
Purification and Characterization of β-Mannanase Derived from Rhizopus microsporus var. rhizopodiformis Expressed in Komagataella phaffii

 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Strains
2.2. Construction and Mutants
2.3. Protein Expression
2.4. Protein Purification
2.5. Protein Analysis
2.6. Determination of Enzyme Properties
2.7. Hydrolysis Properties
2.8. Analytical Methods
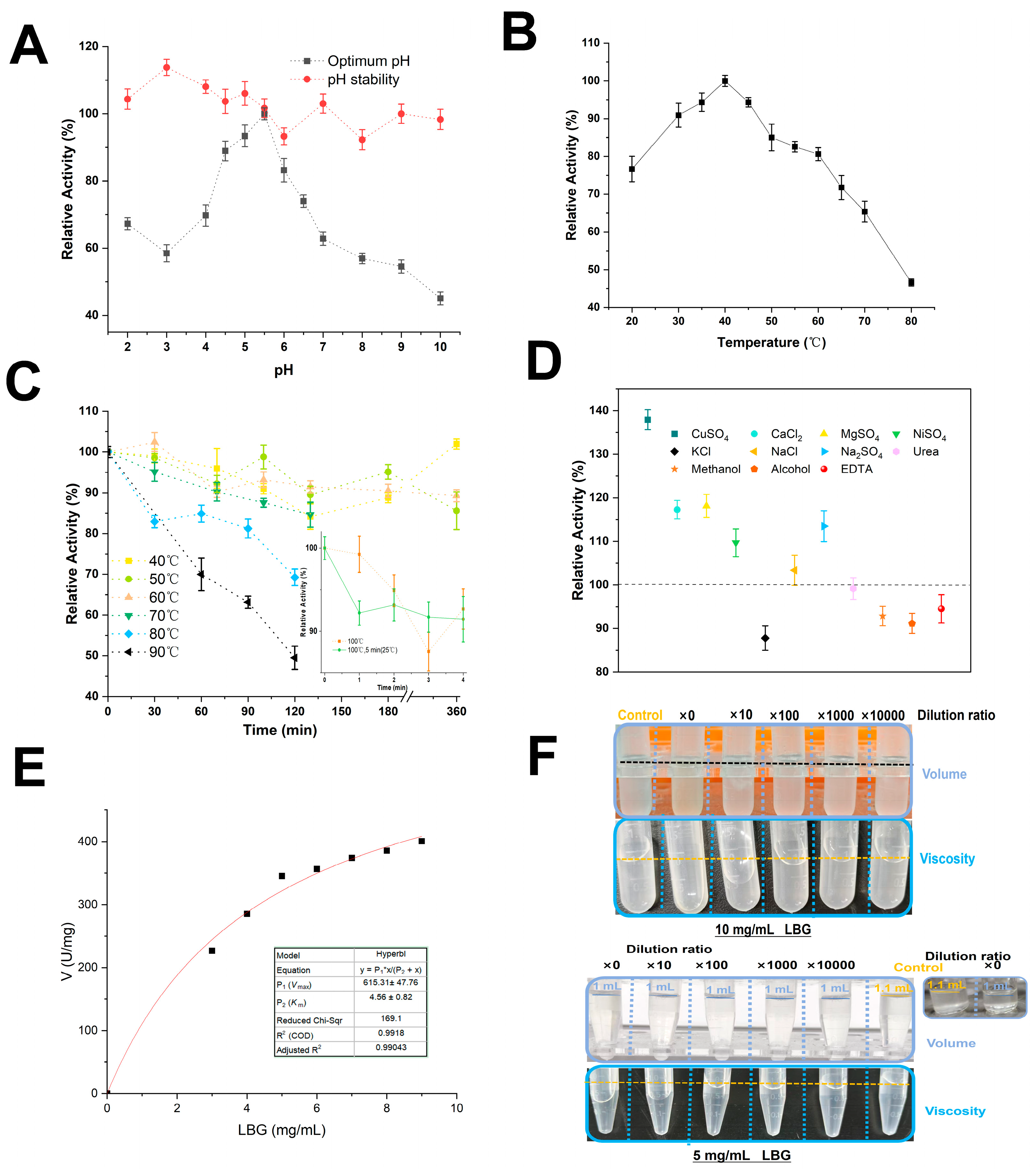
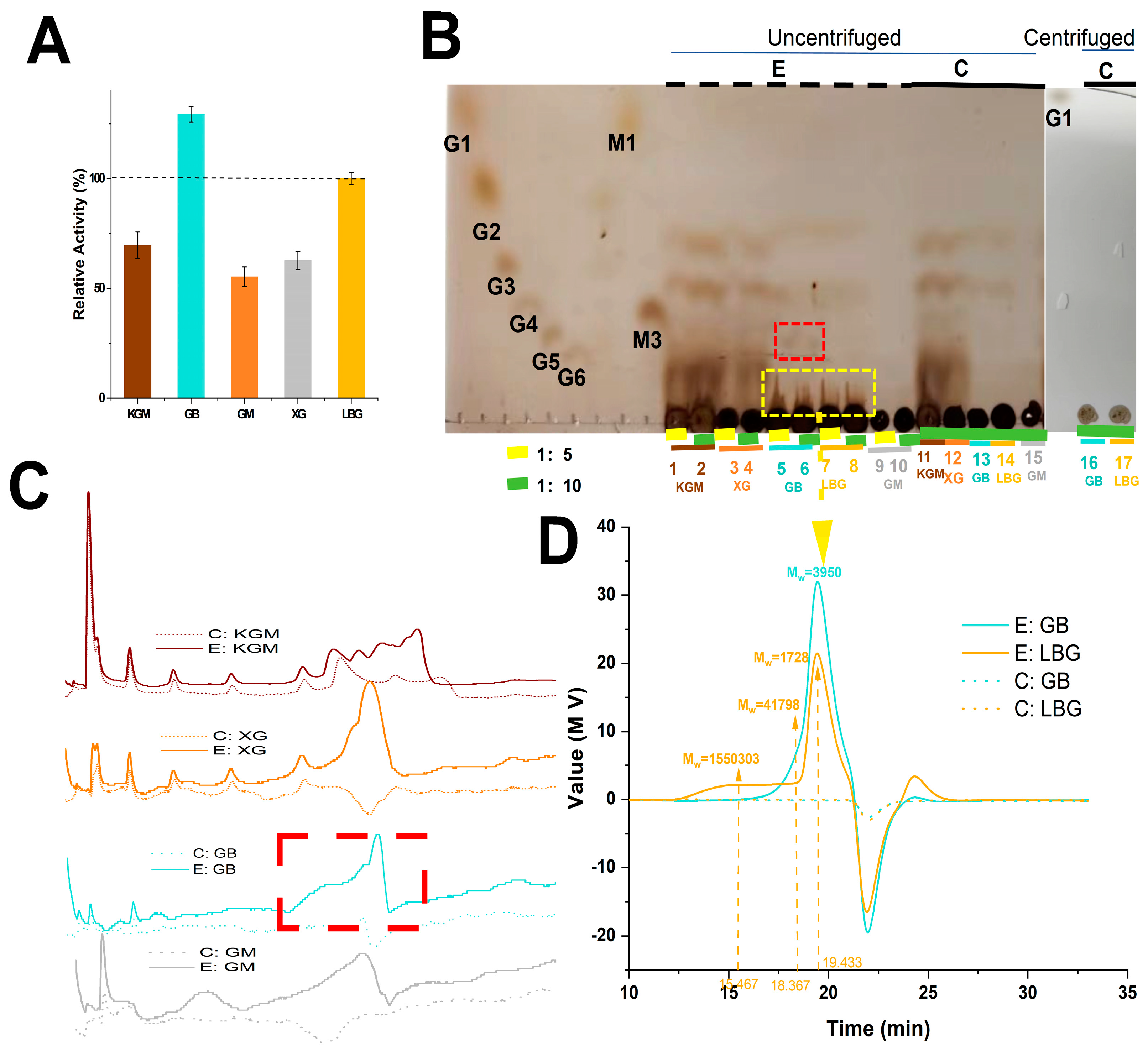
3. Results and Discussion
3.1. Better-Evaluated Conditions for Enzymatic Production of PpRmMan134A
3.2. Enzymatic Properties of PpRmMan134A
3.3. Hydrolysis Properties of PpRmMan134A
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Davy, P.; Kirkman, T.; Scarlett, C.J.; Vuong, Q. Characterisation of a High Fibre Flour Prepared from Soy Milk By-Product and Its Potential Use in White Wheat Bread. Foods 2022, 11, 3921. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Zhou, C.; Li, D.; Pei, J.; Guo, A.; Liu, S.; Li, H. Monitoring the Effects of Hemicellulase on the Different Proofing Stages of Wheat Aleurone-Rich Bread Dough and Bread Quality. Foods 2021, 10, 2427. [Google Scholar] [CrossRef] [PubMed]
- Dawood, A.; Ma, K. Applications of Microbial β-Mannanases. Front. Bioeng. Biotechnol. 2020, 8, 598630. [Google Scholar] [CrossRef] [PubMed]
- Rueckel, M.; Janson, S.; Solbak, A.; Fickler, A. Spatial Activity Mapping of SS-Mannanase on Soybean Seeds. Sci. Rep. 2024, 14, 1037. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Wiemann, M.; Stålbrand, H. β-Mannanase BoMan26B from Bacteroides Ovatus Produces Mannan-Oligosaccharides with Prebiotic Potential from Galactomannan and Softwood β-Mannans. LWT 2021, 151, 112215. [Google Scholar] [CrossRef]
- Mary, P.R.; Prashanth, K.V.H.; Vasu, P.; Kapoor, M. Structural Diversity and Prebiotic Potential of Short Chain β-Manno-Oligosaccharides Generated from Guar Gum by Endo-β-Mannanase (ManB-1601). Carbohydr. Res. 2019, 486, 107822. [Google Scholar] [CrossRef]
- Mohammadzadeh, R.; Karbalaei, M.; Soleimanpour, S.; Mosavat, A.; Rezaee, S.A.; Ghazvini, K.; Farsiani, H. Practical Methods for Expression of Recombinant Protein in the Pichia Pastoris System. Curr. Protoc. 2021, 1, e155. [Google Scholar] [CrossRef]
- Li, Y.; Yi, P.; Liu, J.; Yan, Q.; Jiang, Z. High-Level Expression of an Engineered β-Mannanase (mRmMan5A) in Pichia Pastoris for Manno-Oligosaccharide Production Using Steam Explosion Pretreated Palm Kernel Cake. Bioresour. Technol. 2018, 256, 30–37. [Google Scholar] [CrossRef]
- Wang, N.-N.; Liu, J.; Li, Y.-X.; Ma, J.-W.; Yan, Q.-J.; Jiang, Z.-Q. High-Level Expression of a Glycoside Hydrolase Family 26 β-Mannanase from Aspergillus Niger in Pichia Pastoris for Production of Partially Hydrolysed Fenugreek Gum. Process Biochem. 2021, 100, 90–97. [Google Scholar] [CrossRef]
- Zhang, S.-B.; Zhang, W.-J.; Li, N.; Zhai, H.-C.; Lv, Y.-Y.; Hu, Y.-S.; Cai, J.-P. Functional Expression and Characterization of an Endo-1,4-β-Mannosidase from Triticum Aestivum in Pichia Pastoris. Biologia 2020, 75, 2073–2081. [Google Scholar] [CrossRef]
- Ren, M.; Zhang, Y.; Wang, X.; Qiu, H. Catalytic Hydrogenation of CO2 to Methanol: A Review. Catalysts 2022, 12, 403. [Google Scholar] [CrossRef]
- Zhu, T.; Sun, H.; Wang, M.; Li, Y. Pichia Pastoris as a Versatile Cell Factory for the Production of Industrial Enzymes and Chemicals: Current Status and Future Perspectives. Biotechnol. J. 2019, 14, 1800694. [Google Scholar] [CrossRef] [PubMed]
- Karbalaei, M.; Rezaee, S.A.; Farsiani, H. Pichia Pastoris: A Highly Successful Expression System for Optimal Synthesis of Heterologous Proteins. J. Cell. Physiol. 2020, 235, 5867–5881. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ning, C.; Yuan, M.; Fu, X.; Yang, S.; Wei, X.; Xiao, M.; Mou, H.; Zhu, C. High-Efficiency Expression of a Superior β-Mannanase Engineered by Cooperative Substitution Method in Pichia Pastoris and Its Application in Preparation of Prebiotic Mannooligosaccharides. Bioresour. Technol. 2020, 311, 123482. [Google Scholar] [CrossRef]
- Peña, D.A.; Gasser, B.; Zanghellini, J.; Steiger, M.G.; Mattanovich, D. Metabolic Engineering of Pichia Pastoris. Metab. Eng. 2018, 50, 2–15. [Google Scholar] [CrossRef]
- Hao, J.; Xu, L.; He, H.; Du, X.; Jia, L. High-Level Expression of Staphylococcal Protein A in Pichia Pastoris and Purification and Characterization of the Recombinant Protein. Protein Expr. Purif. 2013, 90, 178–185. [Google Scholar] [CrossRef]
- Kongsinkaew, C.; Ajariyakhajorn, K.; Boonyaratanakornkit, V.; Sooksai, S.; Pornpukdeewattana, S.; Krusong, W.; Sitanggang, A.B.; Charoenrat, T. Membrane-Based Approach for the Removal of Pigment Impurities Secreted by Pichia Pastoris. Food Bioprod. Process. 2023, 139, 178–189. [Google Scholar] [CrossRef]
- Kastilan, R.; Boes, A.; Spiegel, H.; Voepel, N.; Chudobová, I.; Hellwig, S.; Buyel, J.F.; Reimann, A.; Fischer, R. Improvement of a Fermentation Process for the Production of Two PfAMA1-DiCo-Based Malaria Vaccine Candidates in Pichia Pastoris. Sci. Rep. 2017, 7, 11991. [Google Scholar] [CrossRef]
- Güneş, H.; Çalık, P. Oxygen Transfer as a Tool for Fine-Tuning Recombinant Protein Production by Pichia Pastoris under Glyceraldehyde-3-Phosphate Dehydrogenase Promoter. Bioprocess Biosyst. Eng. 2016, 39, 1061–1072. [Google Scholar] [CrossRef]
- Theron, C.W.; Berrios, J.; Delvigne, F.; Fickers, P. Integrating Metabolic Modeling and Population Heterogeneity Analysis into Optimizing Recombinant Protein Production by Komagataella (Pichia) Pastoris. Appl. Microbiol. Biotechnol. 2018, 102, 63–80. [Google Scholar] [CrossRef]
- White, C.R.; Alton, L.A.; Bywater, C.L.; Lombardi, E.J.; Marshall, D.J. Metabolic Scaling Is the Product of Life-History Optimization. Science 2022, 377, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Trigueros, C.A.; Rillig, M.C.; Crowther, T.W. Applying Allometric Theory to Fungi. ISME J. 2017, 11, 2175–2180. [Google Scholar] [CrossRef] [PubMed]
- Barve, G.; Manjithaya, R. Cross-Talk between Autophagy and Sporulation in Saccharomyces Cerevisiae. Yeast 2021, 38, 401–413. [Google Scholar] [CrossRef]
- Siu, K.-H.; Chen, W. Control of the Yeast Mating Pathway by Reconstitution of Functional α-Factor Using Split Intein-Catalyzed Reactions. ACS Synth. Biol. 2017, 6, 1453–1460. [Google Scholar] [CrossRef]
- Travers Cook, T.J.; Skirgaila, C.; Martin, O.Y.; Buser, C.C. Infection by dsRNA Viruses Is Associated with Enhanced Sporulation Efficiency in Saccharomyces Cerevisiae. Ecol. Evol. 2022, 12, e8558. [Google Scholar] [CrossRef] [PubMed]
- West, G.B.; Brown, J.H.; Enquist, B.J. A General Model for the Origin of Allometric Scaling Laws in Biology. Science 1997, 276, 122–126. [Google Scholar] [CrossRef]
- Gillooly, J.F.; Brown, J.H.; West, G.B.; Savage, V.M.; Charnov, E.L. Effects of Size and Temperature on Metabolic Rate. Science 2001, 293, 2248–2251. [Google Scholar] [CrossRef]
- You, X.; Qin, Z.; Li, Y.-X.; Yan, Q.-J.; Li, B.; Jiang, Z.-Q. Structural and Biochemical Insights into the Substrate-Binding Mechanism of a Novel Glycoside Hydrolase Family 134 β-Mannanase. Biochim. Et Biophys. Acta (BBA)-Gen. Subj. 2018, 1862, 1376–1388. [Google Scholar] [CrossRef]
- Cabral, K.M.S.; Almeida, M.S.; Valente, A.P.; Almeida, F.C.L.; Kurtenbach, E. Production of the Active Antifungal Pisum Sativum Defensin 1 (Psd1) in Pichia Pastoris: Overcoming the Inefficiency of the STE13 Protease. Protein Expr. Purif. 2003, 31, 115–122. [Google Scholar] [CrossRef]
- Zhang, B.; Zhao, X.; Wang, Z.; Wang, H.; Zhou, J.; Du, G.; Chen, J.; Li, J. Efficient Secretory Expression and Purification of Food-Grade Porcine Myoglobin in Komagataella Phaffii. J. Agric. Food Chem. 2021, 69, 10235–10245. [Google Scholar] [CrossRef]
- Bahrami, A.; Shojaosadati, S.A.; Khalilzadeh, R.; Mohammadian, J.; Farahani, E.V.; Masoumian, M.R. Prevention of Human Granulocyte Colony-Stimulating Factor Protein Aggregation in Recombinant Pichia Pastoris Fed-Batch Fermentation Using Additives. Biotechnol. Appl. Biochem. 2009, 52, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Çelik, E.; Çalık, P.; Oliver, S.G. Metabolic Flux Analysis for Recombinant Protein Production by Pichia Pastoris Using Dual Carbon Sources: Effects of Methanol Feeding Rate. Biotechnol. Bioeng. 2010, 105, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhang, J.; Liu, B.; Du, G.; Chen, J. High-Level Extracellular Production of Glucose Oxidase by Recombinant Pichia Pastoris Using a Combined Strategy. Appl. Biochem. Biotechnol. 2015, 175, 1429–1447. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, H.; Shi, Y.; Yan, Q.; You, X.; Jiang, Z. Preparation, Characterization, and Prebiotic Activity of Manno-Oligosaccharides Produced from Cassia Gum by a Glycoside Hydrolase Family 134 β-Mannanase. Food Chem. 2020, 309, 125709. [Google Scholar] [CrossRef] [PubMed]
- Ciriminna, R.; Meneguzzo, F.; Delisi, R.; Pagliaro, M. Citric Acid: Emerging Applications of Key Biotechnology Industrial Product. Chem. Cent. J. 2017, 11, 22. [Google Scholar] [CrossRef]
- WHO. Safety Evaluation of Certain Food Additives and Contaminants; WHO Food Additives Series; WHO: Geneva, Switzerland, 2004. [Google Scholar]
- Inan, M.; Meagher, M.M. Non-Repressing Carbon Sources for Alcohol Oxidase (AOX1) Promoter of Pichia Pastoris. J. Bioscience Bioeng. 2001, 92, 585–589. [Google Scholar] [CrossRef]
- Matthews, C.B.; Kuo, A.; Love, K.R.; Love, J.C. Development of a General Defined Medium for Pichia Pastoris. Biotechnol. Bioeng. 2018, 115, 103–113. [Google Scholar] [CrossRef]
- Elzoghby, A.O.; El-Fotoh, W.S.A.; Elgindy, N.A. Casein-Based Formulations as Promising Controlled Release Drug Delivery Systems. J. Control. Release 2011, 153, 206–216. [Google Scholar] [CrossRef]
- Chhanwal, N.; Anishaparvin, A.; Indrani, D.; Raghavarao, K.S.M.S.; Anandharamakrishnan, C. Computational Fluid Dynamics (CFD) Modeling of an Electrical Heating Oven for Bread-Baking Process. J. Food Eng. 2010, 100, 452–460. [Google Scholar] [CrossRef]
- Srivastava, P.K.; Kapoor, M. Production, Properties, and Applications of Endo-β-Mannanases. Biotechnol. Adv. 2017, 35, 1–19. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qu, J.; Long, J.; Li, X.; Zhou, X.; Chen, L.; Qiu, C.; Jin, Z. Purification and Characterization of β-Mannanase Derived from Rhizopus microsporus var. rhizopodiformis Expressed in Komagataella phaffii. Foods 2024, 13, 3324. https://doi.org/10.3390/foods13203324
Qu J, Long J, Li X, Zhou X, Chen L, Qiu C, Jin Z. Purification and Characterization of β-Mannanase Derived from Rhizopus microsporus var. rhizopodiformis Expressed in Komagataella phaffii. Foods. 2024; 13(20):3324. https://doi.org/10.3390/foods13203324
Chicago/Turabian StyleQu, Jinghua, Jie Long, Xingfei Li, Xing Zhou, Long Chen, Chao Qiu, and Zhengyu Jin. 2024. "Purification and Characterization of β-Mannanase Derived from Rhizopus microsporus var. rhizopodiformis Expressed in Komagataella phaffii" Foods 13, no. 20: 3324. https://doi.org/10.3390/foods13203324
APA StyleQu, J., Long, J., Li, X., Zhou, X., Chen, L., Qiu, C., & Jin, Z. (2024). Purification and Characterization of β-Mannanase Derived from Rhizopus microsporus var. rhizopodiformis Expressed in Komagataella phaffii. Foods, 13(20), 3324. https://doi.org/10.3390/foods13203324