


Hypercoordinating Stannanes with C,N-Donor Ligands: A Structural, Computational, and Polymerization Study

 , ,
, ,
Abstract

1. Introduction
2. Results
2.1. Triphenylstannanes
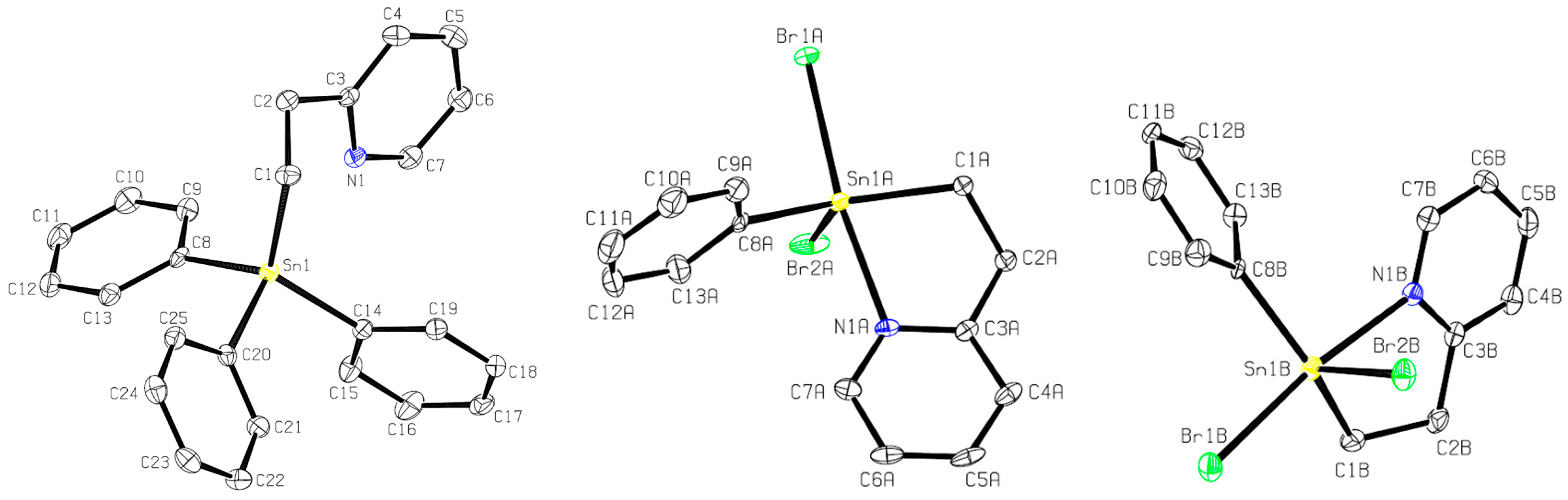
2.2. Dihalo- and Dihydrido-Pyridyl Stannanes

{kind=link}
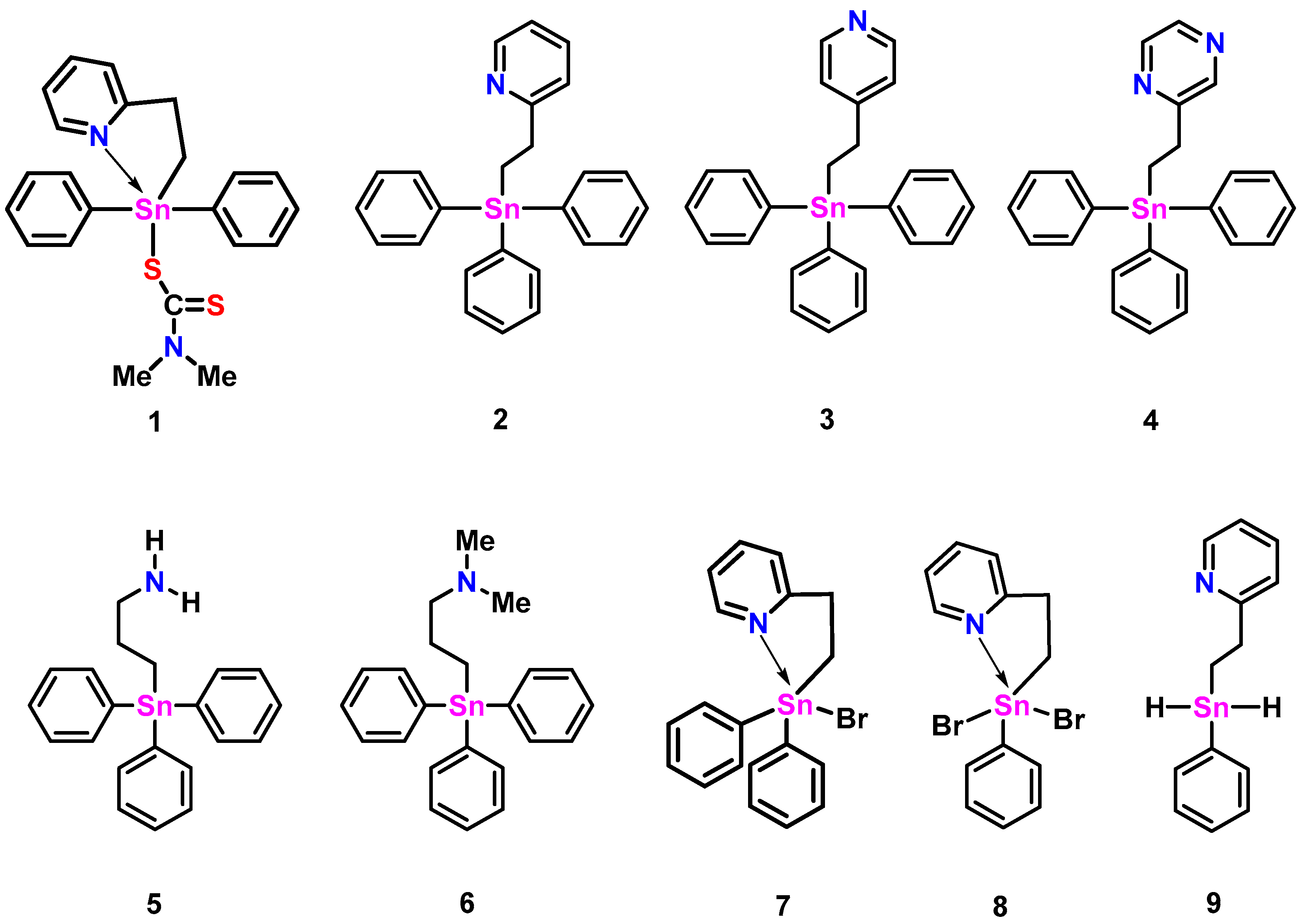
{kind=link}
{kind=link}
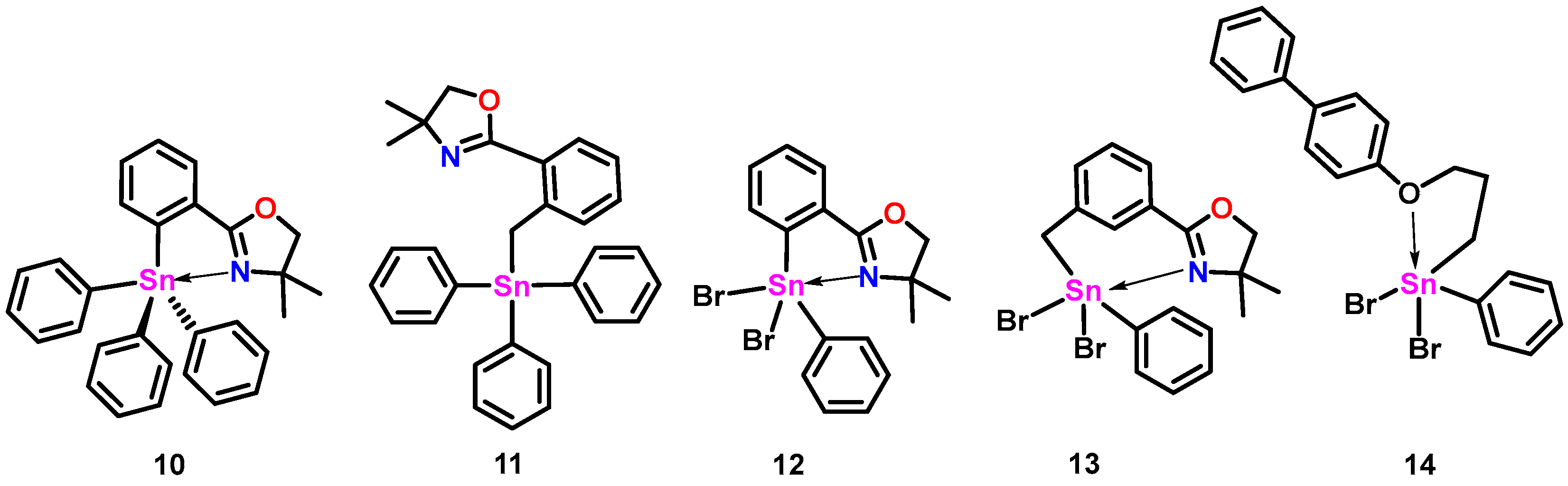{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
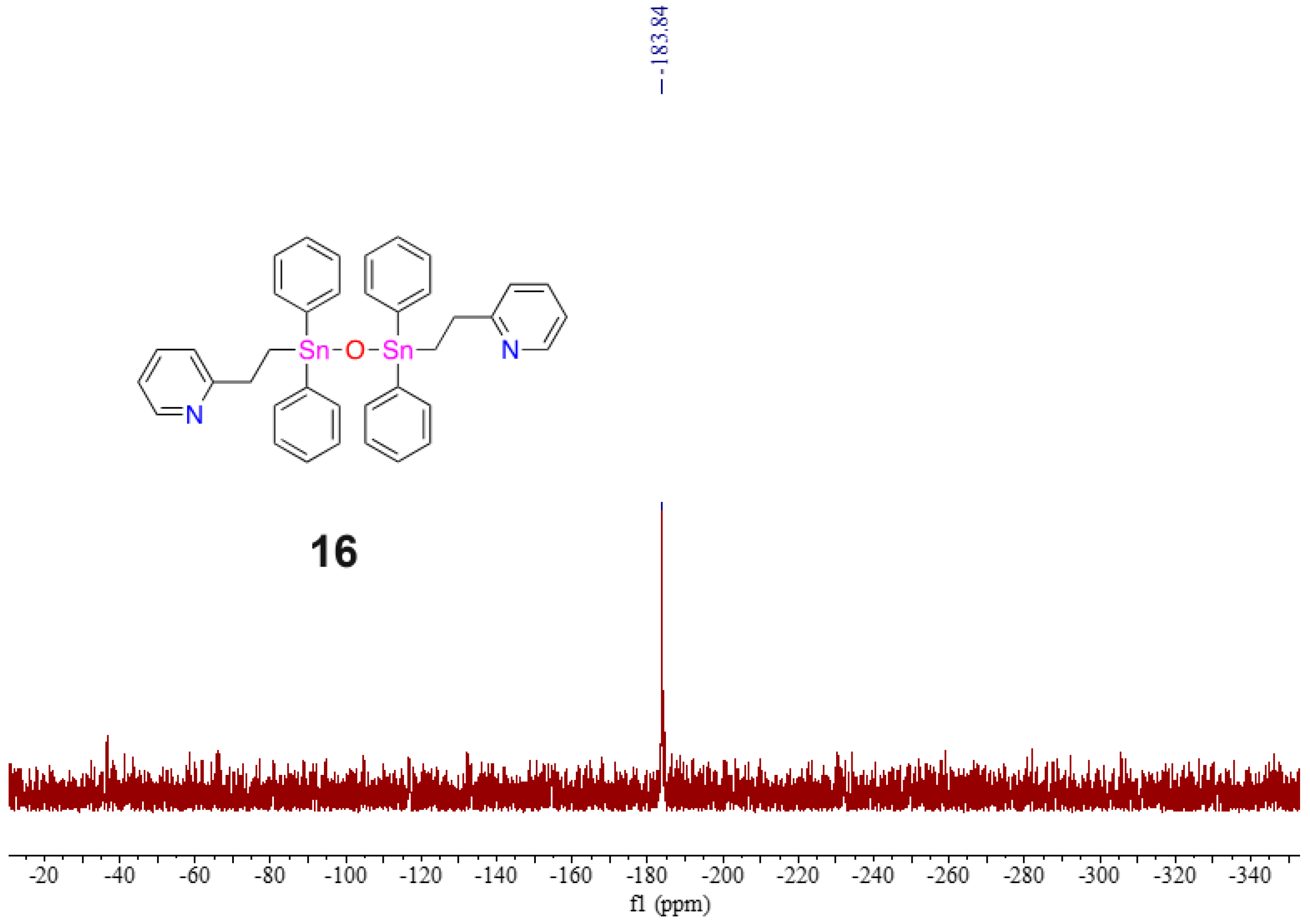{kind=link}
{kind=link}
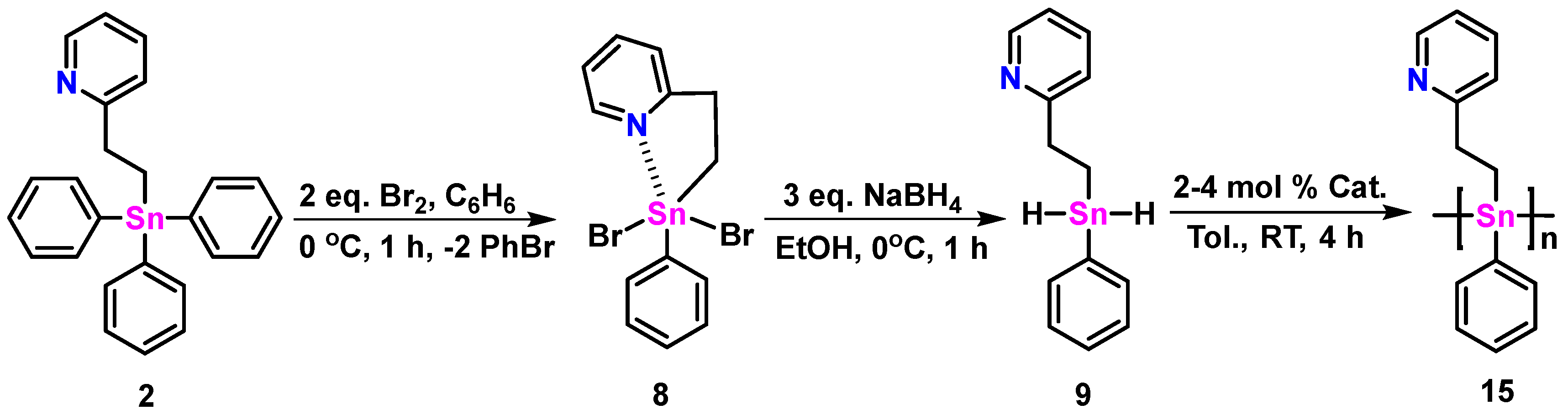{kind=link}
| 119Sn δ ppm (CDCl3) a, (C6D6) b | Calc. Sn-N or Sn-O Distance (Å) c | Expt. Sn-N or Sn-O Distance (Å) | |
|---|---|---|---|
| 1 | −202.0 a | 2.56 | 2.486 (7) [12] |
| 2 | −108.3 a | 2.94 (2.74 d) | 2.888 (2) |
| 3 | −101.2 a | 5.39 | - |
| 4 | −104.3 b | 3.34 | - |
| 5 | −100.8 a | 2.88 | 2.740 (11) [13] |
| 6 | −102.7 a | 3.04 | - |
| 7 | −143.5 a [12] | 2.57 (2.52 d) | - |
| 8 | −183.9 a | 2.50 (2.43 d) | 2.382 (5), 2.363 (5) |
| 9 | −221.6 b | 2.87 | |
| 10 | −157.1 a | 2.73 d | 2.762 (1) [16] |
| 11 | −126.0 a | 2.96 d | 3.176 (4), 3.234 (1) [16] |
| 12 | −290.6 a | 2.41 d | 2.383 (3) [16] |
| 13 | −248.2 a | 2.49 d | 2.424 (2) [16] |
| 14 | −53.3 a | - | 2.918 (7) [14] |
2.3. Computational Studies
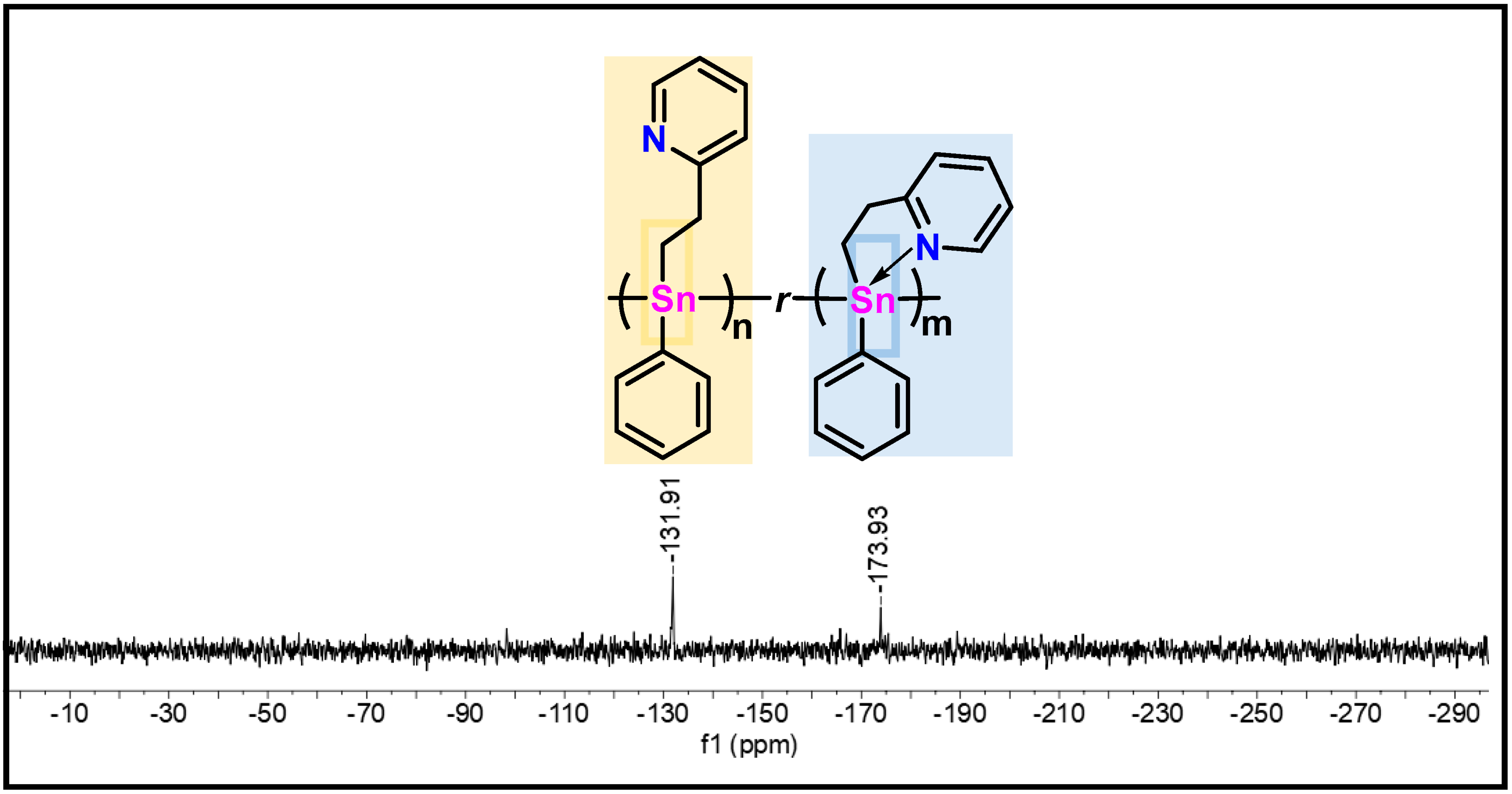
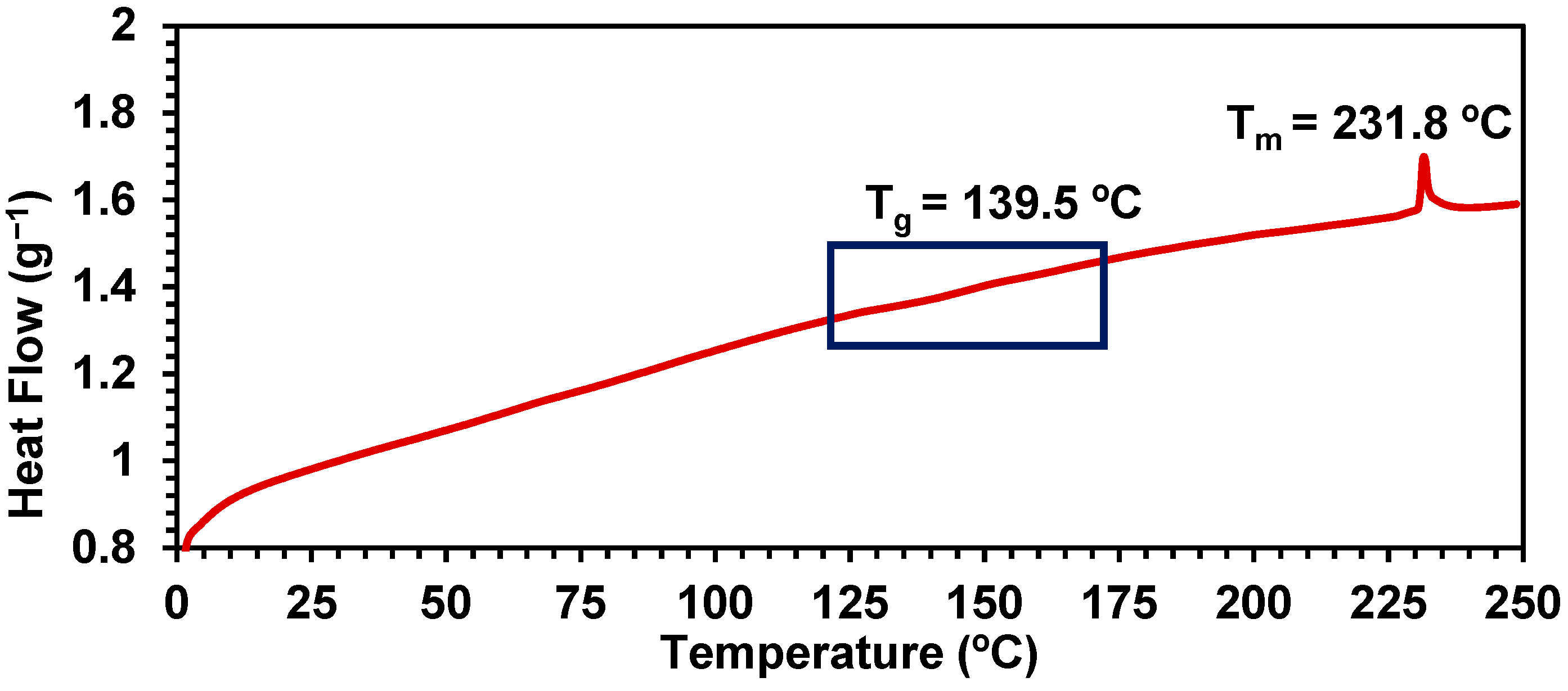
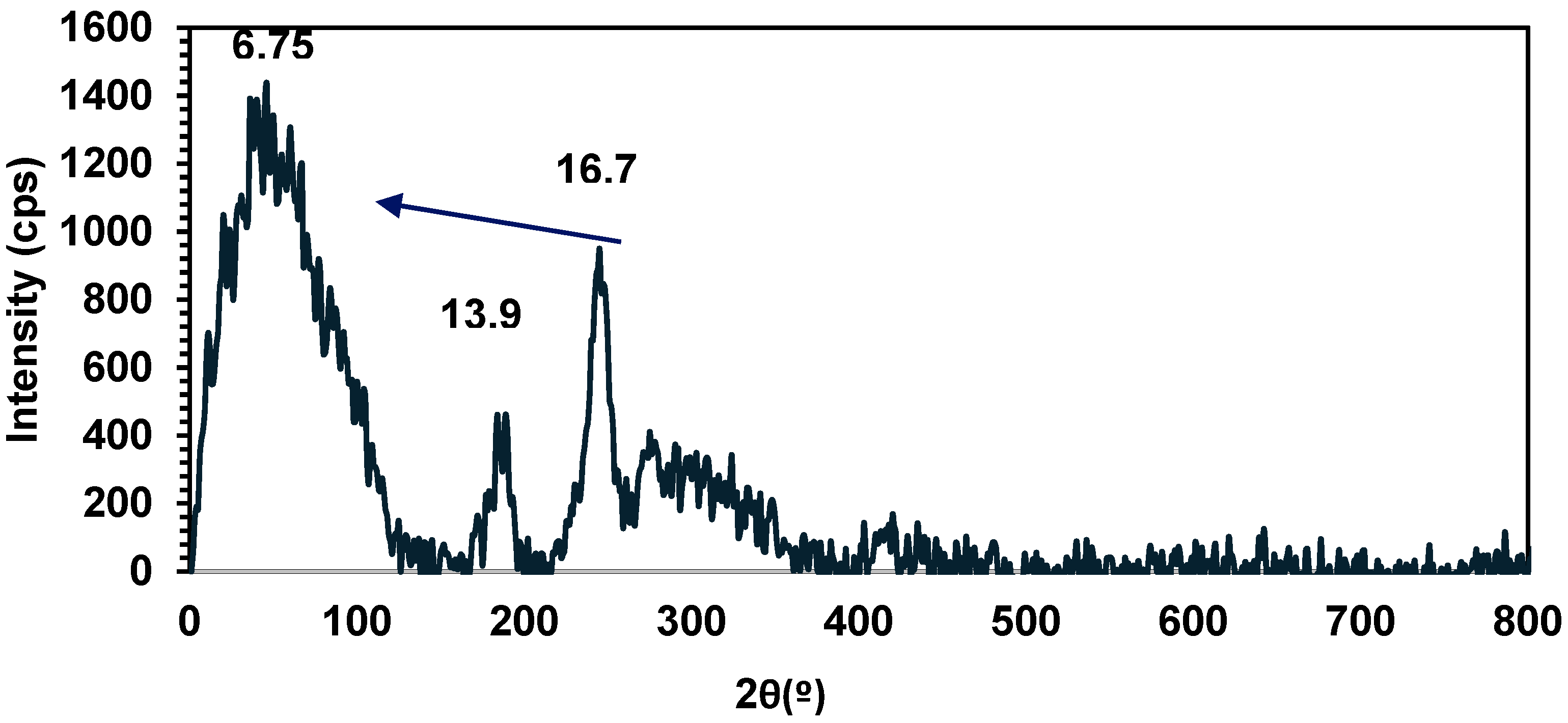
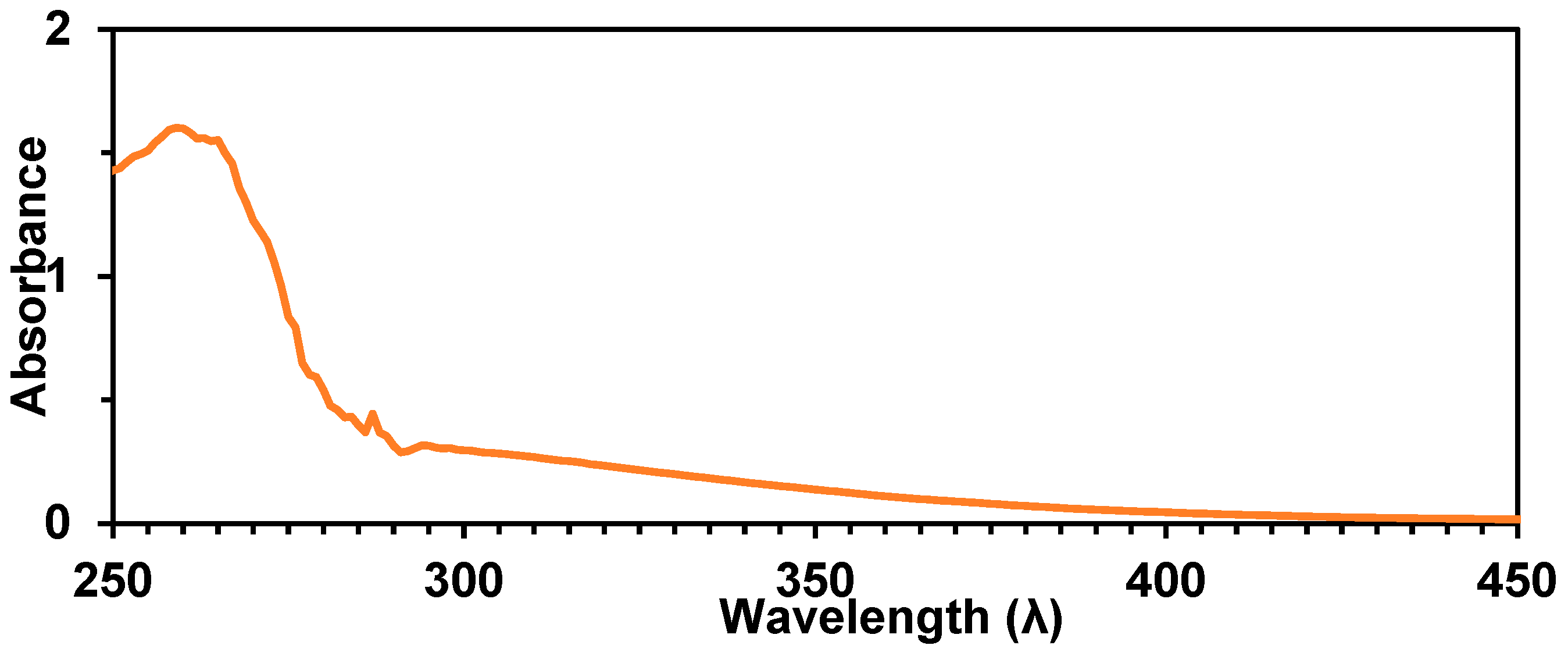
2.4. Synthesis of Polymer 15, Characterization, and Stability
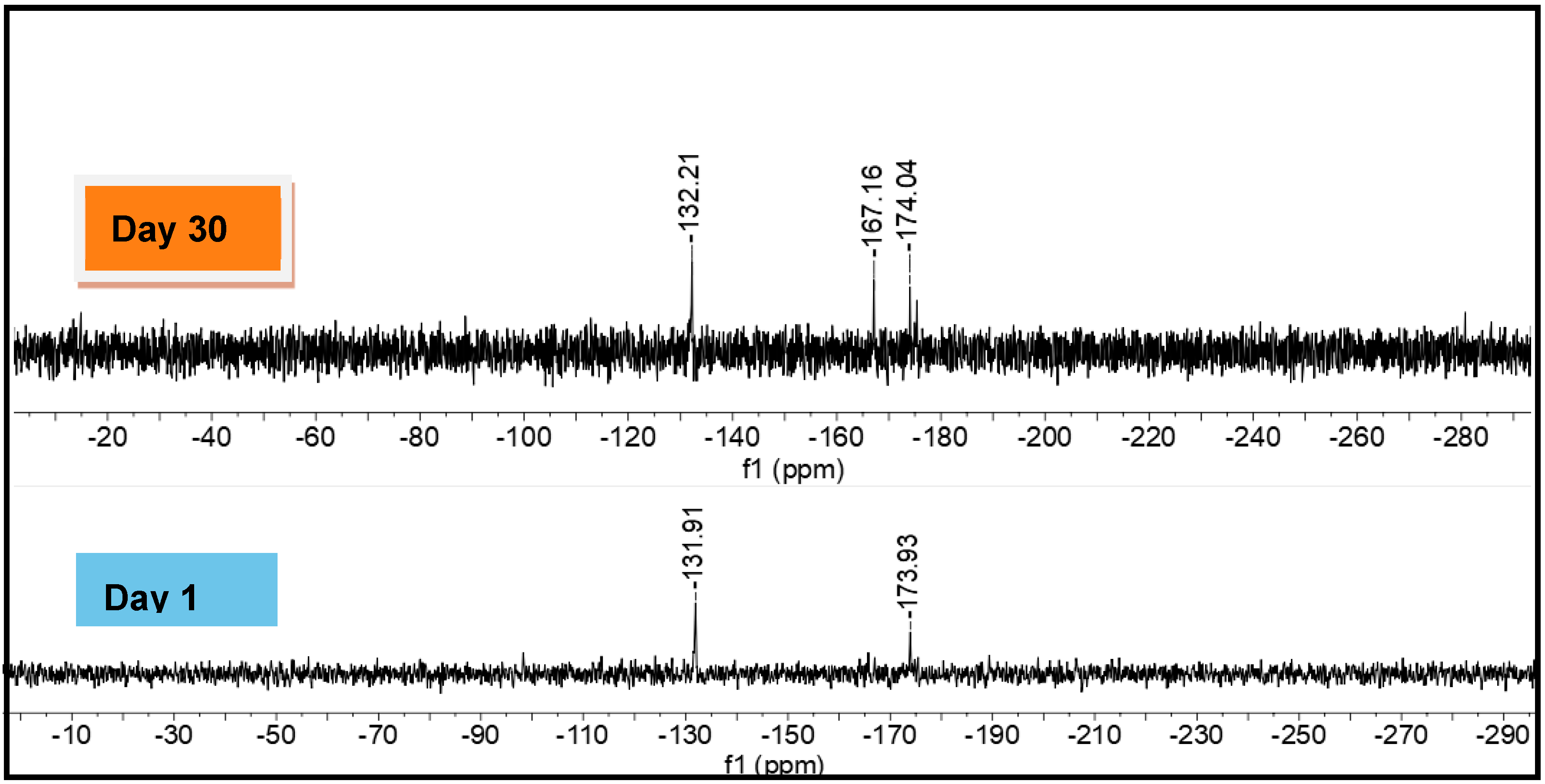
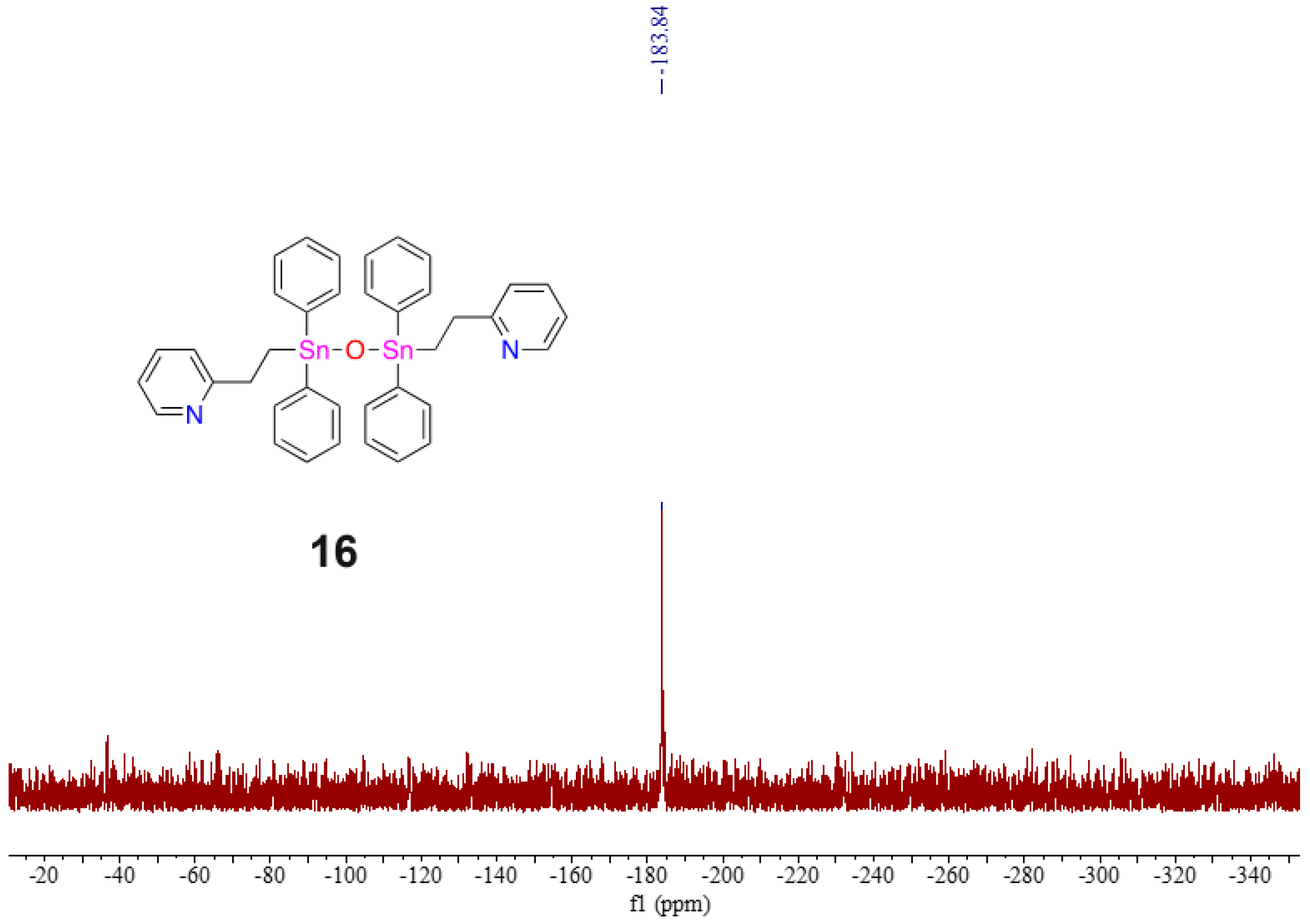
2.5. Evidence of Redistribution and Distannoxane Formation from 15
3. Materials and Methods
3.1. General Considerations
3.2. Synthesis of 2-(pyC2H4)SnPh3 (2)
3.3. Synthesis of 4-(pyC2H4)SnPh3 (3)
3.4. Synthesis of 2-(pzC2H4)SnPh3 (4)
3.5. Synthesis of Me2N(CH2)3SnPh3 (6)
3.6. Synthesis of 2-(pyC2H4)SnPhBr2 (8)
3.7. Synthesis of 2-(pyC2H4)SnPhH2 (9)
3.8. Synthesis of Polymer 15 Prepared with 2 mol% Wilkinson Catalyst
3.9. Synthesis of Polymer 15 Prepared with 4 mol% Wilkinson Catalyst
3.10. Isolation of Pyridyl Distannoxane 16
3.11. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Norman, N.C.; Pringle, P.G. Hypervalence: A Useful Concept or One That Should Be Gracefully Retired? Chemistry 2022, 4, 1226–1249. [Google Scholar] [CrossRef]
- Musher, J.I. The Chemistry of Hypervalent Molecules. Angew. Chem. Int. Ed. 1969, 8, 54–68. [Google Scholar] [CrossRef]
- Pimentel, G.C. The Bonding of Trihalide and Bifluoride Ions by the Molecular Orbital Method. J. Chem. Phys. 1951, 19, 446–448. [Google Scholar] [CrossRef]
- Hach, R.J.; Rundle, R.E. The Structure of Tetramethylammonium Pentaiodide. J. Am. Chem. Soc. 1951, 73, 4321–4324. [Google Scholar] [CrossRef]
- Reed, A.E.; Schleyer, P.V.R. Chemical Bonding in Hypervalent Molecules. The Dominance of Ionic Bonding and Negative Hyperconjugation over d-Orbital Participation. J. Am. Chem. Soc. 1990, 112, 1434–1445. [Google Scholar] [CrossRef]
- Kutzelnigg, W. Chemical Bonding in Higher Main Group Elements. Angew. Chem. Int. Ed. 1984, 23, 272–295. [Google Scholar] [CrossRef]
- Green, M.L.H.; Parkin, G. The classification and representation of main group element compounds that feature three-center four-electron interactions. Dalton Trans. 2016, 45, 18784–18795. [Google Scholar] [CrossRef]
- Zickgraf, A.; Beuter, M.; Kolb, U.; Dräger, M.; Tozer, R.; Dakternieks, D.; Jurkschat, K. Nucleophilic Attack within Ge, Sn and Pb Complexes Containing Me2N(CH2)3- As a Potential Intramolecular Donor Ligand. Inorg. Chim. Acta 1998, 275–276, 203–214. [Google Scholar] [CrossRef]
- Scheiner, S. Origins and properties of the tetrel bond. Phys. Chem. Chem. Phys. 2021, 23, 5702–5717. [Google Scholar] [CrossRef]
- Bauzá, A.; Seth, S.K.; Frontera, A. Tetrel bonding interactions at work: Impact on tin and lead coordination compounds. Coord. Chem. Rev. 2019, 384, 107–125. [Google Scholar] [CrossRef]
- Ullah, H.; Twamley, B.; Waseem, A.; Rauf, M.K.; Tahir, M.N.; Platts, J.A.; Baker, R.J. Tin⋯Oxygen Tetrel Bonding: A Combined Structural, Spectroscopic, and Computational Study. Cryst. Growth Des. 2017, 17, 4021–4027. [Google Scholar] [CrossRef]
- Mahon, M.F.; Molloy, K.C.; Waterfield, P.C. Synthesis, Characterization, and Reaction Chemistry of [2-(2-Pyridyl)Ethyl]-, [2-(4-Pyridyl)Ethyl]-, and [2-(2-Oxo-4-Pyrrolidinyl)Ethyl]Triphenyltin(IV). Organometallics 1993, 12, 769–774. [Google Scholar] [CrossRef]
- Pichler, J.; Torvisco, A.; Bottke, P.; Wilkening, M.; Uhlig, F. Novel Amino Propyl Substituted Organo Tin Compounds. Can. J. Chem. 2014, 92, 565–573. [Google Scholar] [CrossRef]
- Deacon, P.R.; Devylder, N.; Hill, M.S.; Mahon, M.F.; Molloy, K.C.; Price, G.J. Organotin Compounds Bearing Mesogenic Sidechains: Synthesis, X-Ray Structures and Polymerisation Chemistry. J. Organomet. Chem. 2003, 687, 46–56. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; van Rijn, J.; Verschoor, G.C. Synthesis, Structure, and Spectroscopic Properties of Copper(II) Compounds Containing Nitrogen-Sulphur Donor Ligands; the Crystal and Molecular Structure of Aqua[l,7-Bis(N-Methylbenzimidazol-2′-Yl)-2,6-Dithiaheptane]Copper(II) Perchlorate. J. Chem. Soc. Dalton Trans. 1984, 7, 1349–1356. [Google Scholar] [CrossRef]
- Bender, D.N.; Lough, A.J.; Wylie, R.S.; Gossage, R.A.; Foucher, D.A. Preparation and DFT Studies of κ2C,N-Hypercoordinated Oxazoline Organotins: Monomer Constructs for Stable Polystannanes. Inorganics 2020, 8, 35. [Google Scholar] [CrossRef]
- Komsta, Z.; Cmoch, P.; Stalinski, K. Intramolecularly Sn-N Coordinated Organotin Hydrides. Pol. J. Chem. 2006, 80, 1259–1292. [Google Scholar] [CrossRef]
- Matkowska, D.; Gola, M.; Śniezek, M.; Cmoch, P.; Staliński, K. Structural Assignment of Organotin Hydrides Containing the Oxazoline Ligand. J. Organomet. Chem. 2007, 692, 2036–2045. [Google Scholar] [CrossRef]
- Pau, J.; D’Amaral, G.M.; Lough, A.J.; Wylie, R.S.; Foucher, D.A. Synthesis and Characterization of Readily Modified Poly(Aryl)(Alkoxy)Stannanes by Use of Hypercoordinated Sn Monomers. Chem. Eur. J. 2018, 24, 18762–18771. [Google Scholar] [CrossRef]
- Khan, A.; Pau, J.; Loungxay, J.; Magobenny, T.; Wylie, R.S.; Lough, A.J.; Foucher, D.J. Hypercoordinated organotin(IV) compounds containing C,O- and C,N- chelating ligands: Synthesis, characterisation, DFT studies and polymerization behaviour. Organomet. Chem. 2019, 900, 120910. [Google Scholar] [CrossRef]
- Pau, J.; Choi, J.-W.; Silverthorne, K.; Ranne, M.; Wylie, R.S.; Gossage, R.A.; Lough, A.J.; Foucher, D.A. New Hypercoordinating Organostannanes for the Modular Functionalization of Mono- and Polystannanes: Synthetic and Computational Studies. Eur. J. Inorg. Chem. 2022, 2022, e202100937. [Google Scholar] [CrossRef]
- Luque, F.J.; Zhang, Y.; Alemán, C.; Bachs, M.; Gao, J.; Orozco, M. Solvent Effects in Chloroform Solution: Parametrization of the MST/SCRF Continuum Model. J. Phys. Chem. 1996, 100, 4269–4276. [Google Scholar] [CrossRef]
- Xu, L.; Coote, M.L. Improving the Accuracy of PCM-UAHF and PCM-UAKS Calculations Using Optimized Electrostatic Scaling Factors. J. Chem. Theory Comput. 2019, 15, 6958–6967, Correction in J. Chem. Theory Comput. 2020, 16, 816–817. [Google Scholar] [CrossRef] [PubMed]
- Bagno, A.; Casella, G.; Saielli, G. Relativistic DFT Calculation of 119Sn Chemical Shifts and Coupling Constants in Tin Compounds. J. Chem. Theory Comput. 2006, 2, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Sindlinger, C.P.; Aicher, F.S.W.; Weseman, L. Cationic Stannylenes: In Situ Generation and NMR Spectroscopic Characterization. Inorg. Chem. 2017, 56, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Stückrath, J.B.; Gasevic, T.; Bursch, M.; Grimme, S. Benchmark Study on the Calculation of 119Sn NMR Chemical Shifts. Inorg. Chem. 2022, 61, 3903–3917. [Google Scholar] [CrossRef] [PubMed]
- Pracht, P.; Bohle, F.; Grimme, S. Automated exploration of the low-energy chemical space with fast quantum chemical methods. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Hansen, A.; Ehlert, S.; Mewes, J.-M. r2SCAN-3c: A “Swiss army knife” composite electronic-structure method. J. Chem. Phys. 2021, 154, 064103. [Google Scholar] [CrossRef]
- Weinhold, F. Natural Bond Orbital Analysis: A Critical Overview of Relationships to Alternative Bonding Perspectives. J. Comput. Chem. 2012, 33, 2363–2379. [Google Scholar] [CrossRef]
- Pau, J.; Lough, A.J.; Wylie, R.S.; Gossage, R.A.; Foucher, D.A. Proof of Concept Studies Directed Towards Designed Molecular Wires: Property Driven Synthesis of Air and Moisture-Stable Polystannanes. Chem. Eur. J. 2017, 57, 14367–14374. [Google Scholar] [CrossRef]
- Trummer, M.; Choffat, F.; Smith, P.; Caseri, W. Polystannanes: Synthesis, Properties, and Outlook. Macromol. Rapid. Commun. 2012, 33, 448–460. [Google Scholar] [CrossRef]
- Khan, A.; Komejan, S.; Patel, A.; Lombardi, C.; Lough, A.J.; Foucher, D.A. Reduction of C,O-Chelated Organotin(IV) Dichlorides and Dihydrides Leading to Protected Polystannanes. J. Organomet. Chem. 2015, 776, 180–191. [Google Scholar] [CrossRef]
- Imori, T.; Lu, V.; Cai, H.; Tilley, T.D. Metal-Catalyzed Dehydropolymerization of Secondary Stannanes to High Molecular Weight Polystannanes. J. Am. Chem. Soc. 1995, 117, 9931–9940. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. [Google Scholar] [CrossRef]
- Gaussian Release 2016: Gaussian 16, Rev. C 01; Gaussian Inc.: Wallingford, CT, USA, 2016; Available online: https://gaussian.com/citation (accessed on 1 April 2024).
- Neese, F. Software update: The ORCA program system—Version 5.0. WIRES Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of Density Functionals by Combining the Method of Constraining Satisfaction with Parametrization for Thermochemistry. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef]
- Perdew, J.P. Unified Theory of Exchange and Correlation Beyond the Local Density Approximation. In Electronic Structure of Solids ’91; Ziesche, P., Eschig, H., Eds.; Akademie Verlag: Berlin, Germany, 1991; pp. 11–20. [Google Scholar]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Caldeweyher, E.; Bannwarth, C.; Grimme, S. Extension of the D3 dispersion coefficient model. J. Chem. Phys. 2017, 147, 034112. [Google Scholar] [CrossRef] [PubMed]
- Caldeweyher, E.; Ehlert, S.; Hansen, A.; Neugebauer, H.; Spicher, S.; Bannwarth, C.; Grimme, S. A generally applicable atomic-charge dependent London dispersion correction. J. Chem. Phys. 2019, 150, 154122. [Google Scholar] [CrossRef] [PubMed]
- Kruse, H.; Grimme, S. A geometrical correction for the inter- and intra-molecular basis set superposition error in Hartree-Fock and density functional theory calculations for large systems. J. Chem. Phys. 2012, 136, 154101. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Karafiloglou, P.; Landis, C.R.; Weinhold, F. NBO 7.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2018. [Google Scholar]




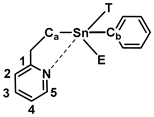 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Conformer | Gref a–Gconf/kJ mol−1 | Sn-N dist./Å | NBO N Lone Pair Donor Anti-Bond Acceptor Interaction Energy/kJ mol−1 | |||||
| Sn-T b | Sn-C a | Sn-C b | Sn-E c | C1-C2 | C4-C5 | |||
| 1-A | 28.2 | 2.56 | 79.1 | 28.7 | 19.3 | 13.8 | 41.1 | 35.0 |
| 2-A | 15.6 | 2.94 | 18.2 | 9.5 | 7.8 | 4.8 | 44.6 | 38.2 |
| 3-A | 7.0 | 5.39 | d | d | d | d | 41.2 | 41.8 |
| 4-A | 12.6 | 3.34 | 3.4 | d | d | d | 41.0 | 38.4 |
| 5-A | 10.3 | 2.88 | 22.9 | 11.5 | 9.9 | 8.5 | - | - |
| 6-A | 10.9 | 3.04 | 12.6 | 7.5 | 5.8 | 3.8 | - | - |
| 7-A | 37.9 | 2.58 | 80.1 | 26.7 | 19.5 | 13.7 | 41.2 | 35.7 |
| 8-A | 37.3 | 2.50 | 86.2 | 36.6 | 27.3 | 21.3 | 40.3 | 34.0 |
| 9-A | 11.2 | 3.15 | 5.9 | 2.5 | d | 2.3 | 46.4 | 40.0 |
| 9-B | 10.8 | 2.87 | 19.2 | 10.0 | 9.0 | 7.3 | 44.2 | 37.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Amaral, G.M.; Bender, D.N.; Piccolo, N.; Lough, A.J.; Gossage, R.A.; Foucher, D.A.; Wylie, R.S. Hypercoordinating Stannanes with C,N-Donor Ligands: A Structural, Computational, and Polymerization Study. Inorganics 2024, 12, 122. https://doi.org/10.3390/inorganics12040122
D’Amaral GM, Bender DN, Piccolo N, Lough AJ, Gossage RA, Foucher DA, Wylie RS. Hypercoordinating Stannanes with C,N-Donor Ligands: A Structural, Computational, and Polymerization Study. Inorganics. 2024; 12(4):122. https://doi.org/10.3390/inorganics12040122
Chicago/Turabian StyleD’Amaral, Gloria M., Desiree N. Bender, Nicola Piccolo, Alan J. Lough, Robert A. Gossage, Daniel A. Foucher, and R. Stephen Wylie. 2024. "Hypercoordinating Stannanes with C,N-Donor Ligands: A Structural, Computational, and Polymerization Study" Inorganics 12, no. 4: 122. https://doi.org/10.3390/inorganics12040122
APA StyleD’Amaral, G. M., Bender, D. N., Piccolo, N., Lough, A. J., Gossage, R. A., Foucher, D. A., & Wylie, R. S. (2024). Hypercoordinating Stannanes with C,N-Donor Ligands: A Structural, Computational, and Polymerization Study. Inorganics, 12(4), 122. https://doi.org/10.3390/inorganics12040122