascular calcification. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1753–1758. [Google Scholar] [CrossRef]
- Hjortnaes, J.; Butcher, J.; Figueiredo, J.L.; Riccio, M.; Kohler, R.H.; Kozloff, K.M.; Weissleder, R.; Aikawa, E. Arterial and aortic valve calcification inversely correlates with osteoporotic bone remodelling: A role for inflammation. Eur. Heart J. 2010, 31, 1975–1984. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef]
- Aikawa, M.; Rabkin, E.; Sugiyama, S.; Voglic, S.J.; Fukumoto, Y.; Furukawa, Y.; Shiomi, M.; Schoen, F.J.; Libby, P. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation 2001, 103, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, J.O.; Aikawa, M.; Tung, C.H.; Aikawa, E.; Kim, D.E.; Ntziachristos, V.; Weissleder, R.; Libby, P. Inflammation in atherosclerosis: Visualizing matrix metalloproteinase action in macrophages in vivo. Circulation 2006, 114, 55–62. [Google Scholar] [CrossRef]
- Bakhshian Nik, A.; Kaiser, K.; Sun, P.; Khomtchouk, B.B.; Hutcheson, J.D. Altered Caveolin-1 Dynamics Result in Divergent Mineralization Responses in Bone and Vascular Calcification. Cell. Mol. Bioeng. 2023, 16, 299–308. [Google Scholar] [CrossRef]
- Bakhshian Nik, A.; Ng, H.H.; Ashbrook, S.K.; Sun, P.; Iacoviello, F.; Shearing, P.R.; Bertazzo, S.; Mero, D.; Khomtchouk, B.B.; Hutcheson, J.D. Epidermal growth factor receptor inhibition prevents vascular calcifying extracellular vesicle biogenesis. Am. J. Physiol. Heart Circ. Physiol. 2023, 324, H553–H570. [Google Scholar] [CrossRef]
- Kelly-Arnold, A.; Maldonado, N.; Laudier, D.; Aikawa, E.; Cardoso, L.; Weinbaum, S. Revised microcalcification hypothesis for fibrous cap rupture in human coronary arteries. Proc. Natl. Acad. Sci. USA 2013, 110, 10741–10746. [Google Scholar] [CrossRef] [PubMed]
- Vengrenyuk, Y.; Cardoso, L.; Weinbaum, S. Micro-CT based analysis of a new paradigm for vulnerable plaque rupture: Cellular microcalcifications in fibrous caps. Mol. Cell Biomech. 2008, 5, 37–47. [Google Scholar]
- Imoto, K.; Hiro, T.; Fujii, T.; Murashige, A.; Fukumoto, Y.; Hashimoto, G.; Okamura, T.; Yamada, J.; Mori, K.; Matsuzaki, M. Longitudinal structural determinants of atherosclerotic plaque vulnerability: A computational analysis of stress distribution using vessel models and three-dimensional intravascular ultrasound imaging. J. Am. Coll. Cardiol. 2005, 46, 1507–1515. [Google Scholar] [CrossRef]
- Wang, Y.; Osborne, M.T.; Tung, B.; Li, M.; Li, Y. Imaging Cardiovascular Calcification. J. Am. Heart Assoc. 2018, 7, e008564. [Google Scholar] [CrossRef] [PubMed]
- Ritman, E.L. Small-animal CT—Its Difference from, and Impact on, Clinical CT. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrometers Detect. Assoc. Equip. 2007, 580, 968–970. [Google Scholar] [CrossRef] [PubMed]
- Motoyama, S.; Kondo, T.; Sarai, M.; Sugiura, A.; Harigaya, H.; Sato, T.; Inoue, K.; Okumura, M.; Ishii, J.; Anno, H.; et al. Multislice computed tomographic characteristics of coronary lesions in acute coronary syndromes. J. Am. Coll. Cardiol. 2007, 50, 319–326. [Google Scholar] [CrossRef]
- Liu, H.; Wingert, A.; Wang, J.; Zhang, J.; Wang, X.; Sun, J.; Chen, F.; Khalid, S.G.; Jiang, J.; Zheng, D. Extraction of Coronary Atherosclerotic Plaques From Computed Tomography Imaging: A Review of Recent Methods. Front. Cardiovasc. Med. 2021, 8, 597568. [Google Scholar] [CrossRef]
- Sakuma, H. Coronary CT versus MR angiography: The role of MR angiography. Radiology 2011, 258, 340–349. [Google Scholar] [CrossRef]
- Yoon, Y.E.; Hong, Y.J.; Kim, H.K.; Kim, J.A.; Na, J.O.; Yang, D.H.; Kim, Y.J.; Choi, E.Y. 2014 Korean guidelines for appropriate utilization of cardiovascular magnetic resonance imaging: A joint report of the Korean Society of Cardiology and the Korean Society of Radiology. Korean J. Radiol. 2014, 15, 659–688. [Google Scholar] [CrossRef]
- Kadoglou, N.P.; Stasinopoulou, M.; Velidakis, N.; Khattab, E.; Christodoulou, E.; Gkougkoudi, E.; Valsami, G. The Complex Mechanisms and the Potential Effects of Statins on Vascular Calcification: A Narrative Review. Rev. Cardiovasc. Med. 2024, 25, 51. [Google Scholar] [CrossRef]
- Shreya, D.; Zamora, D.I.; Patel, G.S.; Grossmann, I.; Rodriguez, K.; Soni, M.; Joshi, P.K.; Patel, S.C.; Sange, I. Coronary Artery Calcium Score—A Reliable Indicator of Coronary Artery Disease? Cureus 2021, 13, e20149. [Google Scholar] [CrossRef] [PubMed]
- Puri, R.; Nicholls, S.J.; Shao, M.; Kataoka, Y.; Uno, K.; Kapadia, S.R.; Tuzcu, E.M.; Nissen, S.E. Impact of statins on serial coronary calcification during atheroma progression and regression. J. Am. Coll. Cardiol. 2015, 65, 1273–1282. [Google Scholar] [CrossRef]
- Lee, J.M.; Choi, G.; Hwang, D.; Park, J.; Kim, H.J.; Doh, J.H.; Nam, C.W.; Na, S.H.; Shin, E.S.; Taylor, C.A.; et al. Impact of Longitudinal Lesion Geometry on Location of Plaque Rupture and Clinical Presentations. JACC Cardiovasc. Imaging 2017, 10, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.Z.; Ahmed, M.E.; Huang, Y.; Hakim, D.; Maynard, C.; Cefalo, N.V.; Coskun, A.U.; Costopoulos, C.; Maehara, A.; Stone, G.W.; et al. Comprehensive biomechanical and anatomical atherosclerotic plaque metrics predict major adverse cardiovascular events: A new tool for clinical decision making. Atherosclerosis 2024, 390, 117449. [Google Scholar] [CrossRef] [PubMed]
- Pallon, J.; Homman, P.; Pinheiro, T.; Halpern, M.; Malmqvist, K. A view on elemental distribution alterations of coronary artery walls in atherogenesis. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 1995, 104, 344–350. [Google Scholar] [CrossRef]
- Otsuka, F.; Sakakura, K.; Yahagi, K.; Joner, M.; Virmani, R. Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler. Thromb. Vasc. Biol. 2014, 34, 724–736. [Google Scholar] [CrossRef]
- Hutcheson, J.D.; Maldonado, N.; Aikawa, E. Small entities with large impact: Microcalcifications and atherosclerotic plaque vulnerability. Curr. Opin. Lipidol. 2014, 25, 327–332. [Google Scholar] [CrossRef]
- Pries, A.R.; Reglin, B.; Secomb, T.W. Remodeling of blood vessels: Responses of diameter and wall thickness to hemodynamic and metabolic stimuli. Hypertension 2005, 46, 725–731. [Google Scholar] [CrossRef]
- Montanaro, M.; Scimeca, M.; Anemona, L.; Servadei, F.; Giacobbi, E.; Bonfiglio, R.; Bonanno, E.; Urbano, N.; Ippoliti, A.; Santeusanio, G.; et al. The Paradox Effect of Calcification in Carotid Atherosclerosis: Microcalcification is Correlated with Plaque Instability. Int. J. Mol. Sci. 2021, 22, 395. [Google Scholar] [CrossRef]
- Richardson, P.D.; Davies, M.J.; Born, G.V. Influence of plaque configuration and stress distribution on fissuring of coronary atherosclerotic plaques. Lancet 1989, 2, 941–944. [Google Scholar] [CrossRef]
- Maldonado, N.; Kelly-Arnold, A.; Cardoso, L.; Weinbaum, S. The explosive growth of small voids in vulnerable cap rupture; cavitation and interfacial debonding. J. Biomech. 2013, 46, 396–401. [Google Scholar] [CrossRef]
- Massarwa, E.; Aronis, Z.; Eliasy, R.; Einav, S.; Haj-Ali, R. Nonlinear multiscale analysis of coronary atherosclerotic vulnerable plaque artery: Fluid-structural modeling with micromechanics. Biomech. Model. Mechanobiol. 2021, 20, 1889–1901. [Google Scholar] [CrossRef] [PubMed]
- Saremi, F.; Achenbach, S. Coronary plaque characterization using CT. AJR Am. J. Roentgenol. 2015, 204, W249–W260. [Google Scholar] [CrossRef] [PubMed]
- Vancheri, F.; Longo, G.; Vancheri, S.; Danial, J.S.H.; Henein, M.Y. Coronary Artery Microcalcification: Imaging and Clinical Implications. Diagnostics 2019, 9, 125. [Google Scholar] [CrossRef]
- Dweck, M.R.; Aikawa, E.; Newby, D.E.; Tarkin, J.M.; Rudd, J.H.; Narula, J.; Fayad, Z.A. Noninvasive Molecular Imaging of Disease Activity in Atherosclerosis. Circ. Res. 2016, 119, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Yock, P. Intravascular ultrasound: Novel pathophysiological insights and current clinical applications. Circulation 2001, 103, 604–616. [Google Scholar] [CrossRef]
- Ban, X.; Li, Z.; Duan, Y.; Xu, K.; Xiong, J.; Tu, Y. Advanced imaging modalities provide new insights into coronary artery calcification. Eur. J. Radiol. 2022, 157, 110601. [Google Scholar] [CrossRef]
- Hienz, S.A.; Paliwal, S.; Ivanovski, S. Mechanisms of Bone Resorption in Periodontitis. J. Immunol. Res. 2015, 2015, 615486. [Google Scholar] [CrossRef]
- Udagawa, N.; Takahashi, N.; Akatsu, T.; Tanaka, H.; Sasaki, T.; Nishihara, T.; Koga, T.; Martin, T.J.; Suda, T. Origin of osteoclasts: Mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc. Natl. Acad. Sci. USA 1990, 87, 7260–7264. [Google Scholar] [CrossRef]
- Chinetti-Gbaguidi, G.; Daoudi, M.; Rosa, M.; Vinod, M.; Louvet, L.; Copin, C.; Fanchon, M.; Vanhoutte, J.; Derudas, B.; Belloy, L.; et al. Human Alternative Macrophages Populate Calcified Areas of Atherosclerotic Lesions and Display Impaired RANKL-Induced Osteoclastic Bone Resorption Activity. Circ. Res. 2017, 121, 19–30. [Google Scholar] [CrossRef]
- Qiao, J.H.; Mishra, V.; Fishbein, M.C.; Sinha, S.K.; Rajavashisth, T.B. Multinucleated giant cells in atherosclerotic plaques of human carotid arteries: Identification of osteoclast-like cells and their specific proteins in artery wall. Exp. Mol. Pathol. 2015, 99, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Hutcheson, J.D.; Blaser, M.C.; Aikawa, E. Giving Calcification Its Due: Recognition of a Diverse Disease: A First Attempt to Standardize the Field. Circ. Res. 2017, 120, 270–273. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valentin Cabrera, A.M.; Ashbrook, S.K.; Hutcheson, J.D. Considerations on the Development of Therapeutics in Vascular Calcification. J. Cardiovasc. Dev. Dis. 2025, 12, 206. https://doi.org/10.3390/jcdd12060206
Valentin Cabrera AM, Ashbrook SK, Hutcheson JD. Considerations on the Development of Therapeutics in Vascular Calcification. Journal of Cardiovascular Development and Disease. 2025; 12(6):206. https://doi.org/10.3390/jcdd12060206
Chicago/Turabian StyleValentin Cabrera, Ana M., Sophie K. Ashbrook, and Joshua D. Hutcheson. 2025. "Considerations on the Development of Therapeutics in Vascular Calcification" Journal of Cardiovascular Development and Disease 12, no. 6: 206. https://doi.org/10.3390/jcdd12060206
APA StyleValentin Cabrera, A. M., Ashbrook, S. K., & Hutcheson, J. D. (2025). Considerations on the Development of Therapeutics in Vascular Calcification. Journal of Cardiovascular Development and Disease, 12(6), 206. https://doi.org/10.3390/jcdd12060206