Abstract
Propargyl alcohol is a useful synthon in synthetic organic chemistry. We found that the ruthenium(II) complex [Cp*RuCl(diene)] (Cp* = η5-C5Me5; diene = isoprene or 1,5-cyclooctadiene (cod)) catalyzes dimerization of 1,1-diphenylprop-2-yn-1-ol (1,1-diphenylpropargyl alcohol, 1a) at room temperature to afford an alkylidenebenzocyclobutenyl alcohol 2a quantitatively. Meanwhile, a stoichiometric reaction of the related hydrido complex [Cp*RuH(cod)] with 1a at 50 °C led to isolation of a ruthenocene derivative 4 bearing a cyclopentadienyl ring generated by dehydrogenative trimerization of 1a. Detailed structures of 2a and 4 were determined by X-ray crystallography. The reaction mechanisms for the formation of 2a and 4 were proposed.
1. Introduction
Propargyl alcohol and their derivatives have been attractive starting materials in synthetic organic chemistry [1,2,3,4,5,6,7]. As one of the various transition-metal catalysts, ruthenium complexes with cyclopentadienyl co-ligands have been used effectively for their catalytic transformations involving carbon–carbon and carbon–heteroatom bond formations [8,9]. For example, the Hidai’s thiolato-bridged dinuclear Cp*Ru (Cp* = η5-C5Me5) complexes catalyze nucleophilic propargylic substitution of terminal propargylic alcohols owing to facile and reversible formation of an allenylidene intermediate [10,11,12]. Recently, Fürstner and co-workers described that regioselective hydrometalation [13,14] and ene–yne coupling [15] of propargylic alcohols are catalyzed by Cp*RuCl complexes. The latter reactions are guided by the intramolecular hydrogen bonds between the hydroxy group in the alcohol and the chlorido ligand. As an extension of our study on catalytic transformation of propargylic and structurally related allylic compounds [16,17,18,19], we report here the reactions of the Cp*Ru complexes [Cp*RuX(diene)] (X = Cl, H) with a 1,1-diphenylprop-2-yn-1-ol (1,1-diphenylpropargyl alcohol) HC≡CC(OH)Ph2 (1a). The phenyl substituent in 1a proved to undergo unexpectedly facile C–H bond cleavage or to migrate in the coordination sphere of the Cp*Ru complexes, leading to catalytic formation of a benzocyclobutene as a dimerization product and a novel ruthenocene complex bearing a highly functionalized cyclopentadienyl ring derived from dehydrogenative trimerization of 1a.
2. Results and Discussion
2.1. Ruthenium-Catalytic Dimerization of 1,1-Arylpropargyl Alcohol
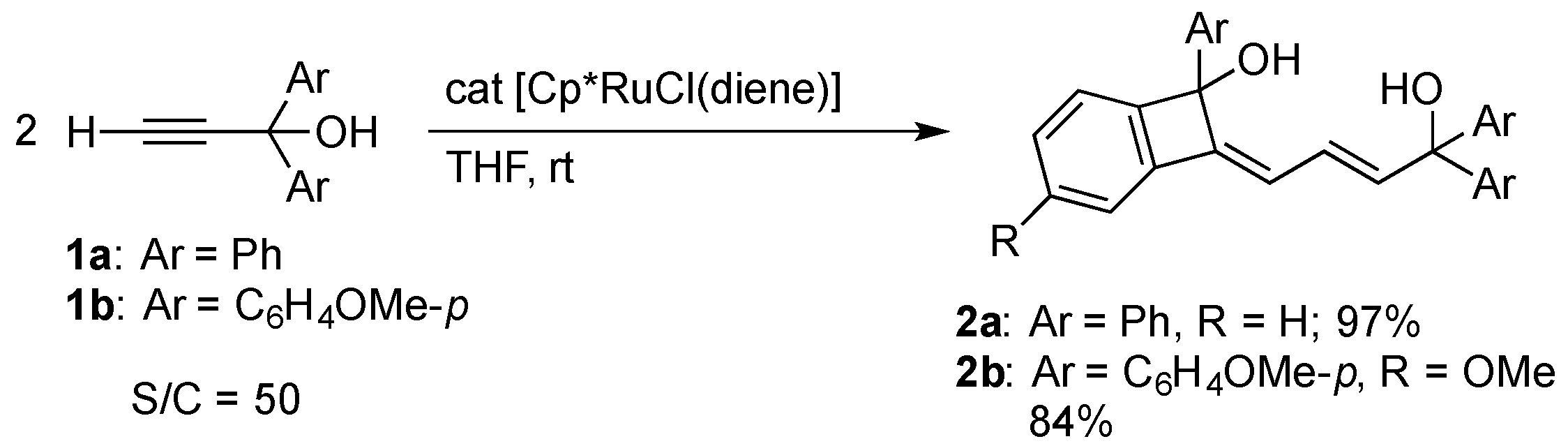
The addition of the ruthenium(II) complex [Cp*RuCl(diene)] (diene = isoprene or 1,5-cyclooctadiene (cod)) to 50 equiv. of 1,1-diphenylpropargyl alcohol 1a in THF at room temperature resulted in full conversion of the alcohol. A subsequent chromatographic workup afforded a novel alkylidenebenzocyclobutenyl alcohol 2a in 97% yield (Scheme 1). The p-methoxyphenyl analogue 1b was also converted to 2b. The products 2 were characterized by X-ray analysis of 2a as well as 1H and 13C{1H} NMR spectroscopy. Figure 1 clearly shows the benzocyclobutene framework of 2a. The C–C bond distances in the six-membered ring of the benzocyclobutene core fall in the range of 1.383(2)–1.398(2) Å, indicating the delocalization of the double bonds. Meanwhile, the short C2–C15 and C16–C17 distances (1.337(2) Å) as well as the long C15–C16 distance (1.456(2) Å) in 2 are in agreement with the butadiene skeleton derived from dimerization of 1. The hydroxy groups is preserved throughout the reaction despite of their ease of dehydration in the coordination sphere [20]. The 1H NMR spectrum of 2a displays three mutually coupled vinyl resonances at δ 6.27, 6.32, and 6.45; these signals are assigned to the H14, H12, and H13 atoms shown in Figure 1, respectively, by HMQC and HMBC experiments.
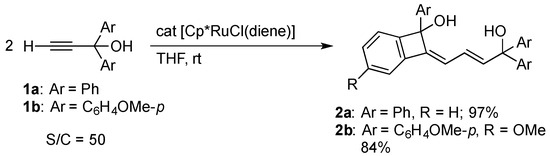
Scheme 1.
Catalytic dimerization of 1,1-diarylpropargyl alcohols 1 with the Cp*RuCl complex.
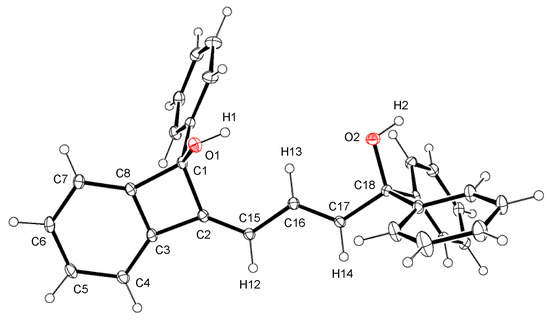
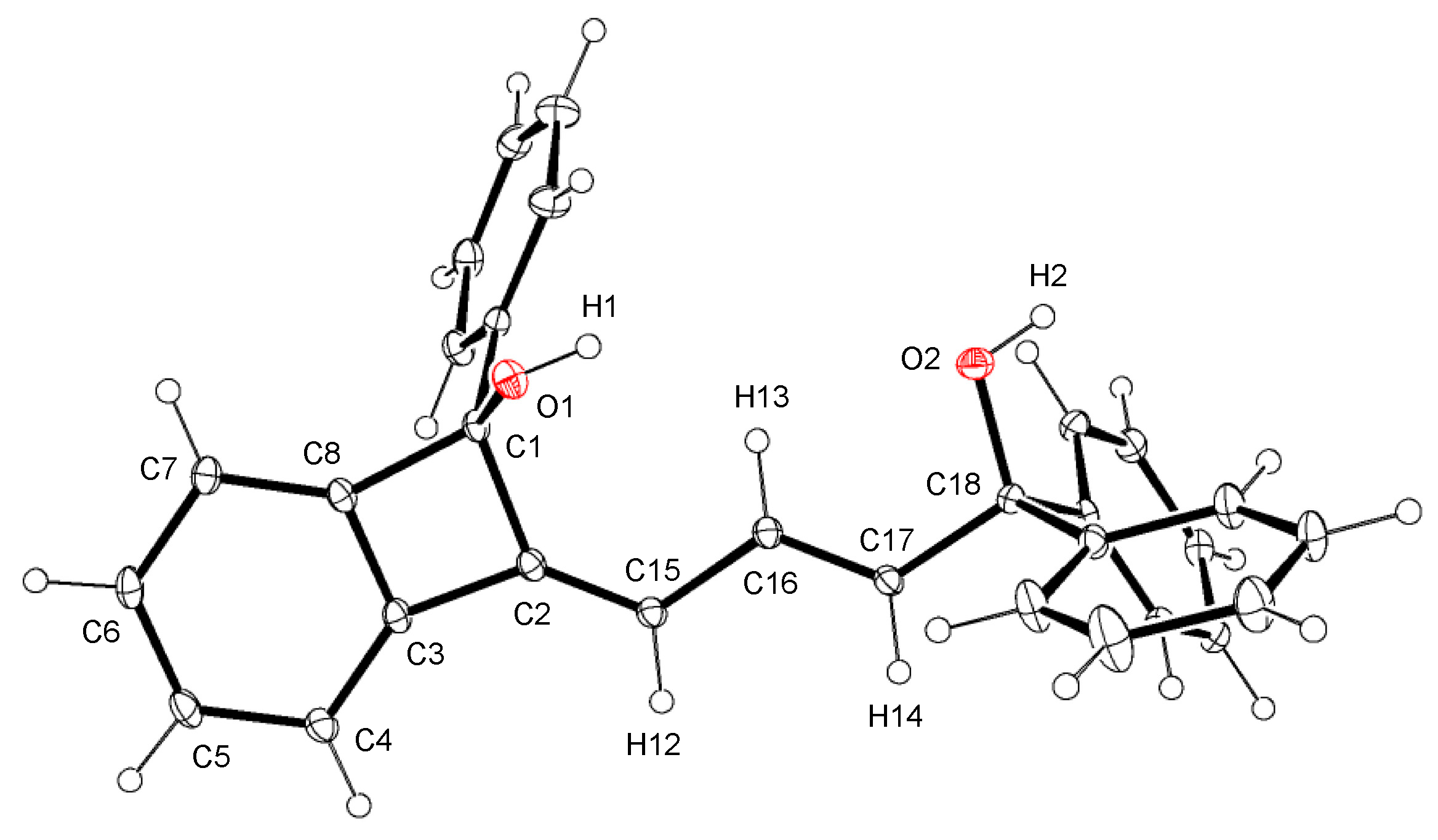
Figure 1.
Crystal structure of 2a·0.5acetone. The solvated molecule is omitted for clarity. Ellipsoids are drawn at the 30% probability level. Selected bond distances (Å) and angles (deg.): C1–C2, 1.565(2); C1–C8, 1.539(2); C2–C3, 1.473(2); C2–C15, 1.337(2); C3–C8, 1.390(2); C15–C16, 1.456(2); C16–C17, 1.337(2); C17–C18, 1.516(2); C2–C1–C8, 84.14(11); C1–C2–C3, 89.27(11); C1–C2–C15, 133.32(14); C3–C2–C15, 137.20(15); C2–C3–C8, 93.11(12); C1–C8–C3, 93.48(12).
The catalytic formation of 2 should entail the orthometalation of one of the aryl group in 1 in addition to the C–C bond formation. We confirmed that a potential intermediate enyne 3 [21], which would be formed by head-to-head dimerization of 1a [22,23], does not undergo the C–H cleavage reaction under the present dimerization conditions (Scheme 2). The result may also exclude the involvement of analogous orthometalation of the terminal monoyne 1 in the catalysis, although a related C–H cleavage reaction of 1a on an osmium complex was known [24]. On the other hand, a labeling experiment using 1a-d1 with a deuterium at the acetylenic position revealed that the hydrogen atom derived from the aromatic C–H bond cleavage is selectively incorporated into the vinyl group adjacent to the C(OH)Ph2 moiety in 2a (Scheme 3).
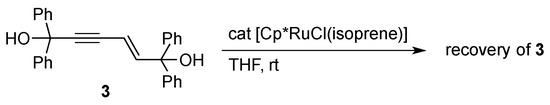
Scheme 2.
Attempted reaction of an enyne with the Cp*RuCl catalyst.
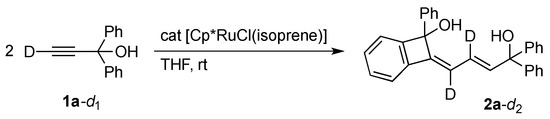
Scheme 3.
Deuterium labeling experiment.
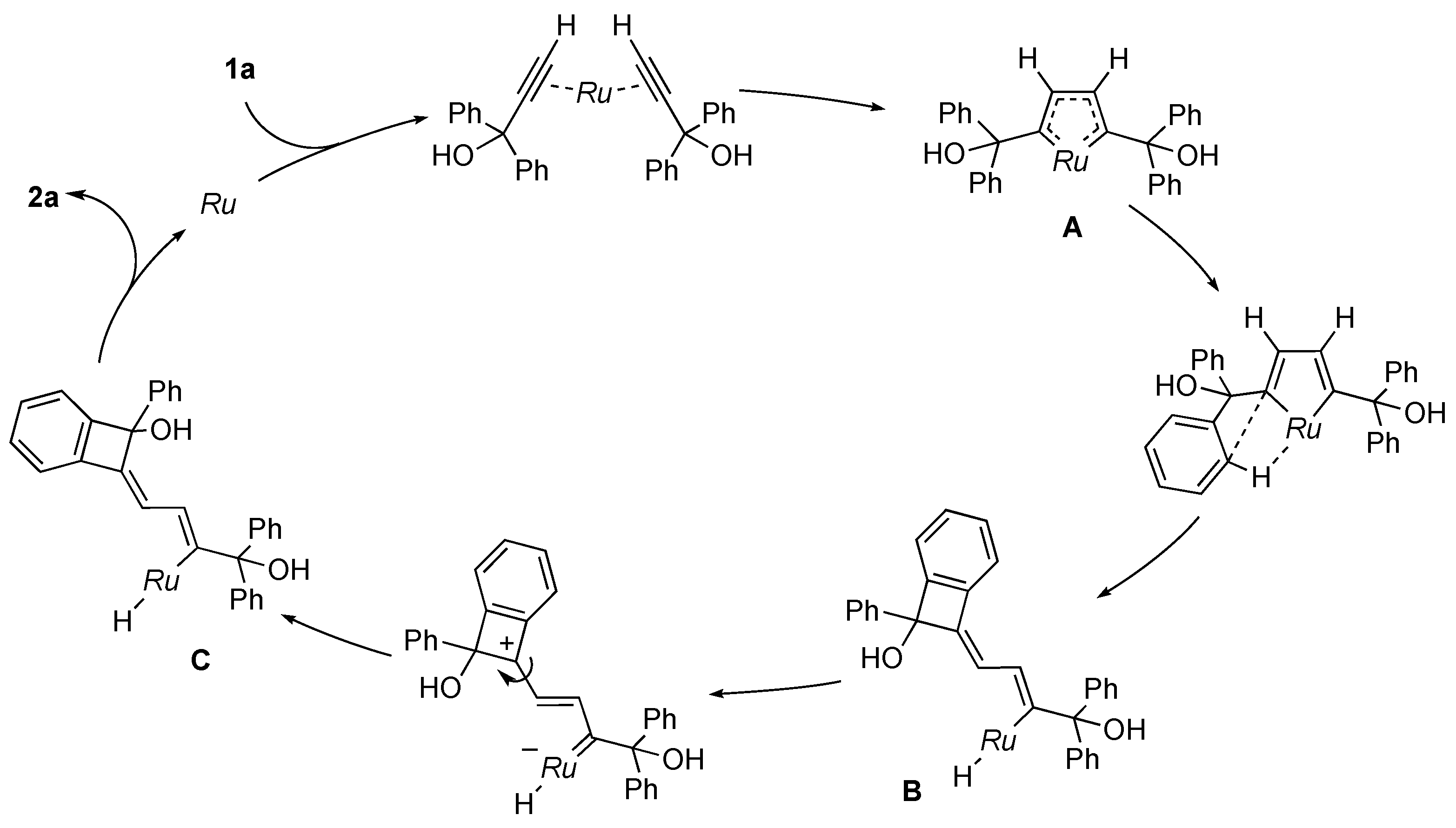
On the basis of these observations, we propose the mechanism for the catalytic dimerization of 1a (Scheme 4). Two molecules of 1a first bind to the ruthenium atom to form a ruthenacycle A [8,9,25]. Subsequent σ-bond metathesis in A would result in the formation of the vinyl intermediate B, which would then undergo E–Z isomerization. Reductive elimination from the hydrido(vinyl) complex C affords the dimerization product 2a. The mechanism is consistent with the deuterium labeling experiment illustrated in Scheme 3. In related reactions of propargylic alcohols without 1-aryl substituents, Dixneuf and co-workers obtained alkylidenecyclobutene derivatives by a three-component dehydrative condensation of propargylic alcohols and carboxylic acid with a Cp*Ru catalyst [26]. They isolated a cyclobutadiene complex, which may be derived from reductive elimination form A, as the reaction intermediate. Trost and co-workers also described a ruthenacycle similar to A as a key intermediate in CpRu-catalyzed reactions of propargylic alcohols [27,28]. On the other hand, Chan and co-workers synthesized indene derivatives by the iron-catalyzed self-condensation of 1-arylpropargyl alcohols involving aromatic C–H bond cleavage [29].
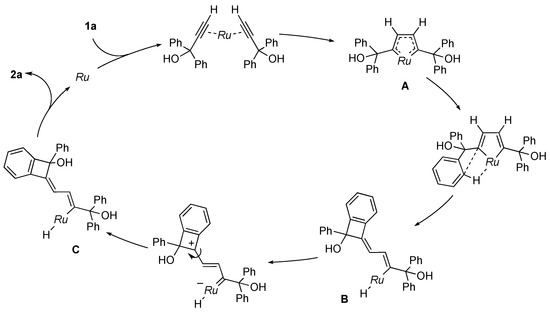
Scheme 4.
Proposed mechanism for catalytic dimerization of 1a. Ru = Cp*RuCl.
2.2. Reaction of a (Hydrido)ruthenium Complex with 1,1-Diphenylpropargyl Alcohol
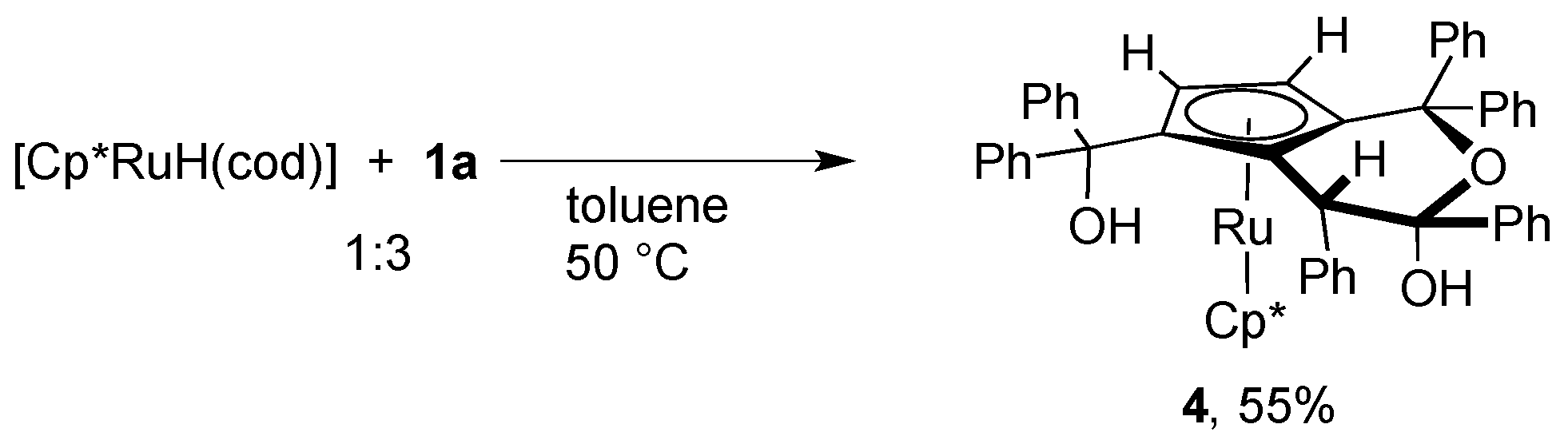
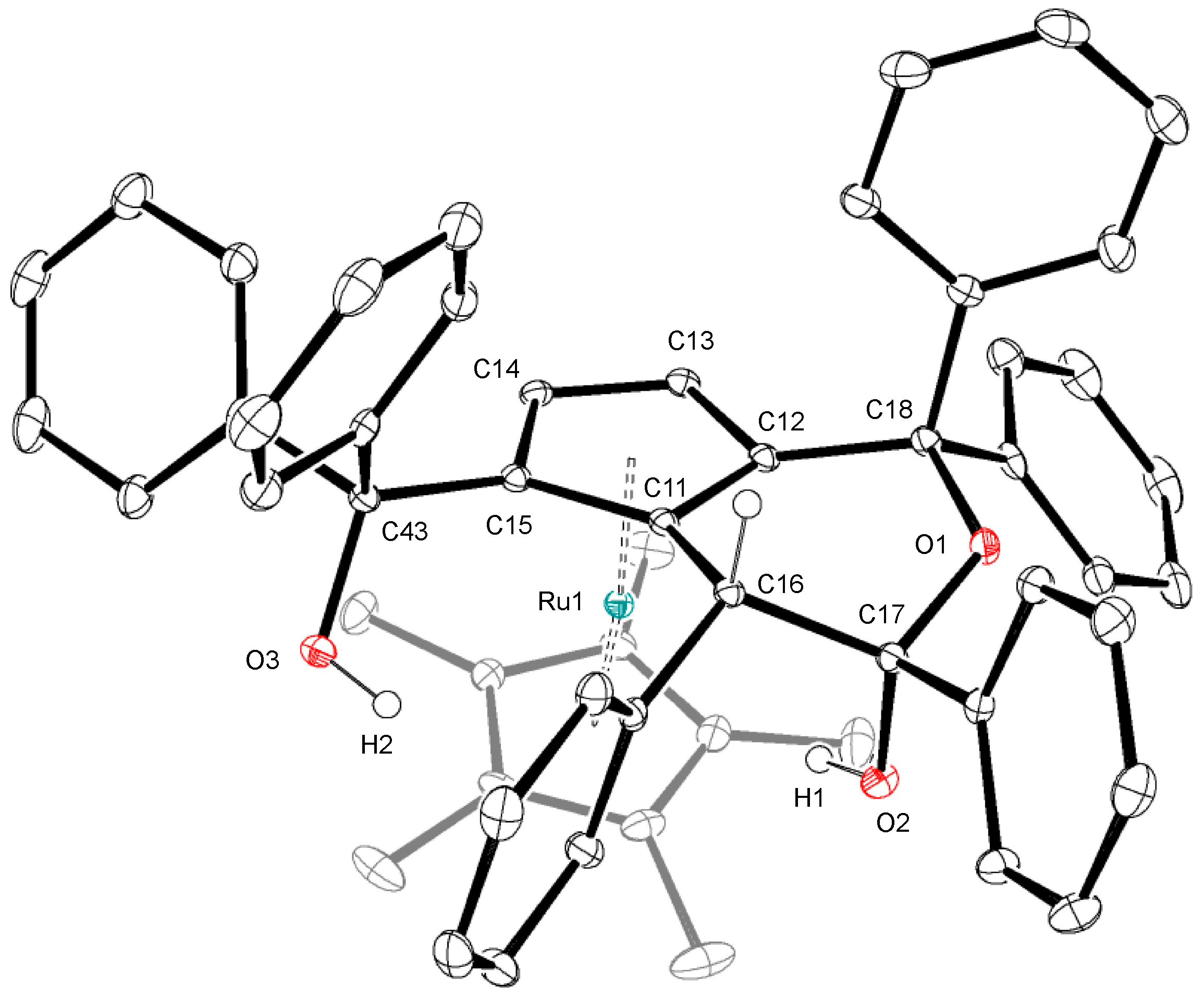
In order to gain further insight into the mechanism of the catalytic dimerization of 1a shown in Scheme 1, we examined the reaction of 1a with a related hydrido complex. When the hydrido complex [Cp*RuH(cod)] was treated with a slight excess of 1a at 50 °C, a novel ruthenocene complex 4 was obtained in moderate yield (Scheme 5). The 1H NMR spectrum of 4 exhibits two mutually coupled doublets at δ 3.56 and 4.24 with a 1H intensity each, which are assignable to the newly formed cyclopentadienyl ligand. Figure 2 depicts the crystal structure of 4 featuring a fused bicyclic hemiketal skelton derived from dehydrative condensation of three molecules of 1a. The two cyclopentadinyl rings in 4 are slightly tilted with a dihedral angle of 12.9°. The Cp-fused six-membered ring adopts a half-chair conformation with an axial hydroxy group, and two vicinal phenyl groups in the ring lie in the anti configuration.
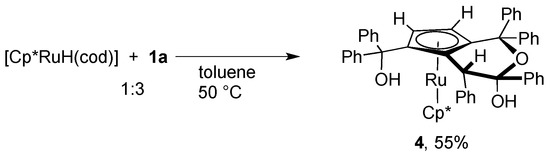
Scheme 5.
Synthesis of 4.
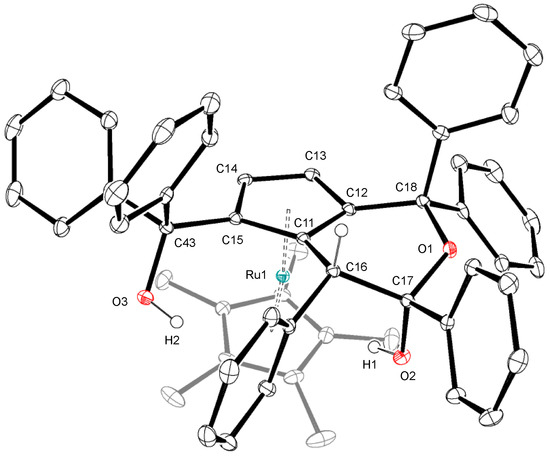
Figure 2.
Crystal structure of 4. Ellipsoids are drawn at the 30% probability level. Selected bond distances (Å): O1–C17, 1.419(2); O1–C18, 1.455(2); C11–C16, 1.513(2); C12–C18, 1.515(2); C16–C17, 1.570(2).
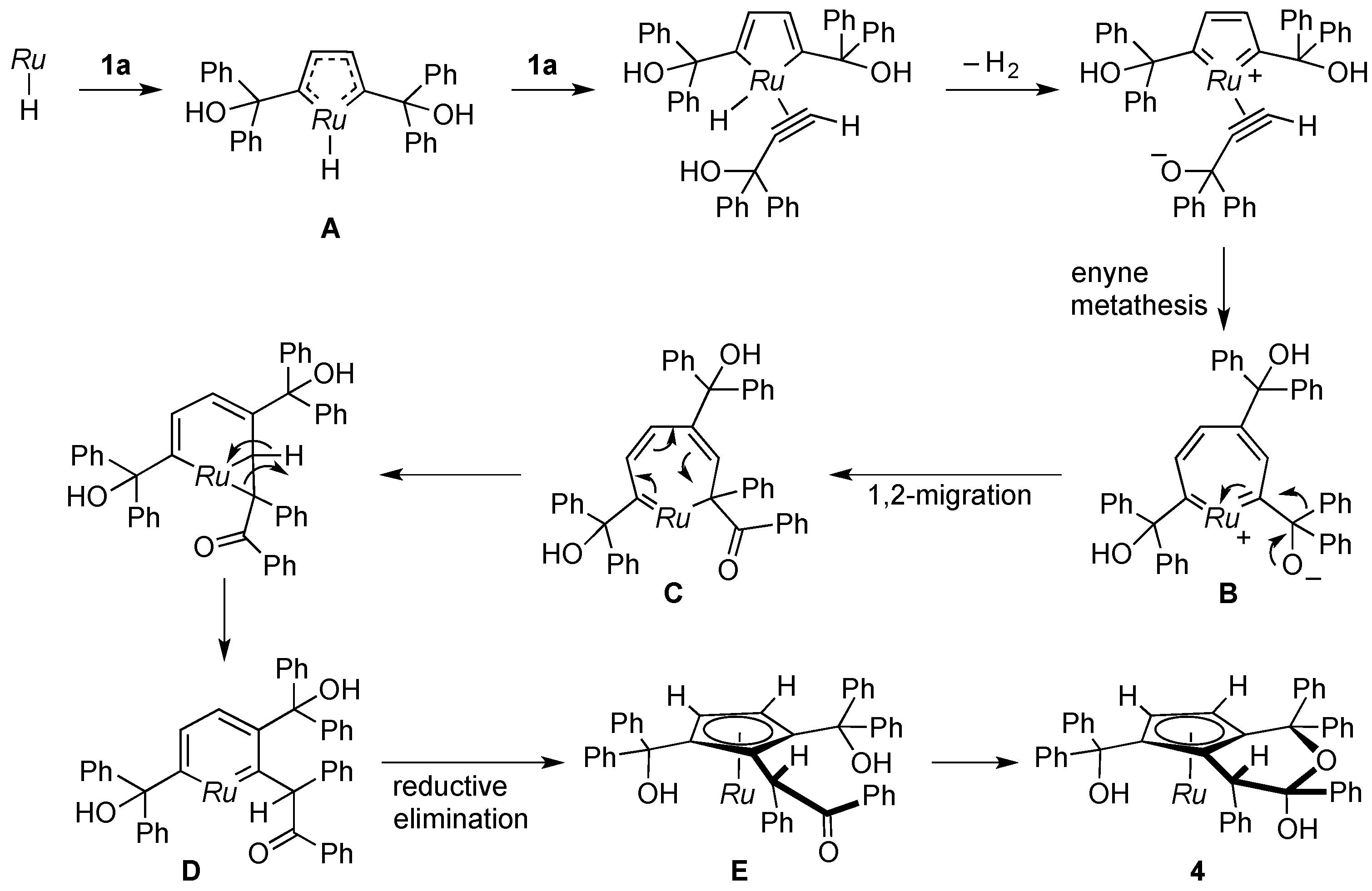
The mechanism for the formation of 4 remains open to speculation; however, the reaction apparently involves 1,2-migration of the phenyl group in the propragylic alcohol. A plausible route is suggested in Scheme 6. Formation of the ruthenacycle A as in the dimerization of 1a (Scheme 4) would be followed by protonation of the hydrido ligand and enyne metathesis to yield the seven-membered ruthenacycle B. Subsequent 1,2-migration of the phenyl group to the electrophilic carbene atom [30] would generate C. Rearrangement of C leads to ring contraction to afford the ruthenabenzene D, which would undergo reductive elimination. Ring closure of the resultant ruthenocene E gives rise to the formation of the hemiketal 4 as the final product.
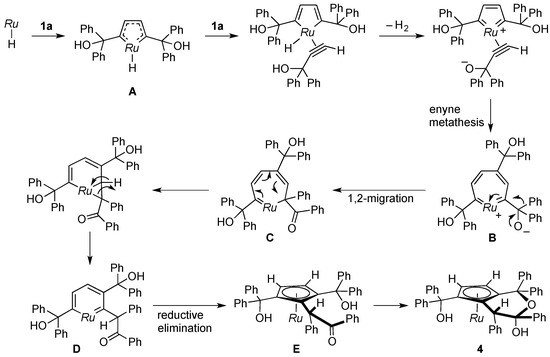
Scheme 6.
Proposed mechanism for formation of 4. Ru = Cp*Ru.
3. Experimental
3.1. General
All manipulations were performed under an atmosphere of argon using standard Schlenk techniques unless otherwise specified. Solvents were dried by refluxing over sodium benzophenone ketyl (THF, diethyl ether, diglyme, toluene, and hexane), P2O5 (dichloromethane and acetonitrile), and Mg(OMe)2 (methanol), and distilled before use. Formic acid was dried over boric oxide and distilled. The ruthenium complexes [Cp*RuCl(diene)] [31] and [Cp*RuH(cod)] [32] were prepared according to the literature. 1H (399.8 MHz) and 13C{1H} (100.53 MHz) NMR spectra were obtained on a JEOL JNM-ECX-400 spectrometer (JEOL Ltd., Tokyo, Japan). 1H NMR shifts are relative to the residual CHCl3 (δ 7.26), while 13C shifts are referenced to CDCl3 (δ 77.0). Elemental analyses were performed on a Perkin–Elmer 2400II CHN analyzer (PerkinElmer, Waltham, MA, USA). ESI-MS spectra were obtained on a JEOL JMS-T100LC spectrometer (JEOL Ltd., Tokyo, Japan) with a positive ionization mode.
3.2. Catalytic Dimerization of 1,1-Diarylprop-2-yn-1-ols to Give 2
To a solution of [Cp*RuCl(isoprene)] (10.2 mg, 0.030 mmol) in THF (6 mL) was added 1,1-diphenylprop-2-yn-1-ol (1a, 312.0 mg, 1.5 mmol), and the mixture was stirred for 2.5 h at room temperature. After removal of the solvent in vacuo, the residue was chromatographed on a column of silica. Elution with hexane/ethyl acetate (9:1 v/v) afforded 2a (302.6 mg, 97%). Single crystals suitable for X-ray analysis were obtained by recrystallization from acetone–pentane. 1H NMR (CDCl3) δ: 2.36, 2.87 (s, 1H each, OH), 6.27 (d, 3JHH = 15.2 Hz, 1H, CH=C(OH)Ph2), 6.32 (d, 3JHH = 11.3 Hz, 1H, CH=C(cyclobutene)), 6.45 (dd, 3JHH = 15.2, 11.3 Hz, 1H, CH–CH=CH), 7.20–7.35 (m, 19H, aryl). 13C{1H} NMR (CDCl3) δ: 79.1 (C(OH)Ph2), 86.2 (C(OH)Ph), 116.2 (C=C(cyclobutene)), 119.3, 121.3, 125.4, 125.6, 126.8 (CH=CHC(OH)Ph2), 126.9, 127.0 (m), 127.9, 128.1, 129.5, 130.0, 131.9, 139.3 (CHC(OH)Ph2), 141.9, 144.8 (CH=C(cyclobutene)), 145.8, 145.9, 149.8, 153.4. Anal. Calcd. for C30H24O2·0.5acetone: C, 84.91; H, 6.11. Found: C, 84.66; H, 6.00.
Data for 2b: Yield 84%. 1H NMR (CDCl3) δ: 2.60, 3.21 (s, 1H each, OH), 3.74 (m, 9H, OMe), 3.77 (s, 3H, OMe), 6.18 (d, 3JHH = 15.2 Hz, 1H, vinyl), 6.20 (d, 3JHH = 11.3 Hz, 1H, vinyl), 6.42 (dd, 3JHH = 15.2, 11.3 Hz, 1H, CH–CH=CH), 6.72–6.78 (m, 8H, aryl), 7.07–7.22 (m, 7H, aryl). ESI-MS (m/z): calcd. for C34H32O6 + Na+, 559.2096; found, 559.2091.
3.3. Synthesis of 4
A mixture of [Cp*RuH(cod)] (52.0 mg, 0.151 mmol) and 1,1-diphenylprop-2-yn-1-ol (1a, 94.0 mg, 0.451 mmol) in toluene (5 mL) was heated at 50 °C for 17 h. After removal of the solvent in vacuo, the resultant solid was recrystallized from THF–diethyl ether to give 4 as yellow crystals (70.9 mg, 0.0824 mmol, 55%). 1H NMR (CDCl3, δ): 1.70 (s, 15H, C5Me5), 2.01, 2.11 (s, 1H each, OH), 3.31 (s, 1H, CH), 3.56, 4.24 (d, 1H each, 3JHH = 2.5 Hz, C5H), 4.91 (d, 1H, JHH = 7.6 Hz, aryl), 6.35 (m, 1H, aryl), 6.91–7.04 (m, 11H, aryl), 7.12–7.43 (m, 13H, aryl), 7.57 (m, 2H, aryl), 8.21 (d, 1H, JHH = 7.9 Hz, aryl). Anal. Calcd. for C55H50O3Ru: C, 76.81; H, 5.86. Found: C, 76.53; H, 5.84.
3.4. Crystallography
Single crystals suitable for X-ray analyses were mounted on a fiber loop. Diffraction experiments were performed on a Rigaku Saturn CCD area detector with graphite monochromated Mo Kα radiation (λ = 0.710 73Å). Intensity data were corrected for Lorentz–polarization effects and for absorption. Structure solution and refinements were carried out by using the Crystal Structure program package [33]. The heavy-atom positions were determined by a direct methods program (SIR92 [34]) and the remaining non-hydrogen atoms were found by subsequent Fourier syntheses and refined by full-matrix least-squares techniques against F2 using the SHELXL-2014/7 program [35]. The hydrogen atoms were included in the refinements with a riding model. The hydroxy hydrogen atoms in 2a·0.5acetone were placed at two disordered positions linked to hydrogen acceptors (carbonyl and hydroxy groups) with 50% occupancies. Details of crystallographic data are summarized in Supplementary Materials.
Crystal Data for 2a·0.5acetone (Supplementary Materials) : C31.5H27O2.5 (M = 445.56 g/mol), triclinic, space group P (no. 2), a = 10.337(6) Å, b = 10.816(6) Å, c = 12.119(7) Å, α = 72.216(16)°, β = 84.13(2)°, γ = 69.554(17)°, V = 1209.0(12) Å3, Z = 2, T = 93 K, μ(Mo Kα) = 0.076 mm−1, Dcalc. = 1.224 g/cm3, 9706 reflections measured (6.2° ≤ 2Θ ≤ 55.0°), 5322 unique (Rint = 0.0336) which were used in all calculations. The final R1 was 0.0510 (I > 2σ(I)) and wR2 was 0.1477 (all data).
Crystal Data for 4: C55H50O3Ru (M = 860.07 g/mol), monoclinic, space group P21/c (no. 14), a = 17.089(4) Å, b = 12.512(3) Å, c = 20.001(4) Å, β = 107.378(2)°, V = 4081.5(15) Å3, Z = 4, T = 123 K, μ(Mo Kα) = 0.431 mm−1, Dcalc. = 1.400 g/cm3, 31910 reflections measured (6.4° ≤ 2Θ ≤ 55.0°), 9326 unique (Rint = 0.0300) which were used in all calculations. The final R1 was 0.0332 (I > 2σ(I)) and wR2 was 0.0795 (all data).
4. Conclusions
We found that the ruthenium(II) complex [Cp*RuCl(diene)] catalyzed a novel mode of dimerization of 1,1-diarylpropargyl alcohols 1 to afford the highly functionalized cyclobutenes 2, which has been unambiguously characterized by NMR spectroscopy and X-ray crystallography. The proposed mechanism for the catalytic formation of 2 involves the ordinary five-membered ruthenacycle as a primary intermediate. However, the presence of the aryl substituents in 1 led to unexpected aromatic C–H bond cleavage to afford the benzocyclobutenes 2 with high efficiency and selectivity. Formation of the five-membered ruthenacycle intermediate before the C–H bond cleavage was supported by the reaction of the related hydrido complex [Cp*RuH(cod)]. The dehydrogenative cyclization of three molecules of 1a therein took place without the C–H bond cleavage to generate the fused cyclopentadienyl ring in the product 4. These results added new aspects to the coordination chemistry and catalysis of the Cp*Ru complexes [8,9,12,36,37].
Supplementary Materials
The following are available online at www.mdpi.com/2304-6740/5/4/80/s1. Cif and cif-checked files. CCDC-1582525 (2a·0.5acetone) and CCDC-1582526 (4) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html.
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research (S) (no. 22225004) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Author Contributions
Hoang Ngan Nguyen and Naoto Tashima performed the experiments; Takao Ikariya supervised the research; Shigeki Kuwata initiated and coordinated the research and wrote the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wang, L.-X.; Tang, Y.-L. Cycloisomerization of Pyridine-Substituted Propargylic Alcohols or Esters to Construct Indolizines and Indolizinones. Eur. J. Org. Chem. 2017, 2207–2213. [Google Scholar] [CrossRef]
- Rintjema, J.; Kleij, A. Substrate-Assisted Carbon Dioxide Activation as a Versatile Approach for Heterocyclic Synthesis. Synthesis 2016, 48, 3863–3878. [Google Scholar]
- Ayers, B.; Chan, P. Harnessing the Versatile Reactivity of Propargyl Alcohols and Their Derivatives for Sustainable Complex Molecule Synthesis. Synlett 2015, 26, 1305–1339. [Google Scholar] [CrossRef]
- Hu, X.-H.; Liu, Z.-T.; Shao, L.; Hu, X.-P. Recent Advances in Catalytic Stereocontrolled Cycloaddition with Terminal Propargylic Compounds. Synthesis 2015, 47, 913–923. [Google Scholar] [CrossRef]
- Zhu, Y.; Sun, L.; Lu, P.; Wang, Y. Recent Advances on the Lewis Acid-Catalyzed Cascade Rearrangements of Propargylic Alcohols and Their Derivatives. ACS Catal. 2014, 4, 1911–1925. [Google Scholar] [CrossRef]
- Bauer, E. Transition-Metal-Catalyzed Functionalization of Propargylic Alcohols and Their Derivatives. Synthesis 2012, 44, 1131–1151. [Google Scholar] [CrossRef]
- Ding, C.-H.; Hou, X.-L. Catalytic Asymmetric Propargylation. Chem. Rev. 2011, 111, 1914–1937. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Itoh, K. Carbon–Carbon Bond Formations via Ruthenacycle Intermediates. In Ruthenium in Organic Synthesis; Murahashi, S.-I., Ed.; Wiley-VCH: Weinheim, Germany, 2004; pp. 95–128. ISBN 3-527-30692-7. [Google Scholar]
- Derien, S.; Dixneuf, P.H. The Versatility of Molecular Ruthenium Catalyst RuCl(COD)(C5Me5). J. Organomet. Chem. 2004, 689, 1382–1392. [Google Scholar] [CrossRef]
- Nishibayashi, Y.; Wakiji, I.; Hidai, M. Novel Propargylic Substitution Reactions Catalyzed by Thiolate-Bridged Diruthenium Complexes via Allenylidene Intermediates. J. Am. Chem. Soc. 2000, 122, 11019–11020. [Google Scholar] [CrossRef]
- Hidai, M.; Mizobe, Y. Research Inspired by the Chemistry of Nitrogenase—Novel Metal Complexes and Their Reactivity Toward Dinitrogen, Nitriles, and Alkynes. Can. J. Chem. 2005, 83, 358–374. [Google Scholar] [CrossRef]
- Nishibayashi, Y. Transition-Metal-Catalyzed Enantioselective Propargylic Substitution Reactions of Propargylic Alcohol Derivatives with Nucleophiles. Synthesis 2012, 44, 489–503. [Google Scholar] [CrossRef]
- Roşca, D.-A.; Radkowski, K.; Wolf, L.M.; Wagh, M.; Goddard, R.; Thiel, W.; Fürstner, A. Ruthenium-Catalyzed Alkyne trans-Hydrometalation: Mechanistic Insights and Preparative Implications. J. Am. Chem. Soc. 2017, 139, 2443–2455. [Google Scholar] [CrossRef] [PubMed]
- Rummelt, S.M.; Radkowski, K.; Roşca, D.-A.; Fürstner, A. Interligand Interactions Dictate the Regioselectivity of trans-Hydrometalations and Related Reactions Catalyzed by [Cp*RuCl]. Hydrogen Bonding to a Chloride Ligand as a Steering Principle in Catalysis. J. Am. Chem. Soc. 2015, 137, 5506–5519. [Google Scholar] [CrossRef] [PubMed]
- Rummelt, S.M.; Cheng, G.-J.; Gupta, P.; Thiel, W.; Fürstner, A. Hydroxy-Directed Ruthenium-Catalyzed Alkene/Alkyne Coupling: Increased Scope, Stereochemical Implications, and Mechanistic Rationale. Angew. Chem. Int. Ed. 2017, 56, 3599–3604. [Google Scholar] [CrossRef] [PubMed]
- Hase, S.; Kayaki, Y.; Ikariya, T. Mechanistic Aspects of the Carboxylative Cyclization of Propargylamines and Carbon Dioxide Catalyzed by Gold(I) Complexes Bearing an N-Heterocyclic Carbene Ligand. ACS Catal. 2015, 5, 5135–5140. [Google Scholar] [CrossRef]
- Kayaki, Y.; Yamamoto, M.; Ikariya, T. N-Heterocyclic Carbenes as Efficient Organocatalysts for CO2 Fixation Reactions. Angew. Chem. Int. Ed. 2009, 48, 4194–4197. [Google Scholar] [CrossRef] [PubMed]
- Kayaki, Y.; Koda, T.; Ikariya, T. Halide-Free Dehydrative Allylation Using Allylic Alcohols Promoted by a Palladium–Triphenyl Phosphite Catalyst. J. Org. Chem. 2004, 69, 2595–2597. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Kuwata, S.; Ikariya, T. Isolation and Interconversion of Protic N-Heterocyclic Carbene and Imidazolyl Complexes: Application to Catalytic Dehydrative Condensation of N-(2-Pyridyl)benzimidazole and Allyl Alcohol. Organometallics 2008, 27, 2176–2178. [Google Scholar] [CrossRef]
- Cadierno, V.; Gimeno, J. Allenylidene and Higher Cumulenylidene Complexes. Chem. Rev. 2009, 109, 3512–3560. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, H.J.; Singer, H. Die Dimerisierung von Endständigen α-Hydroxyacetylenen mit Rhodiumkomplexkatalysatoren. J. Organomet. Chem. 1978, 153, 165–179. [Google Scholar] [CrossRef]
- Katayama, H.; Ozawa, F. Vinylideneruthenium Complexes in Catalysis. Coord. Chem. Rev. 2004, 248, 1703–1715. [Google Scholar] [CrossRef]
- Yi, C.S.; Liu, N. Ruthenium-Catalyzed Coupling Reactions of Alkynes and Alkenes. Synlett 1999, 281–287. [Google Scholar] [CrossRef]
- Crochet, P.; Esteruelas, M.; Gutierrez-Puebla, E. Unusual Activation of 1,1-Diphenyl-2-propyn-1-ol Mediated by the Os(η5-C5H5) Unit. Organometallics 1998, 17, 3141–3142. [Google Scholar] [CrossRef]
- Dutta, B.; Curchod, B.F.E.; Campomanes, P.; Solari, E.; Scopelliti, R.; Rothlisberger, U.; Severin, K. Reactions of Alkynes with [RuCl(cyclopentadienyl)] Complexes: The Important First Steps. Chem. Eur. J. 2010, 16, 8400–8409. [Google Scholar] [CrossRef] [PubMed]
- Le Paih, J.; Derien, S.; Bruneau, C.; Demerseman, B.; Toupet, L.; Dixneuf, P.H. Ruthenium-Catalyzed One-Step Transformation of Propargylic Alcohols Into Alkylidene Cyclobutenes: X-ray Characterization of an Ru(η3-cyclobutenyl) Intermediate. Angew. Chem. Int. Ed. 2001, 40, 2912–2915. [Google Scholar] [CrossRef]
- Trost, B.M.; Rudd, M.T. An Unusual Ruthenium-Catalyzed Dimerization of Propargyl Alcohols. J. Am. Chem. Soc. 2001, 123, 8862–8863. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Rudd, M.T. An Unusual Ruthenium-Catalyzed Cycloisomerization of Alkynes and Propargyl Alcohols. J. Am. Chem. Soc. 2002, 124, 4178–4179. [Google Scholar] [CrossRef] [PubMed]
- Rao, W.; Chan, P.W.H. Unexpected Iron(III) Chloride-Catalysed Dimerisation of 1,1,3-Trisubstituted-prop-2-yn-1-ols as an Expedient Route to Highly Conjugated Indenes. Org. Biomol. Chem. 2010, 8, 4016–4025. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhang, C.; Lin, Y.; He, X.; Zhang, Y.; Wang, J.; Xia, H. 1,2-Migration in the Reactions of Ruthenium Vinyl Carbene with Propargyl Alcohols. Org. Chem. Front. 2014, 1, 1077–1082. [Google Scholar] [CrossRef]
- Fagan, P.J.; Mahoney, W.S.; Calabrese, J.C.; Williams, I.D. Structure and Chemistry of the Complex Tetrakis(η5-pentamethylcyclopentadienyl)tetrakis(μ3-chloro)tetraruthenium(II): A Useful Precursor to (Pentamethylcyclopentadienyl)ruthenium(0), -(II), and -(IV) Complexes. Organometallics 1990, 9, 1843–1852. [Google Scholar] [CrossRef]
- Radkowski, K.; Sundararaju, B.; Fürstner, A. A Functional-Group-Tolerant Catalytic trans Hydrogenation of Alkynes. Angew. Chem. Int. Ed. 2012, 52, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Crystal Structure 4.1: Crystal Structure Analysis Package; Rigaku Corporation: Tokyo, Japan, 2000–2015.
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Burla, M.C.; Polidori, G.; Camallli, M. SIR92—A Program for Automatic Solution of Crystal Structures by Direct Methods. J. Appl. Crystallogr. 1994, 27, 435. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Serron, S.A.; Luo, L.; Stevens, E.D.; Nolan, S.P.; Jones, N.L.; Fagan, P.J. Organoruthenium Thermochemistry. Enthalpies of Reaction of Cp’Ru(COD)Cl (Cp’ = η5-C5H5 and η5-C5Me5) with Tertiary Phosphite Ligands. Organometallics 1996, 15, 5209–5215. [Google Scholar] [CrossRef]
- Bosson, J.; Poater, A.; Cavallo, L.; Nolan, S.P. Mechanism of Racemization of Chiral Alcohols Mediated by 16-Electron Ruthenium Complexes. J. Am. Chem. Soc. 2010, 132, 13146–13149. [Google Scholar] [CrossRef] [PubMed]
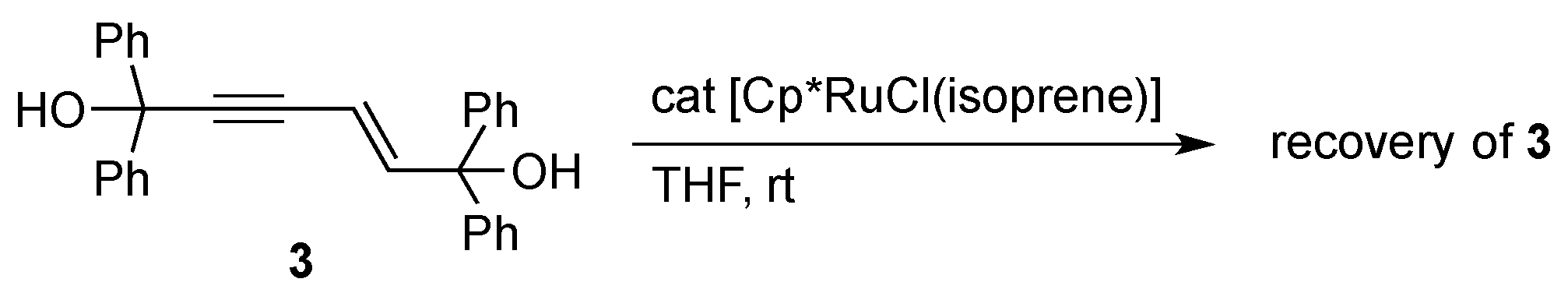
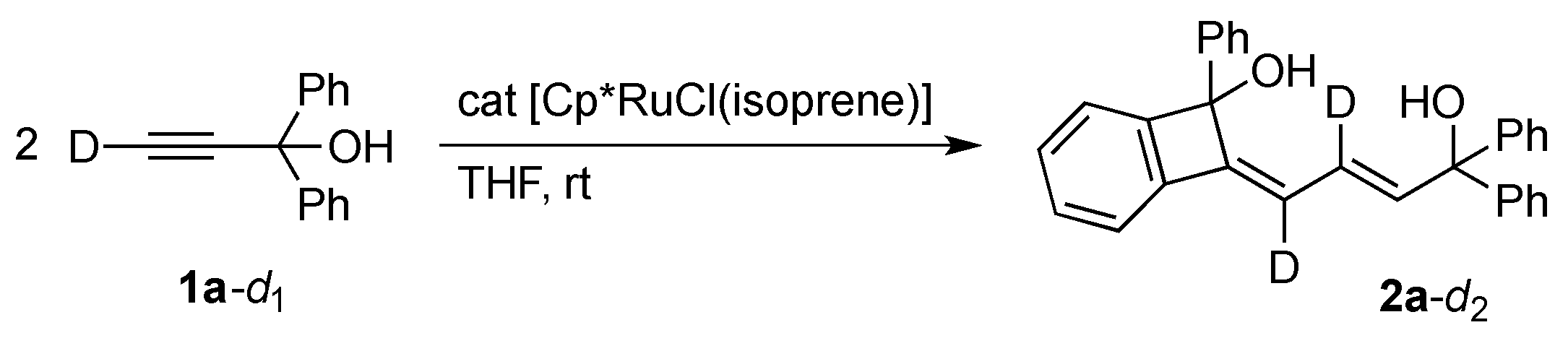
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).