Neuropathological Mechanisms Associated with Pesticides in Alzheimer’s Disease


{kind=link}
Abstract
1. Introduction
2. Epidemiological Links between Pesticide Exposure and Alzheimer’s Disease
3. Pesticides and the Induction of Alzheimer’s Disease Markers in Cell Culture and Animal Models
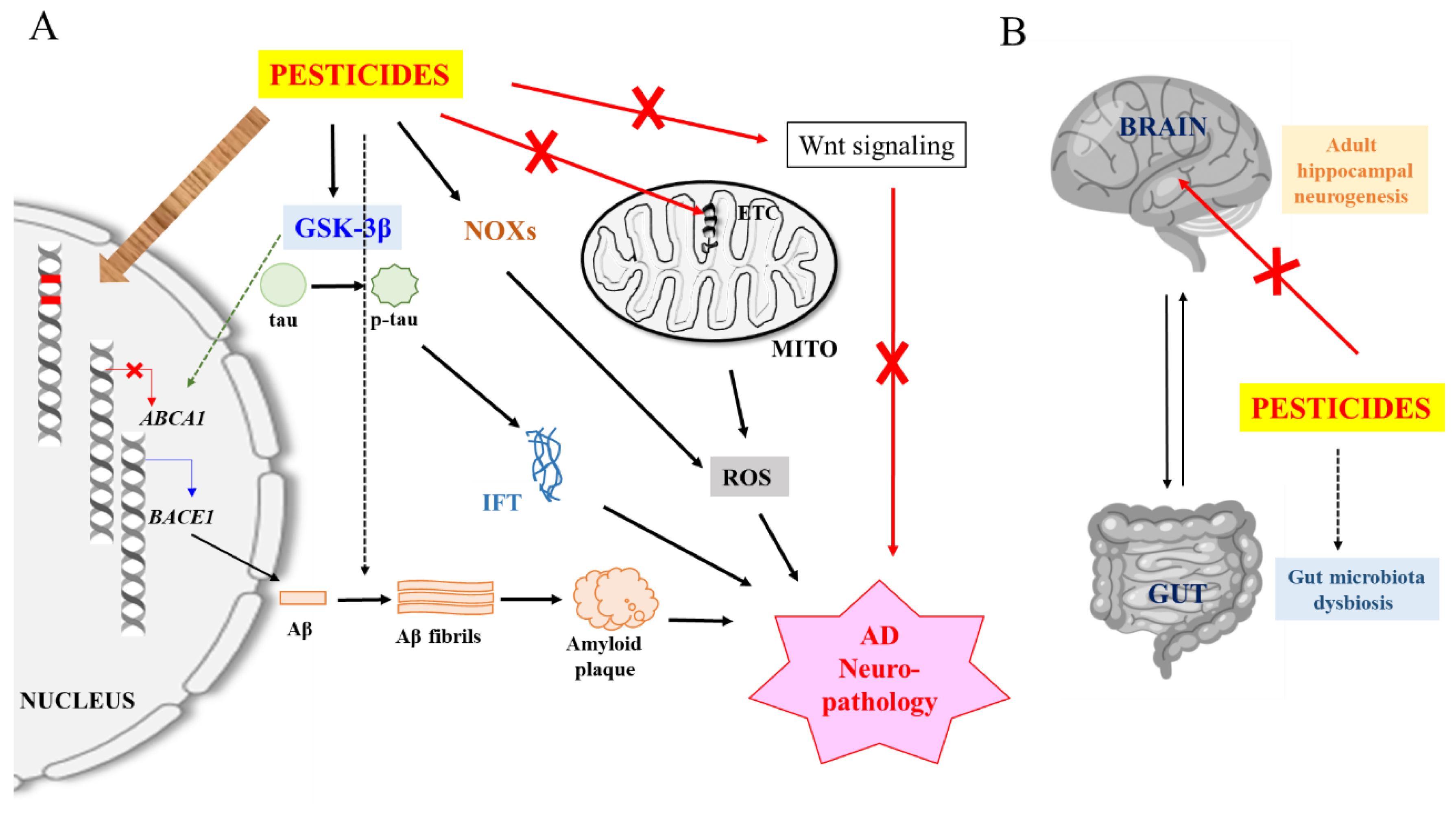
4. Potential Neuropathological Mechanisms of Pesticides
4.1. Induction of Oxidative Stress and Neuroinflammation
4.2. Enhancement of Aβ and tau Expression, Modification and Clearance
4.3. Promotion of Amyloidogenesis
5. Speculative Neuropathological Mechanisms of Pesticides
5.1. DNA Damage and Somatic Mutations
5.2. Epigenetic Mechanisms
5.3. Effect on Adult Neurogenesis
5.4. Dysfunction of the Brain-Gut Axis
6. Epilogue
Funding
Acknowledgments
Conflicts of Interest
References
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer disease: An update on pathobiology and treatment strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement 2016, 12, 459–509. [Google Scholar] [CrossRef]
- Bertram, L.; Tanzi, R.E. Thirty years of Alzheimer’s disease genetics: The implications of systematic meta-analyses. Nat. Rev. Neurosci. 2008, 9, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar] [CrossRef] [PubMed]
- Nhan, H.S.; Chiang, K.; Koo, E.H. The multifaceted nature of amyloid precursor protein and its proteolytic fragments: Friends and foes. Acta Neuropathol. 2015, 129, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, R.; Shen, Y. β-Secretase: Its biology as a therapeutic target in diseases. Trends Pharmacol. Sci. 2013, 34, 215–225. [Google Scholar] [CrossRef]
- De Strooper, B.; Iwatsubo, T.; Wolfe, M.S. Presenilins and γ-secretase: Structure, function, and role in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006304. [Google Scholar] [CrossRef]
- Andrew, R.J.; Kellett, K.A.B.; Thinakaran, G.; Hooper, N.M. A Greek Tragedy: The Growing Complexity of Alzheimer Amyloid Precursor Protein Proteolysis. J. Biol. Chem. 2016, 291, 19235–19244. [Google Scholar] [CrossRef]
- Kuhn, P.H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Rossner, S.; Lichtenthaler, S.F. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef]
- Tang, B.L. Alzheimer’s disease: Channeling APP to non-amyloidogenic processing. Biochem. Biophys. Res. Commun. 2005, 331, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β oligomer hypothesis: Beginning of the third decade. J. Alzheimers Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Tan, L.; Yu, J.T.; Tan, L. Tau in Alzheimer’s disease: Mechanisms and therapeutic strategies. Curr. Alzheimer Res. 2018, 15, 283–300. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef]
- Carmona, S.; Hardy, J.; Guerreiro, R. The genetic landscape of Alzheimer disease. Handb. Clin. Neurol. 2018, 148, 395–408. [Google Scholar]
- Bellenguez, C.; Grenier-Boley, B.; Lambert, J.C. Genetics of Alzheimer’s disease: Where we are, and where we are going. Curr. Opin. Neurobiol. 2019, 61, 40–48. [Google Scholar] [CrossRef]
- Dunn, A.R.; O’Connell, K.M.S.; Kaczorowski, C.C. Gene-by-environment interactions in Alzheimer’s disease and Parkinson’s disease. Neurosci. Biobehav. Rev. 2019, 103, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Eid, A.; Mhatre, I.; Richardson, J.R. Gene-environment interactions in Alzheimer’s disease: A potential path to precision medicine. Pharmacol. Ther. 2019, 199, 173–187. [Google Scholar] [CrossRef]
- Michaelson, D.M. APOE ε4: The most prevalent yet understudied risk factor for Alzheimer’s disease. Alzheimers Dement 2014, 10, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 2015, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Killin, L.O.J.; Starr, J.M.; Shiue, I.J.; Russ, T.C. Environmental risk factors for dementia: A systematic review. BMC Geriatr. 2016, 16, 175. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiao, Q.; Xu, H.; Du, X.; Shi, L.; Jia, F.; Jiang, H. Biometal dyshomeostasis and toxic metal accumulations in the development of Alzheimer’s disease. Front. Mol. Neurosci. 2017, 10, 339. [Google Scholar] [CrossRef]
- Huat, T.J.; Camats-Perna, J.; Newcombe, E.A.; Valmas, N.; Kitazawa, M.; Medeiros, R. Metal toxicity links to Alzheimer’s disease and neuroinflammation. J. Mol. Biol. 2019, 431, 1843–1868. [Google Scholar] [CrossRef]
- Huang, H.; Bihaqi, S.W.; Cui, L.; Zawia, N.H. In vitro Pb exposure disturbs the balance between Aβ production and elimination: The role of AβPP and neprilysin. Neurotoxicology 2011, 32, 300–306. [Google Scholar] [CrossRef]
- Bakulski, K.M.; Rozek, L.S.; Dolinoy, D.C.; Paulson, H.L.; Hu, H. Alzheimer’s disease and environmental exposure to lead: The epidemiologic evidence and potential role of epigenetics. Curr. Alzheimer Res. 2012, 9, 563–573. [Google Scholar] [CrossRef]
- Brown, E.E.; Shah, P.; Pollock, B.G.; Gerretsen, P.; Graff-Guerrero, A. Lead (Pb) in Alzheimer’s dementia: A systematic review of human case- control studies. Curr. Alzheimer Res. 2019, 16, 353–361. [Google Scholar] [CrossRef]
- Weibull, M.G.M.; Simonsen, S.; Oksbjerg, C.R.; Tiwari, M.K.; Hemmingsen, L. Effects of Cu (II) on the aggregation of amyloid-β. J. Biol. Inorg. Chem. 2019, 24, 1197–1215. [Google Scholar] [CrossRef] [PubMed]
- Mathys, Z.K.; White, A.R. Copper and Alzheimer’s disease. Adv. Neurobiol. 2017, 18, 199–216. [Google Scholar] [PubMed]
- Wang, Z.; Wei, X.; Yang, J.; Suo, J.; Chen, J.; Liu, X.; Zhao, X. Chronic exposure to aluminum and risk of Alzheimer’s disease: A meta-analysis. Neurosci. Lett. 2016, 610, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Colomina, M.T.; Peris-Sampedro, F. Aluminum and Alzheimer’s disease. Adv. Neurobiol. 2017, 18, 183–197. [Google Scholar]
- Langston, J.W. The MPTP story. J. Parkinson Dis. 2017, 7, S11–S19. [Google Scholar] [CrossRef]
- Cox, P.A.; Kostrzewa, R.M.; Guillemin, G.J. BMAA and neurodegenerative illness. Neurotox. Res. 2018, 33, 178–183. [Google Scholar] [CrossRef]
- Yegambaram, M.; Manivannan, B.; Beach, T.G.; Halden, R.U. Role of environmental contaminants in the etiology of Alzheimer’s disease: A review. Curr. Alzheimer Res. 2015, 12, 116–146. [Google Scholar] [CrossRef]
- Hernández, A.F.; González-Alzaga, B.; López-Flores, I.; Lacasaña, M. Systematic reviews on neurodevelopmental and neurodegenerative disorders linked to pesticide exposure: Methodological features and impact on risk assessment. Environ. Int. 2016, 92–93, 657–679. [Google Scholar] [CrossRef]
- Sánchez-Santed, F.; Colomina, M.T.; Herrero Hernández, E. Organophosphate pesticide exposure and neurodegeneration. Cortex 2016, 74, 417–426. [Google Scholar] [CrossRef]
- Cassereau, J.; Ferré, M.; Chevrollier, A.; Codron, P.; Verny, C.; Homedan, C.; Lenaers, G.; Procaccio, V.; May-Panloup, P.; Reynier, P. Neurotoxicity of insecticides. Curr. Med. Chem. 2017, 24, 2988–3001. [Google Scholar] [CrossRef]
- Mostafalou, S.; Abdollahi, M. The link of organophosphorus pesticides with neurodegenerative and neurodevelopmental diseases based on evidence and mechanisms. Toxicology 2018, 409, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Zaganas, I.; Kapetanaki, S.; Mastorodemos, V.; Kanavouras, K.; Colosio, C.; Wilks, M.F.; Tsatsakis, A.M. Linking pesticide exposure and dementia: What is the evidence? Toxicology 2013, 307, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.W.; Hong, J.S.; Ohrr, H.; Yi, J.J. Agent Orange exposure and disease prevalence in Korean Vietnam veterans: The Korean veterans health study. Environ. Res. 2014, 133, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Bosma, H.; van Boxtel, M.P.; Ponds, R.W.; Houx, P.J.; Jolles, J. Pesticide exposure and risk of mild cognitive dysfunction. Lancet 2000, 356, 912–913. [Google Scholar] [CrossRef]
- Hayden, K.M.; Norton, M.C.; Darcey, D.; Ostbye, T.; Zandi, P.P.; Breitner, J.C.S.; Welsh-Bohmer, K.A. Cache County Study Investigators Occupational exposure to pesticides increases the risk of incident AD: The Cache County study. Neurology 2010, 74, 1524–1530. [Google Scholar] [CrossRef]
- Parrón, T.; Requena, M.; Hernández, A.F.; Alarcón, R. Association between environmental exposure to pesticides and neurodegenerative diseases. Toxicol. Appl. Pharmacol. 2011, 256, 379–385. [Google Scholar] [CrossRef]
- Richardson, J.R.; Roy, A.; Shalat, S.L.; von Stein, R.T.; Hossain, M.M.; Buckley, B.; Gearing, M.; Levey, A.I.; German, D.C. Elevated serum pesticide levels and risk for Alzheimer disease. JAMA Neurol. 2014, 71, 284–290. [Google Scholar] [CrossRef]
- Graham, J.E.; Rockwood, K.; Beattie, B.L.; McDowell, I.; Eastwood, R.; Gauthier, S. Standardization of the diagnosis of dementia in the Canadian Study of Health and Aging. Neuroepidemiology 1996, 15, 246–256. [Google Scholar] [CrossRef]
- Medehouenou, T.C.M.; Ayotte, P.; Carmichael, P.H.; Kröger, E.; Verreault, R.; Lindsay, J.; Dewailly, É.; Tyas, S.L.; Bureau, A.; Laurin, D. Plasma polychlorinated biphenyl and organochlorine pesticide concentrations in dementia: The Canadian Study of Health and Aging. Environ. Int. 2014, 69, 141–147. [Google Scholar] [CrossRef]
- Medehouenou, T.C.M.; Ayotte, P.; Carmichael, P.H.; Kröger, E.; Verreault, R.; Lindsay, J.; Dewailly, É.; Tyas, S.L.; Bureau, A.; Laurin, D. Exposure to polychlorinated biphenyls and organochlorine pesticides and risk of dementia, Alzheimer’s disease and cognitive decline in an older population: A prospective analysis from the Canadian Study of Health and Aging. Environ. Health 2019, 18, 57. [Google Scholar] [CrossRef]
- Yan, D.; Zhang, Y.; Liu, L.; Yan, H. Pesticide exposure and risk of Alzheimer’s disease: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 32222. [Google Scholar] [CrossRef] [PubMed]
- Gunnarsson, L.G.; Bodin, L. Occupational exposures and neurodegenerative diseases-A systematic literature review and meta-analyses. Int. J. Environ. Res. Public Health 2019, 16, 337. [Google Scholar] [CrossRef] [PubMed]
- Dardiotis, E.; Kosmidis, M.H.; Yannakoulia, M.; Hadjigeorgiou, G.M.; Scarmeas, N. The Hellenic Longitudinal Investigation of Aging and Diet (HELIAD): Rationale, study design, and cohort description. Neuroepidemiology 2014, 43, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Dardiotis, E.; Siokas, V.; Moza, S.; Kosmidis, M.H.; Vogiatzi, C.; Aloizou, A.M.; Geronikola, N.; Ntanasi, E.; Zalonis, I.; Yannakoulia, M.; et al. Pesticide exposure and cognitive function: Results from the Hellenic Longitudinal Investigation of Aging and Diet (HELIAD). Environ. Res. 2019, 177, 108632. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Bozi, M.; Simitsi, A.M.; Koros, C.; Antonelou, R.; Papagiannakis, N.; Maniati, M.; Poula, D.; Stamelou, M.; Vassilatis, D.K.; et al. The relationship between environmental factors and different Parkinson’s disease subtypes in Greece: Data analysis of the Hellenic Biobank of Parkinson’s disease. Parkinsonism Relat. Disord. 2019, 67, 105–112. [Google Scholar] [CrossRef]
- Chaves, R.S.; Melo, T.Q.; Martins, S.A.; Ferrari, M.F.R. Protein aggregation containing β-amyloid, α-synuclein and hyperphosphorylated τ in cultured cells of hippocampus, substantia nigra and locus coeruleus after rotenone exposure. BMC Neurosci. 2010, 11, 144. [Google Scholar] [CrossRef]
- Höglinger, G.U.; Lannuzel, A.; Khondiker, M.E.; Michel, P.P.; Duyckaerts, C.; Féger, J.; Champy, P.; Prigent, A.; Medja, F.; Lombes, A.; et al. The mitochondrial complex I inhibitor rotenone triggers a cerebral tauopathy. J. Neurochem. 2005, 95, 930–939. [Google Scholar] [CrossRef]
- Chen, N.N.; Luo, D.J.; Yao, X.Q.; Yu, C.; Wang, Y.; Wang, Q.; Wang, J.Z.; Liu, G.P. Pesticides induce spatial memory deficits with synaptic impairments and an imbalanced tau phosphorylation in rats. J. Alzheimers Dis. 2012, 30, 585–594. [Google Scholar] [CrossRef]
- Maurya, S.K.; Mishra, J.; Abbas, S.; Bandyopadhyay, S. Cypermethrin stimulates GSK3β-dependent Aβ and p-tau proteins and cognitive loss in young rats: Reduced HB-EGF signaling and downstream neuroinflammation as critical regulators. Mol. Neurobiol. 2016, 53, 968–982. [Google Scholar] [CrossRef]
- Burke, R.D.; Todd, S.W.; Lumsden, E.; Mullins, R.J.; Mamczarz, J.; Fawcett, W.P.; Gullapalli, R.P.; Randall, W.R.; Pereira, E.F.R.; Albuquerque, E.X. Developmental neurotoxicity of the organophosphorus insecticide chlorpyrifos: From clinical findings to preclinical models and potential mechanisms. J. Neurochem. 2017, 142, 162–177. [Google Scholar] [CrossRef]
- Moyano, P.; Frejo, M.T.; Anadon, M.J.; García, J.M.; Díaz, M.J.; Lobo, M.; Sola, E.; García, J.; Del Pino, J. SN56 neuronal cell death after 24 h and 14 days chlorpyrifos exposure through glutamate transmission dysfunction, increase of GSK-3β enzyme, β-amyloid and tau protein levels. Toxicology 2018, 402–403, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.G.; Ribes, D.; Cabré, M.; Domingo, J.L.; Sanchez-Santed, F.; Colomina, M.T. Amyloid β peptide levels increase in brain of AβPP Swedish mice after exposure to chlorpyrifos. Curr. Alzheimer Res. 2011, 8, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Kim, C.; Kim, J.; Yoon, H.; Zhou, H.; Kim, J. Common pesticide, Dichlorodiphenyltrichloroethane (DDT), increases Amyloid-β Levels by impairing the function of ABCA1 and IDE: Implication for Alzheimer’s disease. J. Alzheimers Dis. 2015, 46, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Portelius, E.; Durieu, E.; Bodin, M.; Cam, M.; Pannee, J.; Leuxe, C.; Mabondzo, A.; Oumata, N.; Galons, H.; Lee, J.Y.; et al. Specific Triazine herbicides induce Amyloid-β42 production. J. Alzheimers Dis. 2016, 54, 1593–1605. [Google Scholar] [CrossRef]
- Cam, M.; Durieu, E.; Bodin, M.; Manousopoulou, A.; Koslowski, S.; Vasylieva, N.; Barnych, B.; Hammock, B.D.; Bohl, B.; Koch, P.; et al. Induction of Amyloid-β42 production by Fipronil and other pyrazole insecticides. J. Alzheimers Dis. 2018, 62, 1663–1681. [Google Scholar] [CrossRef]
- Lafon, P.A.; Wang, Y.; Arango-Lievano, M.; Torrent, J.; Salvador-Prince, L.; Mansuy, M.; Mestre-Francès, N.; Givalois, L.; Liu, J.; Mercader, J.V.; et al. Fungicide residues exposure and β-amyloid aggregation in a mouse model of Alzheimer’s disease. Environ. Health Perspect. 2020, 128, 17011. [Google Scholar] [CrossRef]
- Charidimou, A.; Boulouis, G.; Gurol, M.E.; Ayata, C.; Bacskai, B.J.; Frosch, M.P.; Viswanathan, A.; Greenberg, S.M. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain 2017, 140, 1829–1850. [Google Scholar] [CrossRef]
- Malek-Ahmadi, M.; Perez, S.E.; Chen, K.; Mufson, E.J. Braak stage, cerebral amyloid angiopathy, and cognitive decline in early Alzheimer’s disease. J. Alzheimers Dis. 2020, 74, 189–197. [Google Scholar] [CrossRef]
- Hersh, L.B.; Rodgers, D.W. Neprilysin and amyloid beta peptide degradation. Curr. Alzheimer Res. 2008, 5, 225–231. [Google Scholar] [CrossRef]
- Voorhees, J.R.; Remy, M.T.; Erickson, C.M.; Dutca, L.M.; Brat, D.J.; Pieper, A.A. Occupational-like organophosphate exposure disrupts microglia and accelerates deficits in a rat model of Alzheimer’s disease. NPJ Aging Mech. Dis. 2019, 5, 3. [Google Scholar] [CrossRef]
- Farahat, F.M.; Fenske, R.A.; Olson, J.R.; Galvin, K.; Bonner, M.R.; Rohlman, D.S.; Farahat, T.M.; Lein, P.J.; Anger, W.K. Chlorpyrifos exposures in Egyptian cotton field workers. Neurotoxicology 2010, 31, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Ellison, C.A.; Smith, J.N.; Lein, P.J.; Olson, J.R. Pharmacokinetics and pharmacodynamics of chlorpyrifos in adult male Long-Evans rats following repeated subcutaneous exposure to chlorpyrifos. Toxicology 2011, 287, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Lein, P.J.; Bonner, M.R.; Farahat, F.M.; Olson, J.R.; Rohlman, D.S.; Fenske, R.A.; Lattal, K.M.; Lasarev, M.R.; Galvin, K.; Farahat, T.M.; et al. Experimental strategy for translational studies of organophosphorus pesticide neurotoxicity based on real-world occupational exposures to chlorpyrifos. Neurotoxicology 2012, 33, 660–668. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sabarwal, A.; Kumar, K.; Singh, R.P. Hazardous effects of chemical pesticides on human health-Cancer and other associated disorders. Environ. Toxicol. Pharmacol. 2018, 63, 103–114. [Google Scholar] [CrossRef]
- Yu, J.; Zhum, H.; Bhat, A.; El-Sayed, H.; Gudz, T.; Gattoni-Celli, S.; Kindy, M. Influence of chlorpyrifos oxon on the development and progression of Alzheimer’s disease in amyloid precursor protein transgenic mice. Neuroimmunol. Neuroinflamm. 2015, 2, 31–42. [Google Scholar]
- Won, J.H.; Park, S.; Hong, S.; Son, S.; Yu, J.W. Rotenone-induced impairment of mitochondrial electron transport chain confers a selective priming signal for NLRP3 inflammasome activation. J. Biol. Chem. 2015, 290, 27425–27437. [Google Scholar] [CrossRef]
- Elmore, S.E.; La Merrill, M.A. Oxidative Phosphorylation Impairment by DDT and DDE. Front. Endocrinol. 2019, 10, 122. [Google Scholar] [CrossRef]
- Ch, R.; Singh, A.K.; Pathak, M.K.; Singh, A.; Kesavachandran, C.N.; Bihari, V.; Mudiam, M.K.R. Saliva and urine metabolic profiling reveals altered amino acid and energy metabolism in male farmers exposed to pesticides in Madhya Pradesh State, India. Chemosphere 2019, 226, 636–644. [Google Scholar] [CrossRef]
- Yellen, G. Fueling thought: Management of glycolysis and oxidative phosphorylation in neuronal metabolism. J. Cell Biol. 2018, 217, 2235–2246. [Google Scholar] [CrossRef]
- Vlassenko, A.G.; Raichle, M.E. Brain aerobic glycolysis functions and Alzheimer’s disease. Clin. Transl. Imaging 2015, 3, 27–37. [Google Scholar] [CrossRef]
- Atlante, A.; de Bari, L.; Bobba, A.; Amadoro, G. A disease with a sweet tooth: Exploring the Warburg effect in Alzheimer’s disease. Biogerontology 2017, 18, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.A.; Tindale, L.; Lone, A.; Singh, O.; Macauley, S.L.; Stanley, M.; Holtzman, D.M.; Bartha, R.; Cumming, R.C. Aerobic glycolysis in the frontal cortex correlates with memory performance in wild-type mice but not the APP/PS1 mouse model of cerebral amyloidosis. J. Neurosci. 2016, 36, 1871–1878. [Google Scholar] [CrossRef] [PubMed]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.I.; Mook-Jung, I. A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s disease. Cell Metab. 2019, 30, 493–507. [Google Scholar] [CrossRef]
- Rambabu, L.; Megson, I.L.; Eddleston, M. Does oxidative stress contribute to toxicity in acute organophosphorus poisoning?—A systematic review of the evidence. Clin. Toxicol. (Phila.) 2019, 1–16. [Google Scholar] [CrossRef]
- Cristóvão, A.C.; Choi, D.H.; Baltazar, G.; Beal, M.F.; Kim, Y.S. The role of NADPH oxidase 1-derived reactive oxygen species in paraquat-mediated dopaminergic cell death. Antioxid. Redox Signal. 2009, 11, 2105–2118. [Google Scholar] [CrossRef] [PubMed]
- Mangum, L.C.; Borazjani, A.; Stokes, J.V.; Matthews, A.T.; Lee, J.H.; Chambers, J.E.; Ross, M.K. Organochlorine insecticides induce NADPH oxidase-dependent reactive oxygen species in human monocytic cells via phospholipase A2/arachidonic acid. Chem. Res. Toxicol. 2015, 28, 570–584. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.; Hirko, A.C.; King, M.A.; Liu, B. Role of NADPH oxidase in cooperative reactive oxygen species generation in dopaminergic neurons induced by combined treatment with dieldrin and lindane. Toxicol. Lett. 2018, 299, 47–55. [Google Scholar] [CrossRef]
- Tarafdar, A.; Pula, G. The role of NADPH oxidases and oxidative stress in neurodegenerative disorders. Int. J. Mol. Sci. 2018, 19, 3824. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Heid, M.E.; Keyel, P.A.; Kamga, C.; Shiva, S.; Watkins, S.C.; Salter, R.D. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J. Immunol. 2013, 191, 5230–5238. [Google Scholar] [CrossRef]
- Abderrazak, A.; Syrovets, T.; Couchie, D.; El Hadri, K.; Friguet, B.; Simmet, T.; Rouis, M. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015, 4, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Moloudizargari, M.; Moradkhani, F.; Asghari, N.; Fallah, M.; Asghari, M.H.; Moghadamnia, A.A.; Abdollahi, M. NLRP inflammasome as a key role player in the pathogenesis of environmental toxicants. Life Sci. 2019, 231, 116585. [Google Scholar] [CrossRef] [PubMed]
- Walker, K.A.; Ficek, B.N.; Westbrook, R. Understanding the role of systemic inflammation in Alzheimer’s disease. ACS Chem. Neurosci. 2019, 10, 3340–3342. [Google Scholar] [CrossRef] [PubMed]
- Duka, T.; Sidhu, A. The neurotoxin, MPP+, induces hyperphosphorylation of Tau, in the presence of alpha-Synuclein, in SH-SY5Y neuroblastoma cells. Neurotox. Res. 2006, 10, 1–10. [Google Scholar] [CrossRef]
- Lee, S.J.; Chung, Y.H.; Joo, K.M.; Lim, H.C.; Jeon, G.S.; Kim, D.; Lee, W.B.; Kim, Y.S.; Cha, C.I. Age-related changes in glycogen synthase kinase 3beta (GSK3beta) immunoreactivity in the central nervous system of rats. Neurosci. Lett. 2006, 409, 134–139. [Google Scholar] [CrossRef]
- Llorens-Martín, M.; Jurado, J.; Hernández, F.; Avila, J. GSK-3β, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar]
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef]
- Planel, E.; Yasutake, K.; Fujita, S.C.; Ishiguro, K. Inhibition of protein phosphatase 2A overrides tau protein kinase I/glycogen synthase kinase 3 beta and cyclin-dependent kinase 5 inhibition and results in tau hyperphosphorylation in the hippocampus of starved mouse. J. Biol. Chem. 2001, 276, 34298–34306. [Google Scholar] [CrossRef]
- Ly, P.T.T.; Wu, Y.; Zou, H.; Wang, R.; Zhou, W.; Kinoshita, A.; Zhang, M.; Yang, Y.; Cai, F.; Woodgett, J.; et al. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Investig. 2013, 123, 224–235. [Google Scholar] [CrossRef]
- Uemura, K.; Kuzuya, A.; Shimozono, Y.; Aoyagi, N.; Ando, K.; Shimohama, S.; Kinoshita, A. GSK3beta activity modifies the localization and function of presenilin 1. J. Biol. Chem. 2007, 282, 15823–15832. [Google Scholar] [CrossRef]
- Hanger, D.P.; Hughes, K.; Woodgett, J.R.; Brion, J.P.; Anderton, B.H. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 1992, 147, 58–62. [Google Scholar] [CrossRef]
- Mandelkow, E.M.; Drewes, G.; Biernat, J.; Gustke, N.; Van Lint, J.; Vandenheede, J.R.; Mandelkow, E. Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 1992, 314, 315–321. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Chen, G.; Fan, Z.; Luo, J.; Ke, Z.J. GSK3beta and endoplasmic reticulum stress mediate rotenone-induced death of SK-N-MC neuroblastoma cells. Biochem. Pharmacol. 2008, 76, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Hongo, H.; Kihara, T.; Kume, T.; Izumi, Y.; Niidome, T.; Sugimoto, H.; Akaike, A. Glycogen synthase kinase-3β activation mediates rotenone-induced cytotoxicity with the involvement of microtubule destabilization. Biochem. Biophys. Res. Commun. 2012, 426, 94–99. [Google Scholar] [CrossRef]
- Dunning, C.J.; McGauran, G.; Willén, K.; Gouras, G.K.; O’Connell, D.J.; Linse, S. Direct high affinity interaction between Aβ42 and GSK3α stimulates hyperphosphorylation of tau. A new molecular link in Alzheimer’s disease? ACS Chem. Neurosci. 2016, 7, 161–170. [Google Scholar] [CrossRef]
- Zhao, L.; Yan, M.; Wang, X.; Xiong, G.; Wu, C.; Wang, Z.; Zhou, Z.; Chang, X. Modification of Wnt signaling pathway on paraquat-induced inhibition of neural progenitor cell proliferation. Food Chem. Toxicol. 2018, 121, 311–325. [Google Scholar] [CrossRef]
- Stephano, F.; Nolte, S.; Hoffmann, J.; El-Kholy, S.; von Frieling, J.; Bruchhaus, I.; Fink, C.; Roeder, T. Impaired Wnt signaling in dopamine containing neurons is associated with pathogenesis in a rotenone triggered Drosophila Parkinson’s disease model. Sci. Rep. 2018, 8, 2372. [Google Scholar] [CrossRef]
- Wu, Y.; Li, W.; Yuan, M.; Liu, X. The synthetic pyrethroid deltamethrin impairs zebrafish (Danio rerio) swim bladder development. Sci. Total Environ. 2020, 701, 134870. [Google Scholar] [CrossRef]
- Folke, J.; Pakkenberg, B.; Brudek, T. Impaired Wnt signaling in the prefrontal cortex of Alzheimer’s disease. Mol. Neurobiol. 2019, 56, 873–891. [Google Scholar] [CrossRef]
- Palomer, E.; Buechler, J.; Salinas, P.C. Wnt signaling deregulation in the aging and Alzheimer’s brain. Front. Cell. Neurosci. 2019, 13, 227. [Google Scholar] [CrossRef]
- Jia, L.; Piña-Crespo, J.; Li, Y. Restoring Wnt/β-catenin signaling is a promising therapeutic strategy for Alzheimer’s disease. Mol. Brain. 2019, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Parr, C.; Mirzaei, N.; Christian, M.; Sastre, M. Activation of the Wnt/β-catenin pathway represses the transcription of the β-amyloid precursor protein cleaving enzyme (BACE1) via binding of T-cell factor-4 to BACE1 promoter. FASEB J. 2015, 29, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J.Y.; Fuenzalida, M.; Inestrosa, N.C. In vivo activation of Wnt signaling pathway enhances cognitive function of adult mice and reverses cognitive deficits in an Alzheimer’s disease model. J. Neurosci. 2014, 34, 2191–2202. [Google Scholar] [CrossRef] [PubMed]
- Cisternas, P.; Zolezzi, J.M.; Martinez, M.; Torres, V.I.; Wong, G.W.; Inestrosa, N.C. Wnt-induced activation of glucose metabolism mediates the in vivo neuroprotective roles of Wnt signaling in Alzheimer disease. J. Neurochem. 2019, 149, 54–72. [Google Scholar] [CrossRef]
- Chen, R.H.; Ding, W.V.; McCormick, F. Wnt signaling to beta-catenin involves two interactive components. Glycogen synthase kinase-3beta inhibition and activation of protein kinase C. J. Biol. Chem. 2000, 275, 17894–17899. [Google Scholar] [CrossRef]
- Chen, M.; Wang, J.; Lu, J.; Bond, M.C.; Ren, X.R.; Lyerly, H.K.; Barak, L.S.; Chen, W. The anti-helminthic niclosamide inhibits Wnt/Frizzled1 signaling. Biochemistry 2009, 48, 10267–10274. [Google Scholar] [CrossRef]
- Köppen, J.; Schulze, A.; Machner, L.; Wermann, M.; Eichentopf, R.; Guthardt, M.; Hähnel, A.; Klehm, J.; Kriegeskorte, M.C.; Hartlage-Rübsamen, M.; et al. Amyloid-beta peptides trigger aggregation of alpha-Synuclein in vitro. Molecules 2020, 25, 580. [Google Scholar] [CrossRef]
- Bhasne, K.; Mukhopadhyay, S. Formation of heterotypic amyloids: α-Synuclein in co-Aggregation. Proteomics 2018, 18, e1800059. [Google Scholar] [CrossRef]
- Gómez-Martín, A.; Altakroni, B.; Lozano-Paniagua, D.; Margison, G.P.; de Vocht, F.; Povey, A.C.; Hernández, A.F. Increased N7-methyldeoxyguanosine DNA adducts after occupational exposure to pesticides and influence of genetic polymorphisms of paraoxonase-1 and glutathione S-transferase M1 and T1. Environ. Mol. Mutagen. 2015, 56, 437–445. [Google Scholar] [CrossRef]
- Shah, H.K.; Sharma, T.; Banerjee, B.D. Organochlorine pesticides induce inflammation, ROS production, and DNA damage in human epithelial ovary cells: An in vitro study. Chemosphere 2019, 246, 125691. [Google Scholar] [CrossRef]
- Lundqvist, J.; Hellman, B.; Oskarsson, A. Fungicide prochloraz induces oxidative stress and DNA damage in vitro. Food Chem. Toxicol. 2016, 91, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Kashanian, S.; Shariati, Z.; Roshanfekr, H.; Ghobadi, S. DNA binding studies of 3, 5, 6-trichloro-2-pyridinol pesticide metabolite. DNA Cell Biol. 2012, 31, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Hernández, A.F.; Menéndez, P. Linking pesticide exposure with pediatric leukemia: Potential underlying mechanisms. Int. J. Mol. Sci. 2016, 17, 461. [Google Scholar] [CrossRef] [PubMed]
- Van Maele-Fabry, G.; Gamet-Payrastre, L.; Lison, D. Residential exposure to pesticides as risk factor for childhood and young adult brain tumors: A systematic review and meta-analysis. Environ. Int. 2017, 106, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Verheijen, B.M.; Vermulst, M.; van Leeuwen, F.W. Somatic mutations in neurons during aging and neurodegeneration. Acta Neuropathol. 2018, 135, 811–826. [Google Scholar] [CrossRef] [PubMed]
- Leija-Salazar, M.; Piette, C.; Proukakis, C. Somatic mutations in neurodegeneration. Neuropathol. Appl. Neurobiol. 2018, 44, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Veltman, J.A. The role of de novo mutations in adult-onset neurodegenerative disorders. Acta Neuropathol. 2019, 137, 183–207. [Google Scholar] [CrossRef]
- Lee, M.H.; Siddoway, B.; Kaeser, G.E.; Segota, I.; Rivera, R.; Romanow, W.J.; Liu, C.S.; Park, C.; Kennedy, G.; Long, T.; et al. Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature 2018, 563, 639–645. [Google Scholar] [CrossRef]
- Driver, J.A.; Zhou, X.Z.; Lu, K.P. Pin1 dysregulation helps to explain the inverse association between cancer and Alzheimer’s disease. Biochim. Biophys. Acta 2015, 1850, 2069–2076. [Google Scholar] [CrossRef]
- Park, J.S.; Lee, J.; Jung, E.S.; Kim, M.H.; Kim, I.B.; Son, H.; Kim, S.; Kim, S.; Park, Y.M.; Mook-Jung, I.; et al. Brain somatic mutations observed in Alzheimer’s disease associated with aging and dysregulation of tau phosphorylation. Nat. Commun. 2019, 10, 3090. [Google Scholar] [CrossRef]
- Kwok, J.B.J. Role of epigenetics in Alzheimer’s and Parkinson’s disease. Epigenomics 2010, 2, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Bordoni, L.; Nasuti, C.; Fedeli, D.; Galeazzi, R.; Laudadio, E.; Massaccesi, L.; López-Rodas, G.; Gabbianelli, R. Early impairment of epigenetic pattern in neurodegeneration: Additional mechanisms behind pyrethroid toxicity. Exp. Gerontol. 2019, 124, 110629. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.; Koutros, S.; Bonner, M.R.; Barry, K.H.; Alavanja, M.C.R.; Andreotti, G.; Byun, H.M.; Chen, L.; Beane Freeman, L.E.; Hofmann, J.N.; et al. Pesticide use and LINE-1 methylation among male private pesticide applicators in the Agricultural Health Study. Environ. Epigenetics 2017, 3, dvx005. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.M.; Vera, M.K.M.; Bhandari, R.K. Developmental and epigenetic effects of Roundup and glyphosate exposure on Japanese medaka (Oryzias latipes). Aquat. Toxicol. 2019, 210, 215–226. [Google Scholar] [CrossRef]
- Kochmanski, J.; VanOeveren, S.E.; Patterson, J.R.; Bernstein, A.I. Developmental Dieldrin Exposure Alters DNA Methylation at Genes Related to Dopaminergic Neuron Development and Parkinson’s Disease in Mouse Midbrain. Toxicol. Sci. 2019, 169, 593–607. [Google Scholar] [CrossRef]
- Cervellati, C.; Valacchi, G.; Tisato, V.; Zuliani, G.; Marsillach, J. Evaluating the link between Paraoxonase-1 levels and Alzheimer’s disease development. Minerva Med. 2019, 110, 238–250. [Google Scholar] [CrossRef]
- Huen, K.; Solomon, O.; Kogut, K.; Eskenazi, B.; Holland, N. PON1 DNA methylation and neurobehavior in Mexican-American children with prenatal organophosphate exposure. Environ. Int. 2018, 121, 31–40. [Google Scholar] [CrossRef]
- Yan, M.; Dou, T.; Lv, W.; Wang, X.; Zhao, L.; Chang, X.; Zhou, Z. Integrated analysis of paraquat-induced microRNAs-mRNAs changes in human neural progenitor cells. Toxicol. Vitr. 2017, 44, 196–205. [Google Scholar] [CrossRef]
- Zhang, Q.; Zheng, S.; Wang, S.; Wang, W.; Xing, H.; Xu, S. Chlorpyrifos induced oxidative stress to promote apoptosis and autophagy through the regulation of miR-19a-AMPK axis in common carp. Fish Shellfish Immunol. 2019, 93, 1093–1099. [Google Scholar] [CrossRef]
- Zhao, M.W.; Yang, P.; Zhao, L.L. Chlorpyrifos activates cell pyroptosis and increases susceptibility on oxidative stress-induced toxicity by miR-181/SIRT1/PGC-1α/Nrf2 signaling pathway in human neuroblastoma SH-SY5Y cells: Implication for association between chlorpyrifos and Parkinson’s disease. Environ. Toxicol. 2019, 34, 699–707. [Google Scholar]
- Kopf, P.G.; Walker, M.K. Overview of developmental heart defects by dioxins, PCBs, and pesticides. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2009, 27, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, T.A.; Skavicus, S.; Card, J.; Levin, E.D.; Seidler, F.J. Diverse neurotoxicants target the differentiation of embryonic neural stem cells into neuronal and glial phenotypes. Toxicology 2016, 372, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, L.; Rosca, A.; Oniga, A.; Zambrano, A.; Ramos, J.J.; González, M.C.; Liste, I.; Motas, M. Effects of chlorpyrifos on cell death and cellular phenotypic specification of human neural stem cells. Sci. Total Environ. 2019, 683, 445–454. [Google Scholar] [CrossRef]
- Saito, H.; Hara, K.; Tominaga, T.; Nakashima, K.; Tanemura, K. Early-life exposure to low levels of permethrin exerts impairments in learning and memory with the effects on neuronal and glial population in adult male mice. J. Appl. Toxicol. 2019, 39, 1651–1662. [Google Scholar] [CrossRef] [PubMed]
- Toda, T.; Parylak, S.L.; Linker, S.B.; Gage, F.H. The role of adult hippocampal neurogenesis in brain health and disease. Mol. Psychiatry 2019, 24, 67–87. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.S.; Drew, M.R. Functional neurogenesis over the years. Behav. Brain Res. 2020, 382, 112470. [Google Scholar] [CrossRef] [PubMed]
- Nasuti, C.; Fattoretti, P.; Carloni, M.; Fedeli, D.; Ubaldi, M.; Ciccocioppo, R.; Gabbianelli, R. Neonatal exposure to permethrin pesticide causes lifelong fear and spatial learning deficits and alters hippocampal morphology of synapses. J. Neurodev. Disord. 2014, 6, 7. [Google Scholar] [CrossRef]
- Hossain, M.M.; DiCicco-Bloom, E.; Richardson, J.R. Hippocampal ER stress and learning deficits following repeated pyrethroid exposure. Toxicol. Sci. 2015, 143, 220–228. [Google Scholar] [CrossRef]
- Mu, Y.; Gage, F.H. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol. Neurodegener. 2011, 6, 85. [Google Scholar] [CrossRef]
- Hollands, C.; Tobin, M.K.; Hsu, M.; Musaraca, K.; Yu, T.S.; Mishra, R.; Kernie, S.G.; Lazarov, O. Depletion of adult neurogenesis exacerbates cognitive deficits in Alzheimer’s disease by compromising hippocampal inhibition. Mol. Neurodegener. 2017, 12, 64. [Google Scholar] [CrossRef]
- Scopa, C.; Marrocco, F.; Latina, V.; Ruggeri, F.; Corvaglia, V.; La Regina, F.; Ammassari-Teule, M.; Middei, S.; Amadoro, G.; Meli, G.; et al. Impaired adult neurogenesis is an early event in Alzheimer’s disease neurodegeneration, mediated by intracellular Aβ oligomers. Cell Death Differ. 2020, 27, 934–948. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Cheng, X.; Jiang, J.; Wang, J.; Xie, J.; Hu, X.; Huang, Y.; Song, L.; Liu, M.; Cai, L.; et al. The toxic influence of paraquat on hippocampal neurogenesis in adult mice. Food Chem. Toxicol. 2017, 106, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Herzine, A.; Laugeray, A.; Feat, J.; Menuet, A.; Quesniaux, V.; Richard, O.; Pichon, J.; Montécot-Dubourg, C.; Perche, O.; Mortaud, S. Perinatal exposure to Glufosinate ammonium herbicide impairs neurogenesis and neuroblast migration through cytoskeleton destabilization. Front. Cell Neurosci. 2016, 10, 191. [Google Scholar] [CrossRef] [PubMed]
- Feat-Vetel, J.; Larrigaldie, V.; Meyer-Dilhet, G.; Herzine, A.; Mougin, C.; Laugeray, A.; Gefflaut, T.; Richard, O.; Quesniaux, V.; Montécot-Dubourg, C.; et al. Multiple effects of the herbicide glufosinate-ammonium and its main metabolite on neural stem cells from the subventricular zone of newborn mice. Neurotoxicology 2018, 69, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Spielman, L.J.; Gibson, D.L.; Klegeris, A. Unhealthy gut, unhealthy brain: The role of the intestinal microbiota in neurodegenerative diseases. Neurochem. Int. 2018, 120, 149–163. [Google Scholar] [CrossRef]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef]
- Lin, L.; Zheng, L.J.; Zhang, L.J. Neuroinflammation, gut microbiome, and Alzheimer’s disease. Mol. Neurobiol. 2018, 55, 8243–8250. [Google Scholar] [CrossRef]
- Sun, M.; Ma, K.; Wen, J.; Wang, G.; Zhang, C.; Li, Q.; Bao, X.; Wang, H. A review of the brain-gut-microbiome axis and the potential role of microbiota in Alzheimer’s disease. J. Alzheimers Dis. 2020, 73, 849–865. [Google Scholar] [CrossRef]
- Jiang, C.; Li, G.; Huang, P.; Liu, Z.; Zhao, B. The gut microbiota and Alzheimer’s disease. J. Alzheimers Dis. 2017, 58, 1–15. [Google Scholar] [CrossRef]
- Wu, S.C.; Cao, Z.S.; Chang, K.M.; Juang, J.L. Intestinal microbial dysbiosis aggravates the progression of Alzheimer’s disease in Drosophila. Nat. Commun. 2017, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Park, J.; Han, D.; Yang, J.; Kim, A.; Woo, J.; Kim, Y.; Mook-Jung, I. Molecular and functional signatures in a novel Alzheimer’s disease mouse model assessed by quantitative proteomics. Mol. Neurodegener. 2018, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kim, Y.; Choi, H.; Kim, W.; Park, S.; Lee, D.; Kim, D.K.; Kim, H.J.; Choi, H.; Hyun, D.W.; et al. Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer’s disease animal model. Gut 2020, 69, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Jin, C.; Wang, Y.; Fu, Z.; Jin, Y. Exposure to the fungicide propamocarb causes gut microbiota dysbiosis and metabolic disorder in mice. Environ. Pollut. 2018, 237, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Aitbali, Y.; Ba-M’hamed, S.; Elhidar, N.; Nafis, A.; Soraa, N.; Bennis, M. Glyphosate based- herbicide exposure affects gut microbiota, anxiety and depression-like behaviors in mice. Neurotoxicol. Teratol. 2018, 67, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Chi, L.; Tu, P.; Gao, N.; Lu, K. The carbamate Aldicarb altered the gut microbiome, metabolome, and lipidome of C57BL/6J mice. Chem. Res. Toxicol. 2019, 32, 67–79. [Google Scholar] [CrossRef]
- Perez-Fernandez, C.; Morales-Navas, M.; Guardia-Escote, L.; Garrido-Cárdenas, J.A.; Colomina, M.T.; Giménez, E.; Sánchez-Santed, F. Long-term effects of low doses of Chlorpyrifos exposure at the preweaning developmental stage: A locomotor, pharmacological, brain gene expression and gut microbiome analysis. Food Chem. Toxicol. 2020, 135, 110865. [Google Scholar] [CrossRef]
- Guardia-Escote, L.; Basaure, P.; Biosca-Brull, J.; Cabré, M.; Blanco, J.; Pérez-Fernández, C.; Sánchez-Santed, F.; Domingo, J.L.; Colomina, M.T. APOE genotype and postnatal chlorpyrifos exposure modulate gut microbiota and cerebral short-chain fatty acids in preweaning mice. Food Chem. Toxicol. 2020, 135, 110872. [Google Scholar] [CrossRef]

© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, B.L. Neuropathological Mechanisms Associated with Pesticides in Alzheimer’s Disease. Toxics 2020, 8, 21. https://doi.org/10.3390/toxics8020021
Tang BL. Neuropathological Mechanisms Associated with Pesticides in Alzheimer’s Disease. Toxics. 2020; 8(2):21. https://doi.org/10.3390/toxics8020021
Chicago/Turabian StyleTang, Bor Luen. 2020. "Neuropathological Mechanisms Associated with Pesticides in Alzheimer’s Disease" Toxics 8, no. 2: 21. https://doi.org/10.3390/toxics8020021
APA StyleTang, B. L. (2020). Neuropathological Mechanisms Associated with Pesticides in Alzheimer’s Disease. Toxics, 8(2), 21. https://doi.org/10.3390/toxics8020021