
Polycyclic Aromatic Hydrocarbons (PAHs) Exposure Triggers Inflammation and Endothelial Dysfunction in BALB/c Mice: A Pilot Study

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Protocol
2.3. Sampling Extraction
2.4. Cytokine Analysis
2.5. Gene Expression by RT-qPCR
2.6. Western Blotting
2.7. Statistical Analysis
3. Results
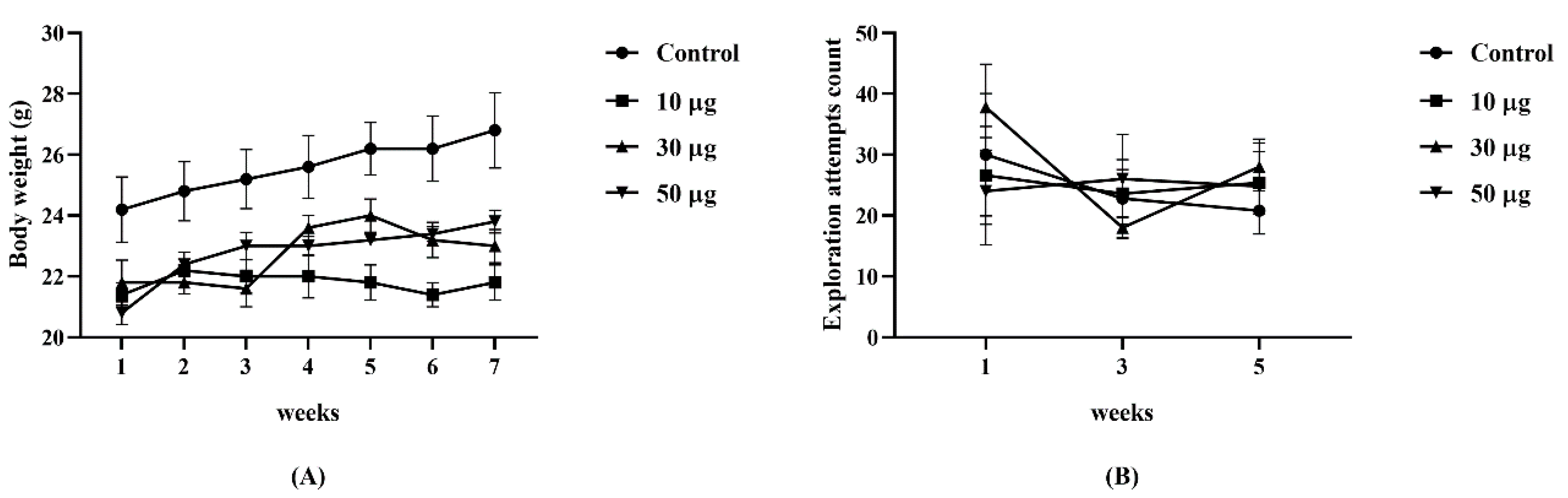
3.1. Animals
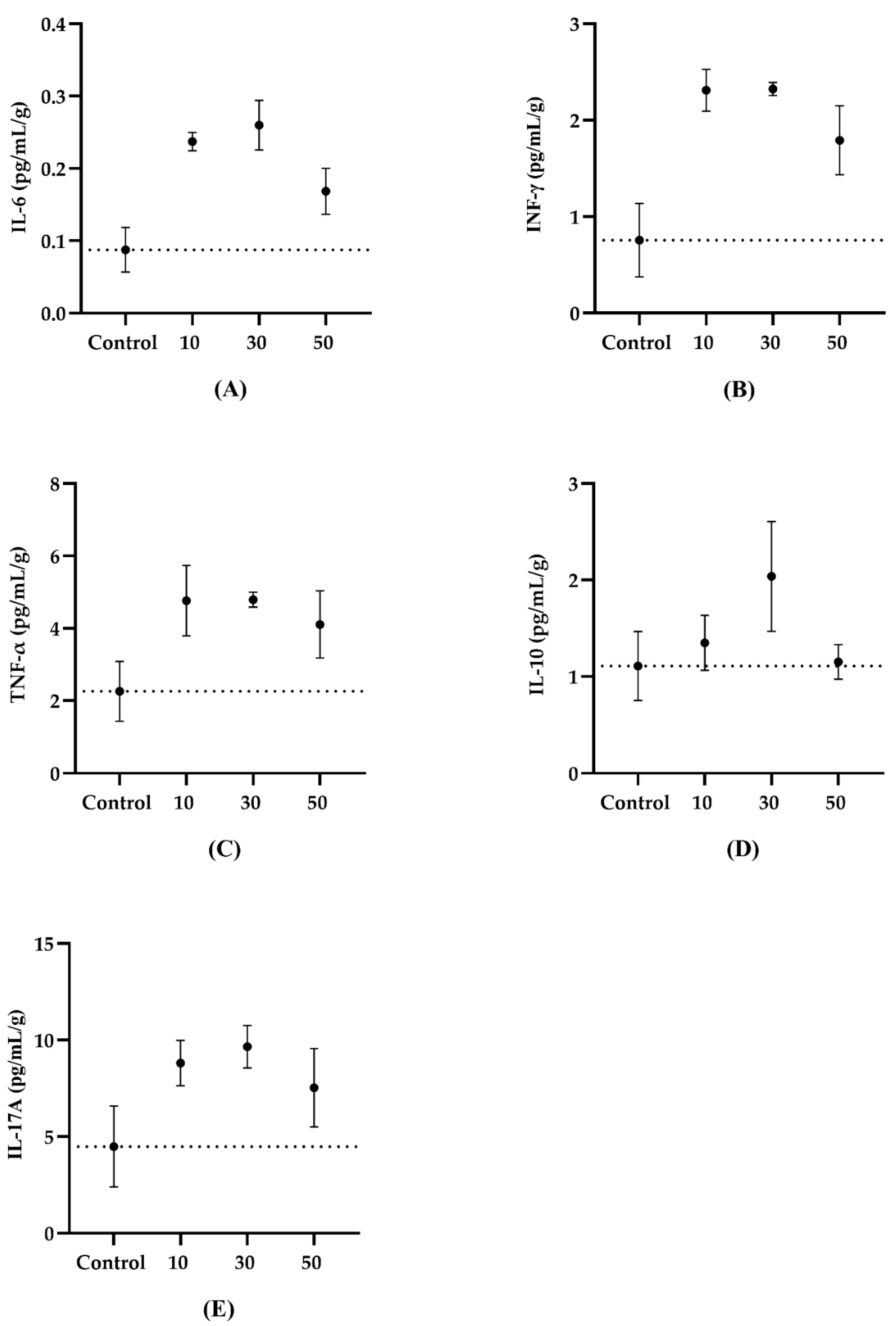
3.2. Serum Cytokines
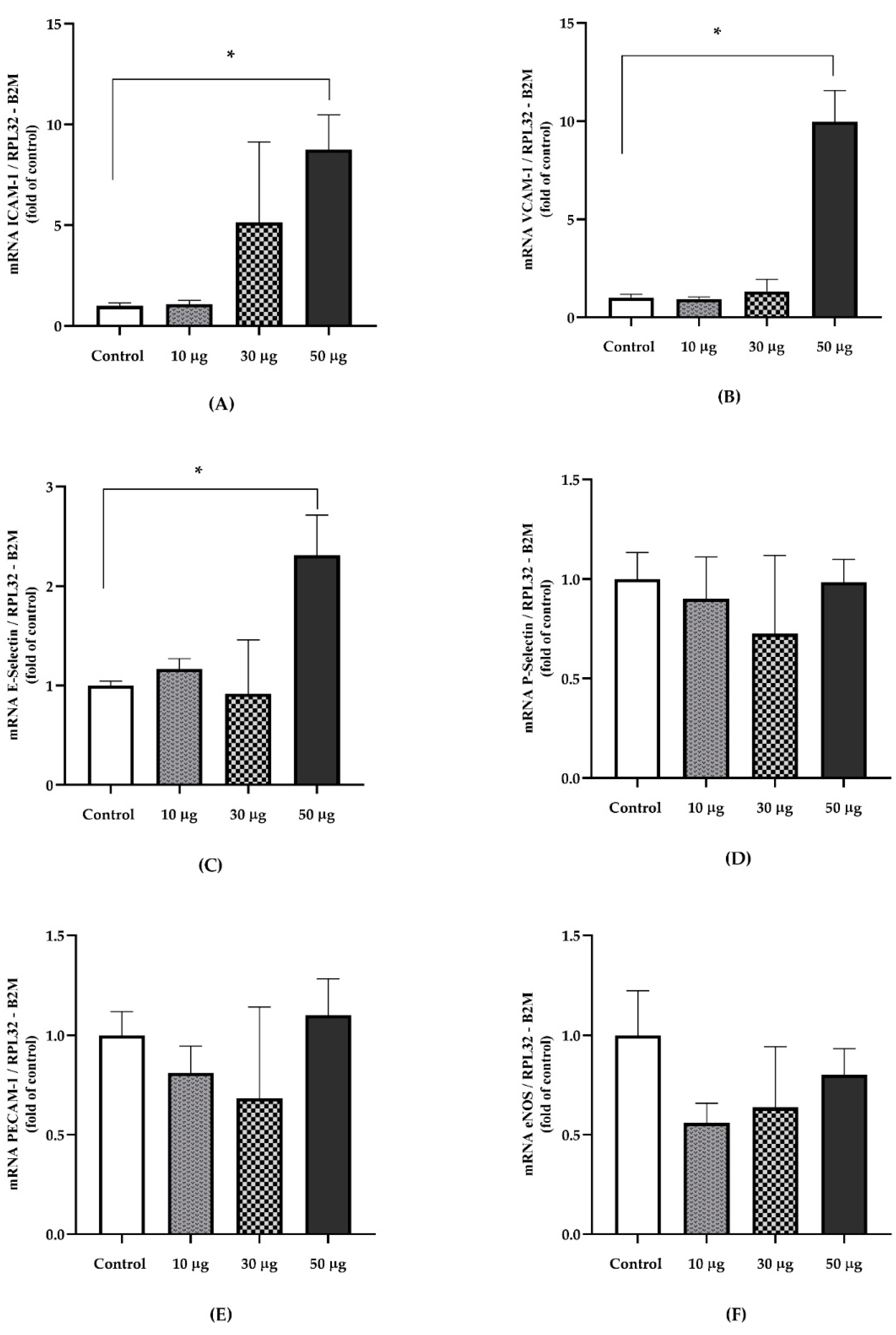
3.3. Gene Expression
3.4. Protein Expression
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- State of Global Air 2019; Health Effects Institute: Boston, MA, USA, 2019; Available online: www.stateofglobalair.org (accessed on 30 March 2022).
- Brook, R.D.; Rajagopalan, S. Stressed About Air Pollution: Time for Personal Action. Circulation 2017, 136, 628–631. [Google Scholar] [CrossRef] [PubMed]
- Wyzga, R.E.; Rohr, A.C. Long-term particulate matter exposure: Attributing health effects to individual PM components. J. Air Waste Manag. Assoc. 2015, 65, 523–543. [Google Scholar] [CrossRef] [PubMed]
- Jankowska-Kieltyka, M.; Roman, A.; Nalepa, I. The Air We Breathe: Air Pollution as a Prevalent Proinflammatory Stimulus Contributing to Neurodegeneration. Front. Cell. Neurosci. 2021, 15, 647643. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.; Slezakova, K.; Delerue-Matos, C.; Pereira, M.C.; Morais, S. Children environmental exposure to particulate matter and polycyclic aromatic hydrocarbons and biomonitoring in school environments: A review on indoor and outdoor exposure levels, major sources and health impacts. Environ. Int. 2019, 124, 180–204. [Google Scholar] [CrossRef] [PubMed]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Some non-heterocyclic polycyclic aromatic hydrocarbons and some related exposures. IARC Monogr. Eval. Carcinog. Risks Hum. 2010, 92, 1–853. [Google Scholar]
- Schulz, H.; Harder, V.; Ibald-Mulli, A.; Khandoga, A.; Koenig, W.; Krombach, F.; Radykewicz, R.; Stampfl, A.; Thorand, B.; Peters, A. Cardiovascular effects of fine and ultrafine particles. J. Aerosol. Med. 2005, 18, 1–22. [Google Scholar] [CrossRef]
- Brook, R.D.; Rajagopalan, S.; Pope, C.A., 3rd; Brook, J.R.; Bhatnagar, A.; Diez-Roux, A.V.; Holguin, F.; Hong, Y.; Luepker, R.V.; Mittleman, M.A.; et al. Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation 2010, 121, 2331–2378. [Google Scholar] [CrossRef]
- Lee, K.K.; Miller, M.R.; Shah, A.S.V. Air Pollution and Stroke. J. Stroke 2018, 20, 2–11. [Google Scholar] [CrossRef]
- van Eeden, S.F.; Tan, W.C.; Suwa, T.; Mukae, H.; Terashima, T.; Fujii, T.; Qui, D.; Vincent, R.; Hogg, J.C. Cytokines involved in the systemic inflammatory response induced by exposure to particulate matter air pollutants (PM(10)). Am. J. Respir. Crit. Care Med. 2001, 164, 826–830. [Google Scholar] [CrossRef]
- Yan, Z.; Jin, Y.; An, Z.; Liu, Y.; Samet, J.M.; Wu, W. Inflammatory cell signaling following exposures to particulate matter and ozone. Biochim. Biophys. Acta 2016, 1860, 2826–2834. [Google Scholar] [CrossRef]
- Zhao, C.N.; Xu, Z.; Wu, G.C.; Mao, Y.M.; Liu, L.N.; Qian, W.; Dan, Y.L.; Tao, S.S.; Zhang, Q.; Sam, N.B.; et al. Emerging role of air pollution in autoimmune diseases. Autoimmun. Rev. 2019, 18, 607–614. [Google Scholar] [CrossRef]
- Fiorito, G.; Vlaanderen, J.; Polidoro, S.; Gulliver, J.; Galassi, C.; Ranzi, A.; Krogh, V.; Grioni, S.; Agnoli, C.; Sacerdote, C.; et al. Oxidative stress and inflammation mediate the effect of air pollution on cardio- and cerebrovascular disease: A prospective study in nonsmokers. Environ. Mol. Mutagenesis 2018, 59, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Breitner, S.; Cascio, W.E.; Devlin, R.B.; Neas, L.M.; Ward-Caviness, C.; Diaz-Sanchez, D.; Kraus, W.E.; Hauser, E.R.; Schwartz, J.; et al. Association between short-term exposure to ambient fine particulate matter and myocardial injury in the CATHGEN cohort. Environ. Pollut. 2021, 275, 116663. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhao, J.; Zhuo, C.; Zheng, L. The Association Between Ambient Air Pollution and Atrial Fibrillation A Systematic Review and Meta-Analysis. Int. Heart J. 2021, 62, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, C.M. Endothelium. Arterioscler. Thromb. Vasc. Biol. 2016, 36, e26–e31. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Steyers, C.M., 3rd; Miller, F.J., Jr. Endothelial dysfunction in chronic inflammatory diseases. Int. J. Mol. Sci. 2014, 15, 11324–11349. [Google Scholar] [CrossRef]
- Sitia, S.; Tomasoni, L.; Atzeni, F.; Ambrosio, G.; Cordiano, C.; Catapano, A.; Tramontana, S.; Perticone, F.; Naccarato, P.; Camici, P.; et al. From endothelial dysfunction to atherosclerosis. Autoimmun. Rev. 2010, 9, 830–834. [Google Scholar] [CrossRef]
- Zhang, Y.; Dong, S.; Wang, H.; Tao, S.; Kiyama, R. Biological impact of environmental polycyclic aromatic hydrocarbons (ePAHs) as endocrine disruptors. Environ. Pollut. 2016, 213, 809–824. [Google Scholar] [CrossRef]
- Pozo, K.; Estellano, V.H.; Harner, T.; Diaz-Robles, L.; Cereceda-Balic, F.; Etcharren, P.; Pozo, K.; Vidal, V.; Guerrero, F.; Vergara-Fernandez, A. Assessing Polycyclic Aromatic Hydrocarbons (PAHs) using passive air sampling in the atmosphere of one of the most wood-smoke-polluted cities in Chile: The case study of Temuco. Chemosphere 2015, 134, 475–481. [Google Scholar] [CrossRef]
- Hanson, L.R.; Fine, J.M.; Svitak, A.L.; Faltesek, K.A. Intranasal administration of CNS therapeutics to awake mice. J. Vis. Exp. 2013, 74, e4440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaar, K.L.; Brenneman, M.M.; Savitz, S.I. Functional assessments in the rodent stroke model. Exp. Transl. Stroke Med. 2010, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Morton, D.B.; Griffiths, P.H. Guidelines on the recognition of pain, distress and discomfort in experimental animals and an hypothesis for assessment. Vet. Rec. 1985, 116, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Ruijter, J.M.; Lorenz, P.; Tuomi, J.M.; Hecker, M.; van den Hoff, M.J. Fluorescent-increase kinetics of different fluorescent reporters used for qPCR depend on monitoring chemistry, targeted sequence, type of DNA input and PCR efficiency. Mikrochim. Acta 2014, 181, 1689–1696. [Google Scholar] [CrossRef]
- Silva-Renno, A.; Baldivia, G.C.; Oliveira-Junior, M.C.; Brandao-Rangel, M.A.R.; El-Mafarjeh, E.; Dolhnikoff, M.; Mauad, T.; Britto, J.M.; Saldiva, P.H.N.; Oliveira, L.V.F.; et al. Exercise Performed Concomitantly with Particulate Matter Exposure Inhibits Lung Injury. Int. J. Sports Med. 2018, 39, 133–140. [Google Scholar] [CrossRef]
- Avila, L.C.; Bruggemann, T.R.; Bobinski, F.; da Silva, M.D.; Oliveira, R.C.; Martins, D.F.; Mazzardo-Martins, L.; Duarte, M.M.; de Souza, L.F.; Dafre, A.; et al. Effects of High-Intensity Swimming on Lung Inflammation and Oxidative Stress in a Murine Model of DEP-Induced Injury. PLoS ONE 2015, 10, e0137273. [Google Scholar] [CrossRef]
- Ye, J.; Zhu, R.; He, X.; Feng, Y.; Yang, L.; Zhu, X.; Deng, Q.; Wu, T.; Zhang, X. Association of plasma IL-6 and Hsp70 with HRV at different levels of PAHs metabolites. PLoS ONE 2014, 9, e92964. [Google Scholar] [CrossRef]
- Totlandsdal, A.I.; Ovrevik, J.; Cochran, R.E.; Herseth, J.I.; Bolling, A.K.; Lag, M.; Schwarze, P.; Lilleaas, E.; Holme, J.A.; Kubatova, A. The occurrence of polycyclic aromatic hydrocarbons and their derivatives and the proinflammatory potential of fractionated extracts of diesel exhaust and wood smoke particles. J. Environ. Sci. Health A Tox. Hazard. Subst. Environ. Eng. 2014, 49, 383–396. [Google Scholar] [CrossRef]
- Tateishi, Y.; Oda, S.; Nakamura, M.; Watanabe, K.; Kuwaki, T.; Moriguchi, T.; Hirasawa, H. Depressed Heart Rate Variability Is Associated with High Il-6 Blood Level and Decline in the Blood Pressure in Septic Patients. Shock 2007, 28, 549–553. [Google Scholar] [CrossRef]
- Mata-Espinosa, D.; Hernández-Pando, R. Interferón gamma aspectos básicos, importancia clínica y usos terapéuticos. Rev. Investig. Clínica 2008, 60, 421–431. [Google Scholar]
- Klumper, C.; Kramer, U.; Lehmann, I.; von Berg, A.; Berdel, D.; Herberth, G.; Beckmann, C.; Link, E.; Heinrich, J.; Hoffmann, B.; et al. Air pollution and cytokine responsiveness in asthmatic and non-asthmatic children. Environ. Res. 2015, 138, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Samadi, M.T.; Shakerkhatibi, M.; Poorolajal, J.; Rahmani, A.; Rafieemehr, H.; Hesam, M. Association of long term exposure to outdoor volatile organic compounds (BTXS) with pro-inflammatory biomarkers and hematologic parameters in urban adults: A cross-sectional study in Tabriz, Iran. Ecotoxicol. Environ. Saf. 2019, 180, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Uski, O.J.; Happo, M.S.; Jalava, P.I.; Brunner, T.; Kelz, J.; Obernberger, I.; Jokiniemi, J.; Hirvonen, M.R. Acute systemic and lung inflammation in C57Bl/6J mice after intratracheal aspiration of particulate matter from small-scale biomass combustion appliances based on old and modern technologies. Inhal. Toxicol. 2012, 24, 952–965. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Zhang, X. Interleukin-10: New perspectives on an old cytokine. Immunol. Rev. 2008, 226, 205–218. [Google Scholar] [CrossRef]
- Dobreva, Z.G.; Kostadinova, G.S.; Popov, B.N.; Petkov, G.S.; Stanilova, S.A. Proinflammatory and anti-inflammatory cytokines in adolescents from Southeast Bulgarian cities with different levels of air pollution. Toxicol. Ind. Health 2015, 31, 1210–1217. [Google Scholar] [CrossRef]
- Xue, M.; Qiqige, C.; Zhang, Q.; Zhao, H.; Su, L.; Sun, P.; Zhao, P. Effects of Tumor Necrosis Factor alpha (TNF-alpha) and Interleukina 10 (IL-10) on Intercellular Cell Adhesion Molecule-1 (ICAM-1) and Cluster of Differentiation 31 (CD31) in Human Coronary Artery Endothelial Cells. Med. Sci. Monit. 2018, 24, 4433–4439. [Google Scholar] [CrossRef]
- Li, R.; Zhou, R.; Zhang, J. Function of PM2.5 in the pathogenesis of lung cancer and chronic airway inflammatory diseases. Oncol. Lett. 2018, 15, 7506–7514. [Google Scholar] [CrossRef]
- Donato, A.J.; Morgan, R.G.; Walker, A.E.; Lesniewski, L.A. Cellular and molecular biology of aging endothelial cells. J. Mol. Cell. Cardiol. 2015, 89, 122–135. [Google Scholar] [CrossRef]
- Brown, J.D.; Lin, C.Y.; Duan, Q.; Griffin, G.; Federation, A.; Paranal, R.M.; Bair, S.; Newton, G.; Lichtman, A.; Kung, A.; et al. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol. Cell 2014, 56, 219–231. [Google Scholar] [CrossRef]
- Ntziachristos, L.; Ning, Z.; Geller, M.D.; Sheesley, R.J.; Schauer, J.J.; Sioutas, C. Fine, ultrafine and nanoparticle trace element compositions near a major freeway with a high heavy-duty diesel fraction. Atmos. Environ. 2007, 41, 5684–5696. [Google Scholar] [CrossRef]
- Lee, C.C.; Huang, S.H.; Yang, Y.T.; Cheng, Y.W.; Li, C.H.; Kang, J.J. Motorcycle exhaust particles up-regulate expression of vascular adhesion molecule-1 and intercellular adhesion molecule-1 in human umbilical vein endothelial cells. Toxicol. In Vitro 2012, 26, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Nemmar, A.; Hoet, P.M.; Vanquickenborne, B.; Dinsdale, D.; Thomeer, M.; Hoylaerts, M.; Vanbilloen, H.; Mortelmans, L.; Nemery, B.J.C. Passage of inhaled particles into the blood circulation in humans. Circulation 2002, 105, 411–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haarmann-Stemmann, T.; Abel, J.; Fritsche, E.; Krutmann, J. The AhR–Nrf2 pathway in keratinocytes: On the road to chemoprevention? J. Investig. Dermatol. 2012, 132, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Lawal, A.O. Air particulate matter induced oxidative stress and inflammation in cardiovascular disease and atherosclerosis: The role of Nrf2 and AhR-mediated pathways. Toxicol. Lett. 2017, 270, 88–95. [Google Scholar] [CrossRef]
- Møller, P.; Christophersen, D.V.; Jacobsen, N.R.; Skovmand, A.; Gouveia, A.C.; Andersen, M.H.; Kermanizadeh, A.; Jensen, D.M.; Danielsen, P.H.; Roursgaard, M.; et al. Atherosclerosis and vasomotor dysfunction in arteries of animals after exposure to combustion-derived particulate matter or nanomaterials. Crit. Rev. Toxicol. 2016, 46, 437–476. [Google Scholar] [CrossRef]
- Brinchmann, B.C.; Le Ferrec, E.; Bisson, W.H.; Podechard, N.; Huitfeldt, H.S.; Gallais, I.; Sergent, O.; Holme, J.A.; Lagadic-Gossmann, D.; Øvrevik, J. Evidence of selective activation of aryl hydrocarbon receptor nongenomic calcium signaling by pyrene. Biochem. Pharm. 2018, 158, 1–12. [Google Scholar] [CrossRef]
- Orakij, W.; Chetiyanukornkul, T.; Chuesaard, T.; Kaganoi, Y.; Uozaki, W.; Homma, C.; Boongla, Y.; Tang, N.; Hayakawa, K.; Toriba, A. Personal inhalation exposure to polycyclic aromatic hydrocarbons and their nitro-derivatives in rural residents in northern Thailand. Environ. Monit. Assess. 2017, 189, 510. [Google Scholar] [CrossRef]
- Mu, G.; Fan, L.; Zhou, Y.; Liu, Y.; Ma, J.; Yang, S.; Wang, B.; Xiao, L.; Ye, Z.; Shi, T.; et al. Personal exposure to PM(2.5)-bound polycyclic aromatic hydrocarbons and lung function alteration: Results of a panel study in China. Sci. Total Environ. 2019, 684, 458–465. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence Forward | Sequence Reverse |
|---|---|---|
| ICAM-1 | TTCTCATGCCGCACAGAACT | TCCTGGCCTCGGAGACATTA |
| VCAM-1 | CTGGGAAGCTGGAACGAAGT | GCCAAACACTTGACCGTGAC |
| E-Selectin | AGCCTGCCATGTGGTTGAAT | CTTTGCATGATGGCGTCTCG |
| P-Selectin | GAAGTGTGACGCTGTGCAAT | CAGCTGGAGTCGTAGGCAAA |
| PECAM-1 | GGAAGTGTCCTCCCTTGAGC | GGAGCCTTCCGTTCTTAGGG |
| eNOS | GCTCCCAACTGGACCATCTC | TCTTGCACGTAGGTCTTGGG |
| Ahr | TAAAGTCCACCCCTGCTGAC | CATTCAGCGCCTGTAACAAGA |
| Keap1 | GGCAGGACCAGTTGAACAGT | CATAGCCTCCGAGGACGTAG |
| RelA | CCTGGAGCAAGCCATTAGC | CGCACTGCATTCAAGTCATAG |
| IKK-β | GTGCCTGTGACAGCTTACCT | CTCCAGTCTAGAGTCGTGAAGC |
| IL-6 | CCCCAATTTCCAATGCTCTCC | CGCACTAGGTTTGCCGAGTA |
| TNF-α | ATGGCCTCCCTCTCATCAGT | TTTGCTACGACGTGGGCTAC |
| RPL32 | TAAGCGAAACTGGCGGAAAC | CATCAGGATCTGGCCCTTGA |
| B2M | ACTGACCGGCCTGTATGCTA | CAATGTGAGGCGGGTGGAA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojas, G.A.; Saavedra, N.; Saavedra, K.; Hevia, M.; Morales, C.; Lanas, F.; Salazar, L.A. Polycyclic Aromatic Hydrocarbons (PAHs) Exposure Triggers Inflammation and Endothelial Dysfunction in BALB/c Mice: A Pilot Study. Toxics 2022, 10, 497. https://doi.org/10.3390/toxics10090497
Rojas GA, Saavedra N, Saavedra K, Hevia M, Morales C, Lanas F, Salazar LA. Polycyclic Aromatic Hydrocarbons (PAHs) Exposure Triggers Inflammation and Endothelial Dysfunction in BALB/c Mice: A Pilot Study. Toxics. 2022; 10(9):497. https://doi.org/10.3390/toxics10090497
Chicago/Turabian StyleRojas, Gabriel A., Nicolás Saavedra, Kathleen Saavedra, Montserrat Hevia, Cristian Morales, Fernando Lanas, and Luis A. Salazar. 2022. "Polycyclic Aromatic Hydrocarbons (PAHs) Exposure Triggers Inflammation and Endothelial Dysfunction in BALB/c Mice: A Pilot Study" Toxics 10, no. 9: 497. https://doi.org/10.3390/toxics10090497
APA StyleRojas, G. A., Saavedra, N., Saavedra, K., Hevia, M., Morales, C., Lanas, F., & Salazar, L. A. (2022). Polycyclic Aromatic Hydrocarbons (PAHs) Exposure Triggers Inflammation and Endothelial Dysfunction in BALB/c Mice: A Pilot Study. Toxics, 10(9), 497. https://doi.org/10.3390/toxics10090497