
AMPA Receptor Antagonists Facilitate NEDD4-2-Mediated GRIA1 Ubiquitination by Regulating PP2B-ERK1/2-SGK1 Pathway in Chronic Epilepsy Rats

Abstract
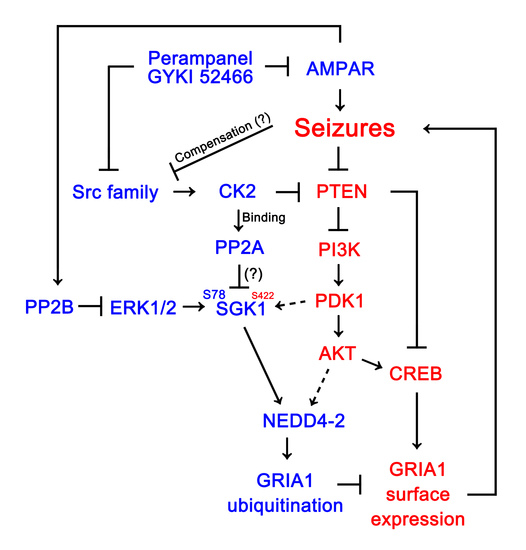
1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Chemicals
2.2. Generation of Chronic Epilepsy Rats
2.3. Surgery
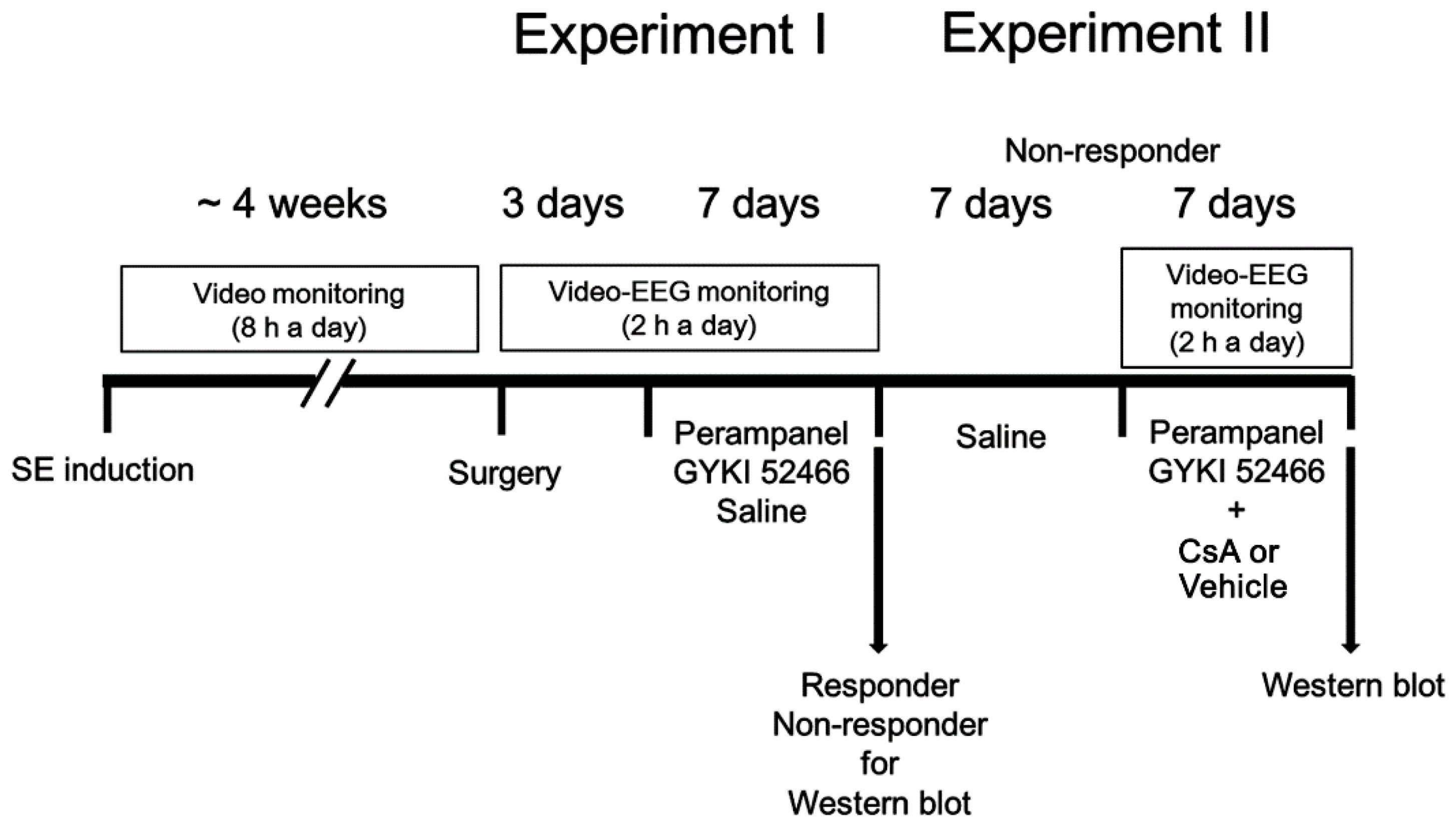
2.4. Drug Trials, EEG Analysis and Quantification of Behavioral Seizure Activity
2.4.1. Experiment I
2.4.2. Experiment II
2.5. Co-Immunoprecipitation
2.6. Western Blot
2.7. Data Analysis
3. Results
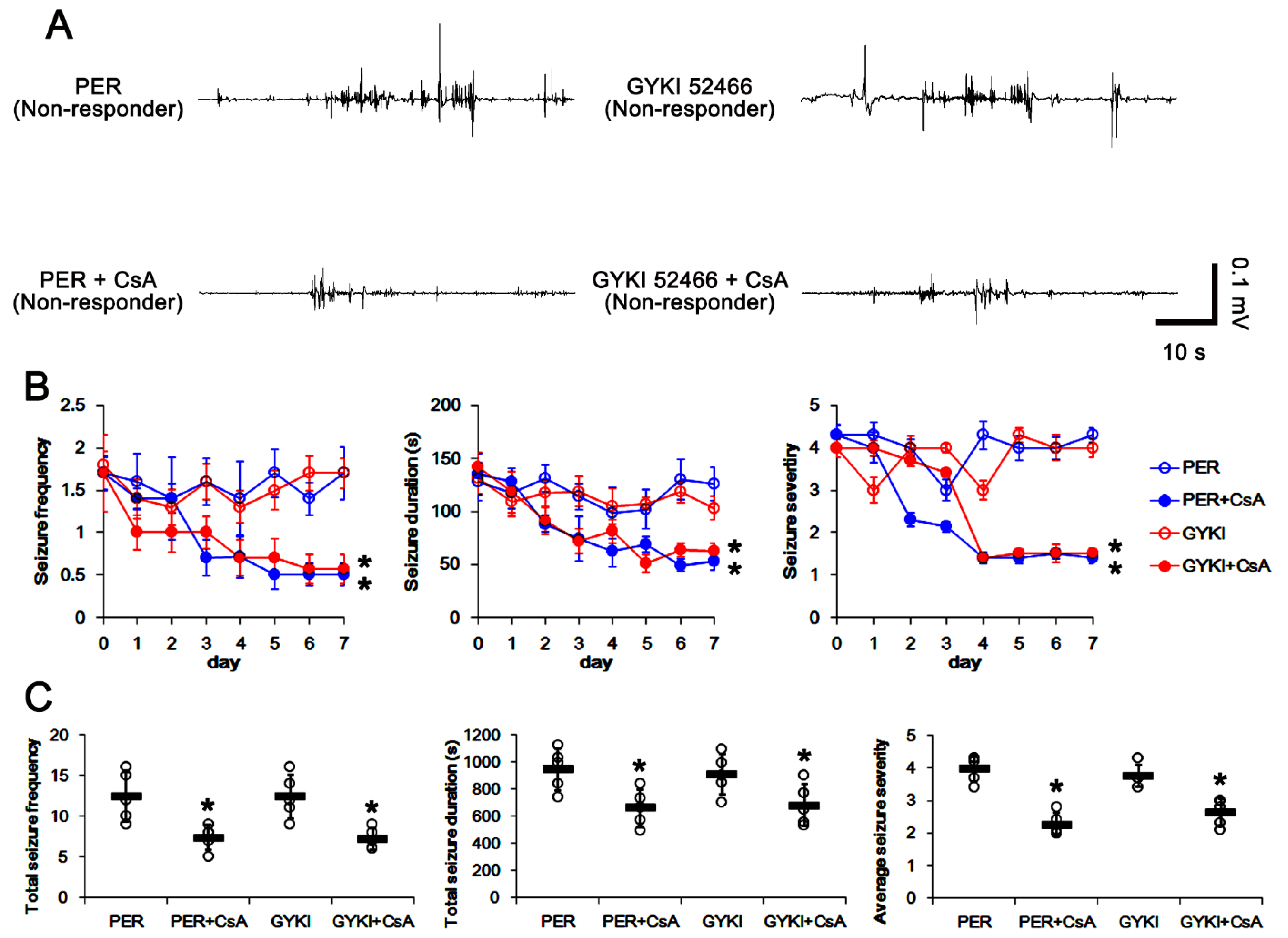
3.1. AMPAR Antagonists Attenuate Spontaneous Seizure Activity in Responders
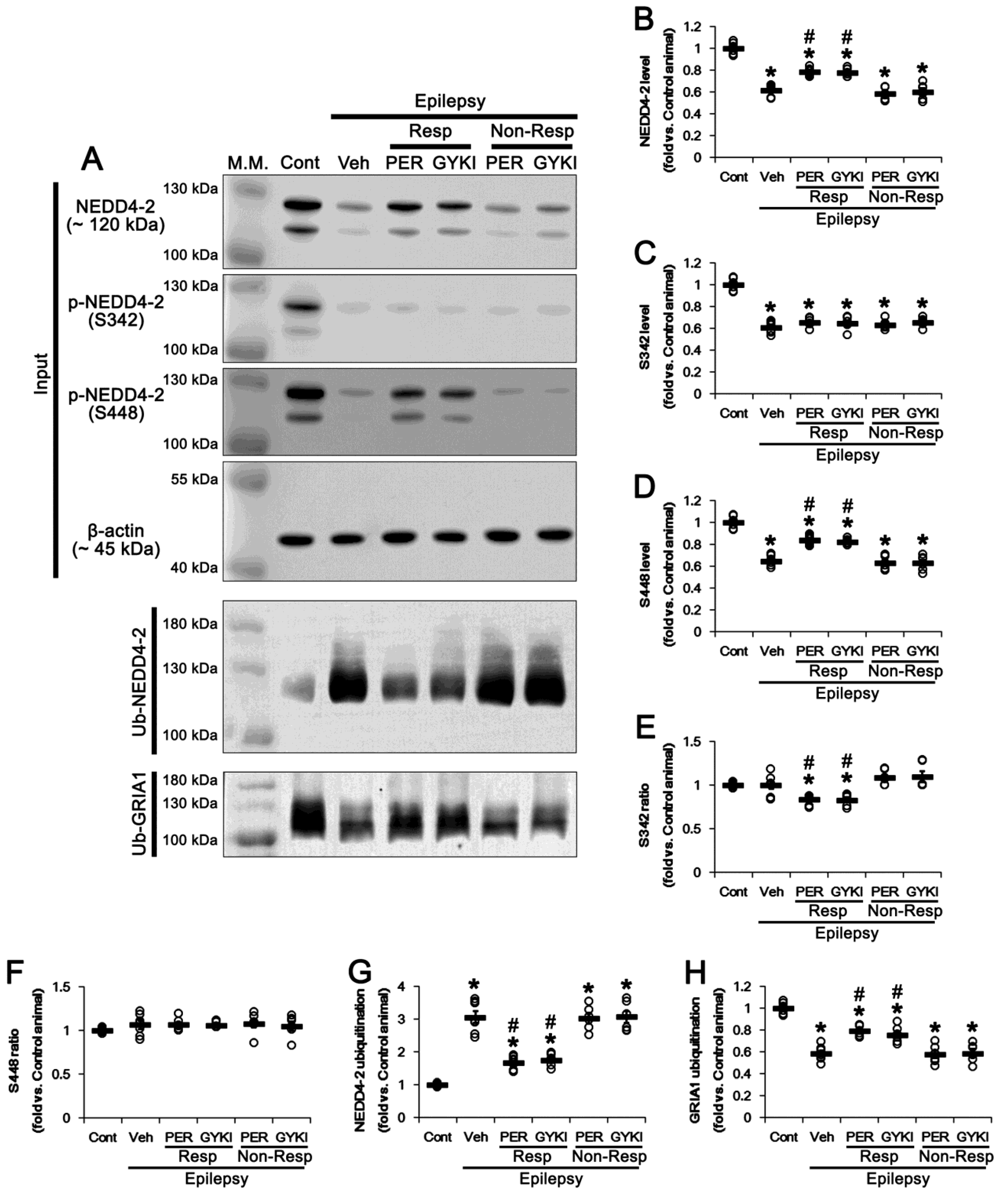
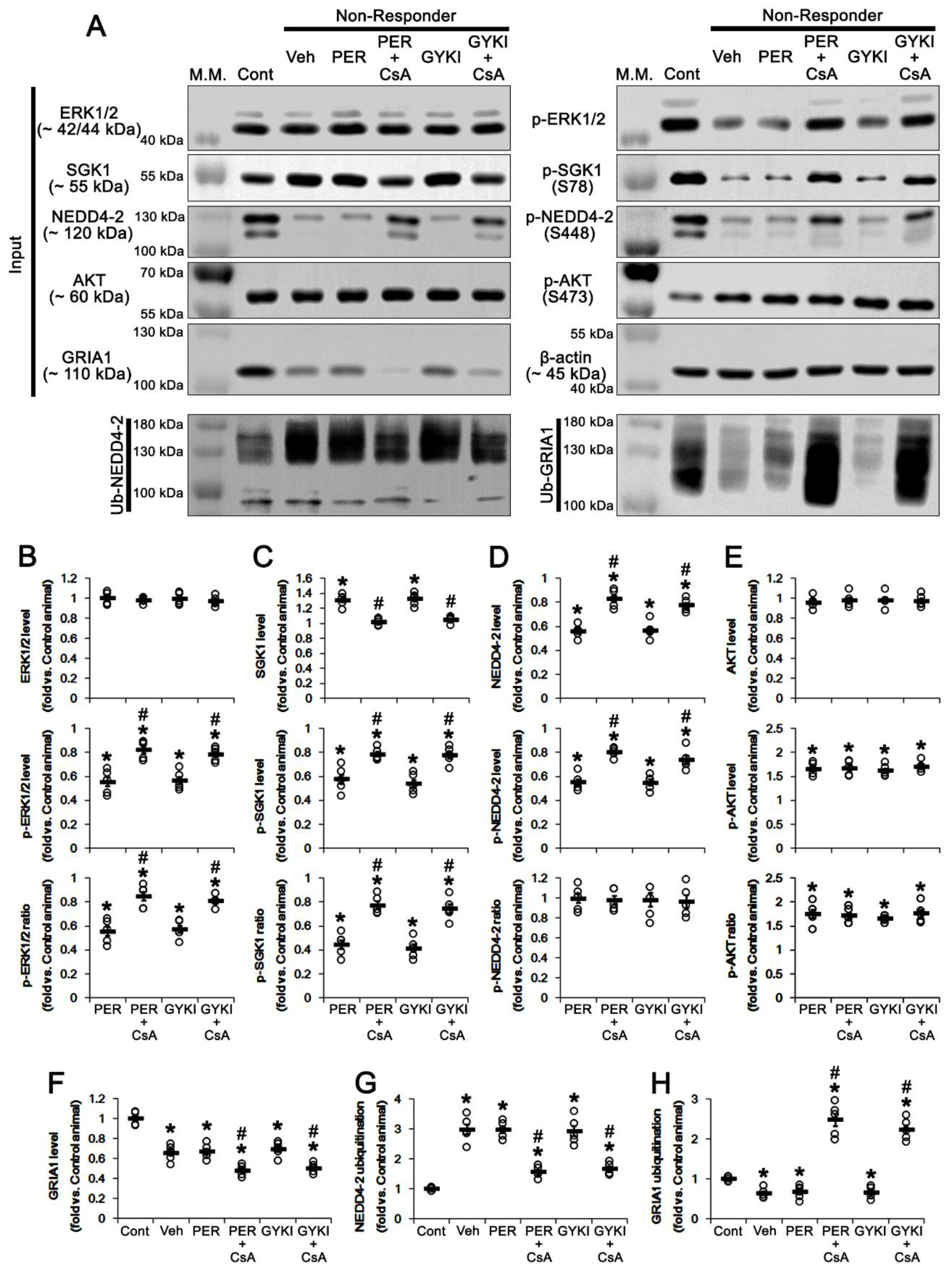
3.2. AMPAR Antagonists Facilitates NEDD4-2-Mediated GRIA1 Ubiquitination by Enhancing NEDD4-2 S448 Phosphorylation in Responders
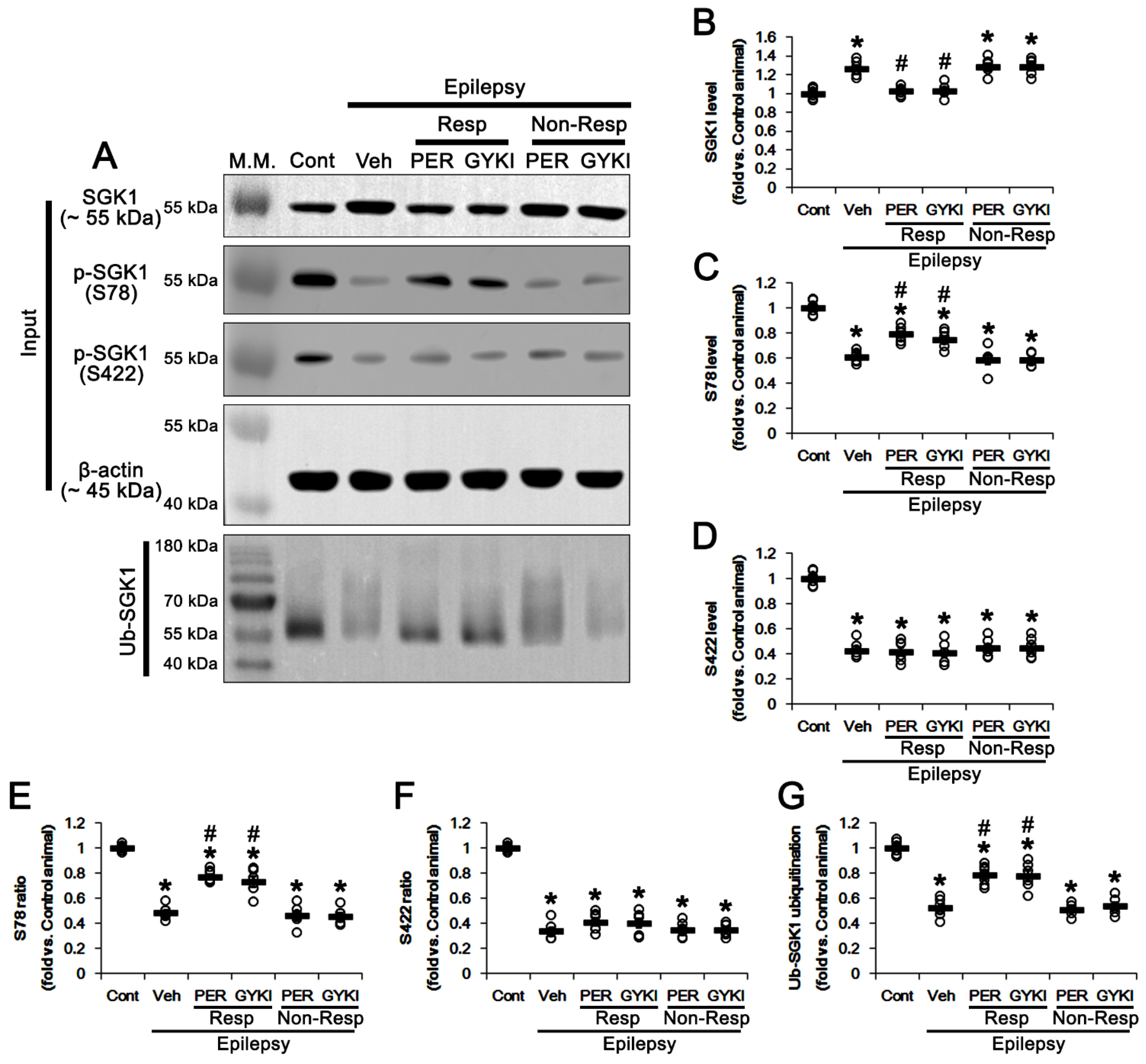
3.3. AMPAR Antagonists Enhance SGK1 S78 Phosphorylation, but Not Protein Level, in Responders
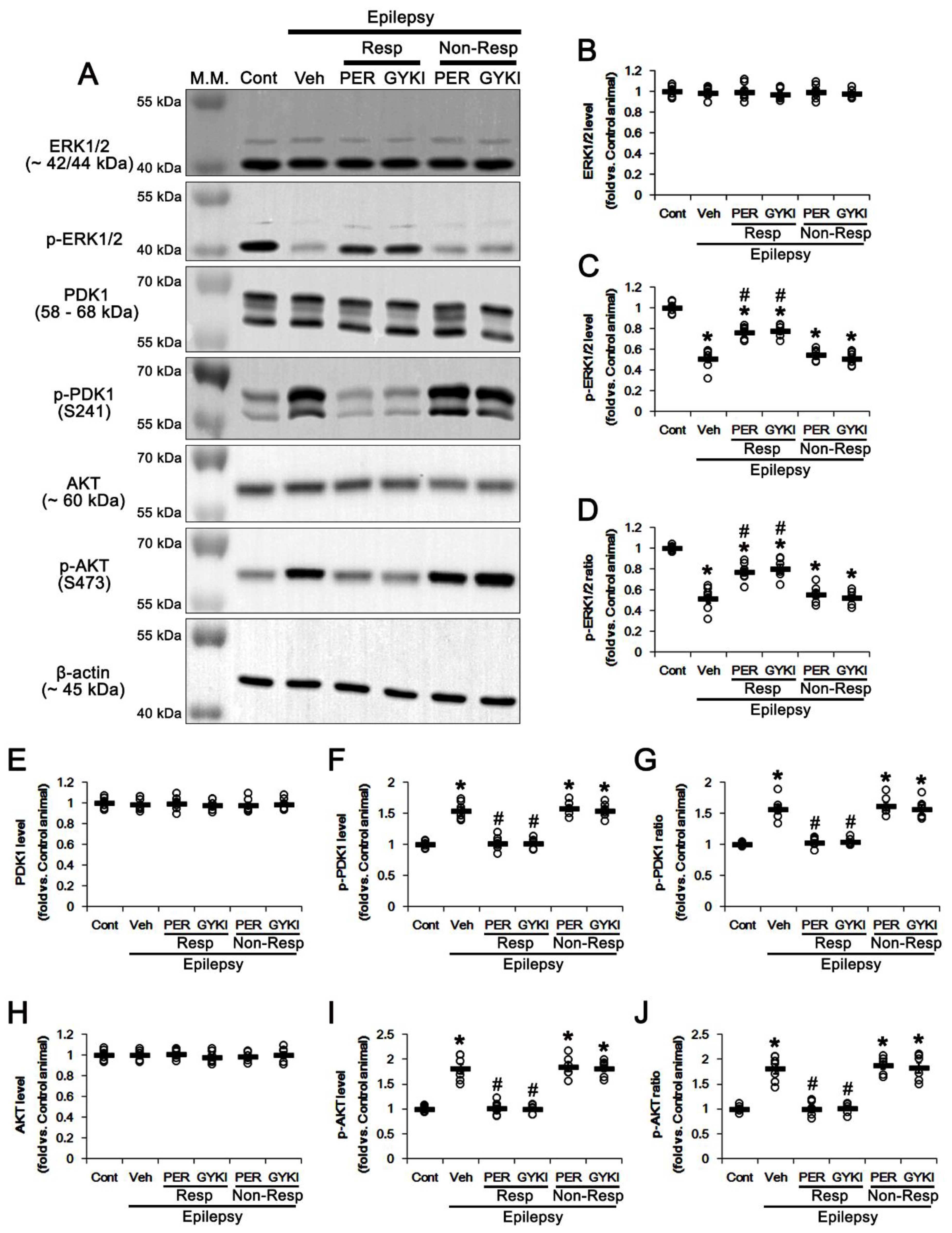
3.4. AMPAR Antagonists Increases ERK1/2, but Reduces PDK1, Phosphorylation in Responders
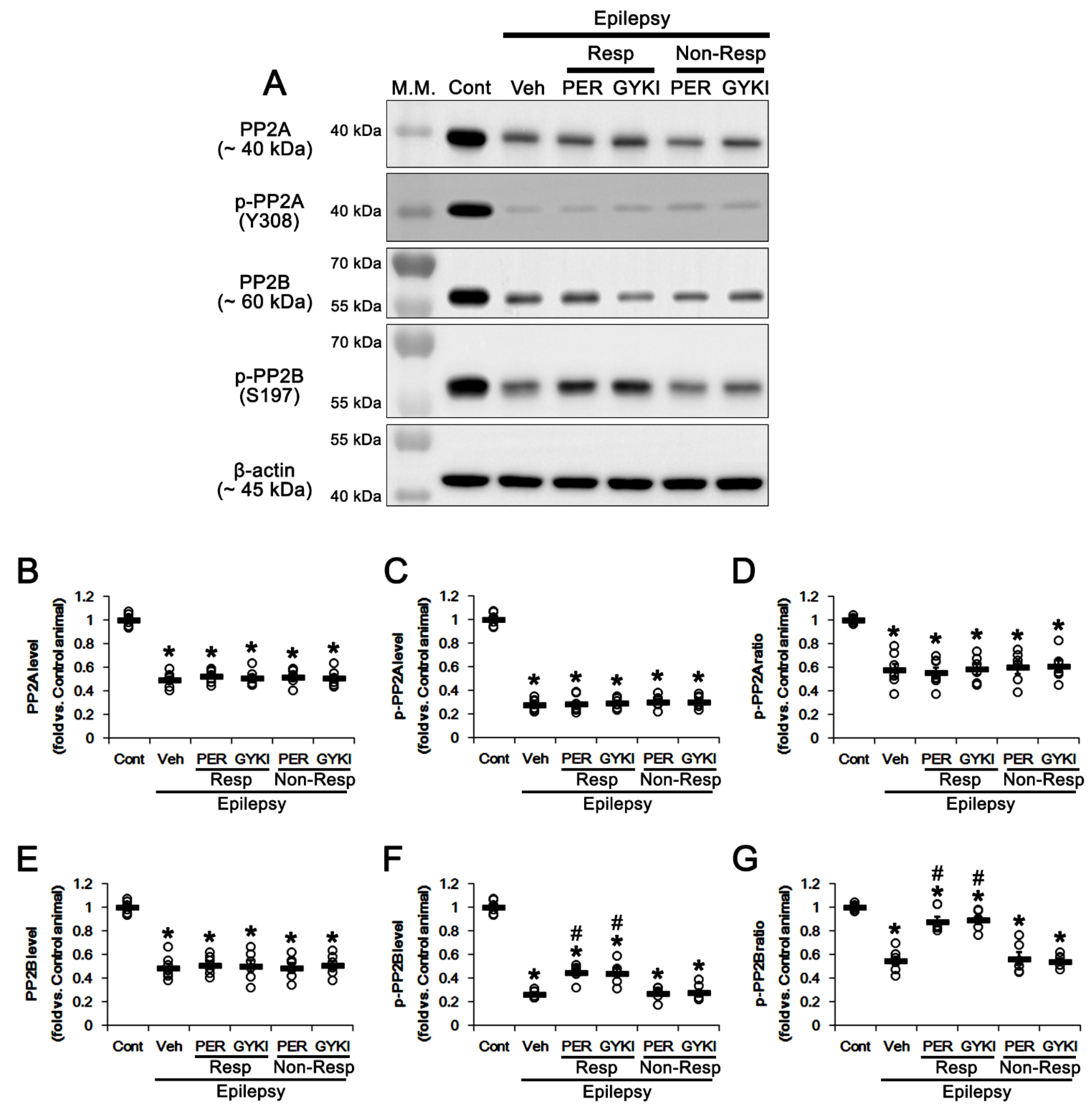
3.5. Effects of AMPAR Antagonists on Protein Phosphatase Phosphorylation
3.6. Co-Treatment of PP2B Inhibitor Increases the Efficacies of AMPAR Antagonists in Non-Responders
3.7. CsA Co-Treatment Facilicates GRIA1 Ubiquitination in Non-Responders to AMPAR Antagonists
3.8. CsA Co-Treatment Reversely Regulates Ubiquitination of NEDD4-2 and GRIA1 in Non-Responders
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Fiest, K.M.; Sauro, K.M.; Wiebe, S.; Patten, S.B.; Kwon, C.S.; Dykeman, J.; Pringsheim, T.; Lorenzetti, D.L.; Jetté, N. Prevalence and incidence of epilepsy: A systematic review and meta-analysis of international studies. Neurology 2017, 88, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Herman, S.T. Epilepsy after brain insult: Targeting epileptogenesis. Neurology 2002, 59, S21–S26. [Google Scholar] [CrossRef] [PubMed]
- Sillanpää, M.; Schmidt, D. Natural history of treated childhood-onset epilepsy: Prospective, long-term population-based study. Brain 2006, 129, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Park, H.; Lee, J.E.; Kim, T.H.; Kang, T.C. PTEN is required for the anti-epileptic effects of AMPA receptor antagonists in chronic epileptic rats. Int. J. Mol. Sci. 2020, 21, 5643. [Google Scholar] [CrossRef]
- Löscher, W. Animal models of epilepsy for the development of antiepileptogenic and disease-modifying drugs. A comparison of the pharmacology of kindling and post-status epilepticus models of temporal lobe epilepsy. Epilepsy Res. 2002, 50, 105–123. [Google Scholar] [CrossRef]
- Egbenya, D.L.; Hussain, S.; Lai, Y.C.; Xia, J.; Anderson, A.E.; Davanger, S. Changes in synaptic AMPA receptor concentration and composition in chronic temporal lobe epilepsy. Mol. Cell. Neurosci. 2018, 92, 93–103. [Google Scholar] [CrossRef]
- Anggono, V.; Huganir, R.L. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr. Opin. Neurobiol. 2012, 22, 461–469. [Google Scholar] [CrossRef]
- Fritsch, B.; Stott, J.J.; Joelle Donofrio, J.; Rogawski, M.A. Treatment of early and late kainic acid-induced status epilepticus with the noncompetitive AMPA receptor antagonist GYKI 52466. Epilepsia 2010, 51, 108–117. [Google Scholar] [CrossRef]
- Mohammad, H.; Sekar, S.; Wei, Z.; Moien-Afshari, F.; Taghibiglou, C. Perampanel but not amantadine prevents behavioral alterations and epileptogenesis in pilocarpine rat model of status epilepticus. Mol. Neurobiol. 2019, 56, 2508–2523. [Google Scholar] [CrossRef]
- Kim, J.E.; Lee, D.S.; Park, H.; Kang, T.C. Src/CK2/PTEN-mediated GluN2B and CREB dephosphorylation regulate the responsiveness to AMPA receptor antagonists in chronic epilepsy rats. Int. J. Mol. Sci. 2020, 21, 9633. [Google Scholar] [CrossRef]
- Zhu, J.; Lee, K.Y.; Jewett, K.A.; Man, H.Y.; Chung, H.J.; Tsai, N.P. Epilepsy-associated gene NEDD4-2 mediates neuronal activity and seizure susceptibility through AMPA receptors. PLoS Genet. 2017, 13, e1006634. [Google Scholar] [CrossRef]
- Ekberg, J.A.; Boase, N.A.; Rychkov, G.; Manning, J.; Poronnik, P.; Kumar, S. Nedd4-2 (NEDD4L) controls intracellular Na(+)-mediated activity of voltage-gated sodium channels in primary cortical neurons. Biochem. J. 2014, 457, 27–31. [Google Scholar] [CrossRef]
- Ekberg, J.; Schuetz, F.; Boase, N.A.; Conroy, S.J.; Manning, J.; Kumar, S.; Poronnik, P.; Adams, D.J. Regulation of the voltage-gated K(+) channels KCNQ2/3 and KCNQ3/5 by ubiquitination. Novel role for Nedd4-2. J. Biol. Chem. 2007, 282, 12135–12142. [Google Scholar] [CrossRef]
- Goel, P.; Manning, J.A.; Kumar, S. NEDD4-2 (NEDD4L): The ubiquitin ligase for multiple membrane proteins. Gene 2015, 557, 1–10. [Google Scholar] [CrossRef]
- Schuetz, F.; Kumar, S.; Poronnik, P.; Adams, D.J. Regulation of the voltage-gated K(+) channels KCNQ2/3 and KCNQ3/5 by serum- and glucocorticoid-regulated kinase-1. Am. J. Physiol. Cell. Physiol. 2008, 295, C73–C80. [Google Scholar] [CrossRef]
- Yu, T.; Calvo, L.; Anta, B.; López-Benito, S.; López-Bellido, R.; Vicente-García, C.; Tessarollo, L.; Rodriguez, R.E.; Arévalo, J.C. In vivo regulation of NGF-mediated functions by Nedd4-2 ubiquitination of TrkA. J. Neurosci. 2014, 34, 6098–6106. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Lee, D.S.; Kim, M.J.; Kang, T.C. PLPP/CIN-mediated NEDD4-2 S448 dephosphorylation regulates neuronal excitability via GluA1 ubiquitination. Cell Death Dis. 2019, 10, 545. [Google Scholar] [CrossRef] [PubMed]
- Epi4K Consortium; Epilepsy Phenome/Genome Project; Allen, A.S.; Berkovic, S.F.; Cossette, P.; Delanty, N.; Dlugos, D.; Eichler, E.E.; Epstein, M.P.; Glauser, T.; et al. De novo mutations in epileptic encephalopathies. Nature 2013, 501, 217–221. [Google Scholar] [PubMed]
- Dibbens, L.M.; Ekberg, J.; Taylor, I.; Hodgson, B.L.; Conroy, S.J.; Lensink, I.L.; Kumar, S.; Zielinski, M.A.; Harkin, L.A.; Sutherland, G.R.; et al. NEDD4-2 as a potential candidate susceptibility gene for epileptic photosensitivity. Genes Brain Behav. 2007, 6, 750–755. [Google Scholar] [CrossRef]
- Vanli-Yavuz, E.N.; Ozdemir, O.; Demirkan, A.; Catal, S.; Bebek, N.; Ozbek, U.; Baykan, B. Investigation of the possible association of NEDD4-2 (NEDD4L) gene with idiopathic photosensitive epilepsy. Acta Neurol. Belg. 2015, 115, 241–245. [Google Scholar] [CrossRef]
- Kim, J.E.; Lee, D.S.; Kim, T.H.; Park, H.; Kim, M.J.; Kang, T.C. PLPP/CIN-mediated Mdm2 dephosphorylation increases seizure susceptibility via abrogating PSD95 ubiquitination. Exp. Neurol. 2020, 331, 113383. [Google Scholar] [CrossRef]
- Kim, J.E.; Lee, D.S.; Kim, T.H.; Park, H.; Kim, M.J.; Kang, T.C. PLPP/CIN-mediated NF2-serine 10 dephosphorylation regulates F-actin stability and Mdm2 degradation in an activity-dependent manner. Cell Death Dis. 2021, 12, 37. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx 2005, 2, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef]
- Tang, F.; Hartz, A.M.S.; Bauer, B. Drug-resistant epilepsy: Multiple hypotheses, few answers. Front. Neurol. 2017, 8, 301. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Lee, D.S.; Park, H.; Kim, T.H.; Kang, T.C. Inhibition of AKT/GSK3β/CREB pathway improves the responsiveness to AMPA receptor antagonists by regulating GRIA1 surface expression in chronic epilepsy rats. Biomedicines 2021, 9, 425. [Google Scholar] [CrossRef] [PubMed]
- Widagdo, J.; Chai, Y.J.; Ridder, M.C.; Chau, Y.Q.; Johnson, R.C.; Sah, P.; Huganir, R.L.; Anggono, V. Activity-Dependent Ubiquitination of GluA1 and GluA2 Regulates AMPA Receptor Intracellular Sorting and Degradation. Cell Rep. 2015, 10, 783–795. [Google Scholar] [CrossRef]
- Lin, A.; Hou, Q.; Jarzylo, L.; Amato, S.; Gilbert, J.; Shang, F.; Man, H.Y. Nedd4-mediated AMPA receptor ubiquitination regulates receptor turnover and trafficking. J. Neurochem. 2011, 119, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, L.A.; Hall, B.J.; Patrick, G.N. Activity-dependent ubiquitination of GluA1 mediates a distinct AMPA receptor endocytosis and sorting pathway. J. Neurosci. 2010, 30, 16718–16729. [Google Scholar] [CrossRef]
- Kim, J.E.; Choi, H.C.; Song, H.K.; Kang, T.C. Perampanel affects up-stream regulatory signaling pathways of GluA1 phosphorylation in normal and epileptic rats. Front. Cell. Neurosci. 2019, 13, 80. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.R.; Kang, T.C. Blockade of endothelin B receptor improves the efficacy of levetiracetam in chronic epileptic rats. Seizure 2015, 31, 133–140. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Racine, R.J. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Lee, K.Y.; Jewett, K.A.; Chung, H.J.; Tsai, N.P. Loss of fragile X protein FMRP impairs homeostatic synaptic downscaling through tumor suppressor p53 and ubiquitin E3 ligase Nedd4-2. Hum. Mol. Genet. 2018, 27, 2805–2816. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Peng, J.; Kong, H.; Yang, P.; He, F.; Deng, X.; Gan, N.; Yin, F. The role of ubiquitin/Nedd4-2 in the pathogenesis of mesial temporal lobe epilepsy. Physiol. Behav. 2015, 143, 104–112. [Google Scholar] [CrossRef]
- Chandran, S.; Li, H.; Dong, W.; Krasinska, K.; Adams, C.; Alexandrova, L.; Chien, A.; Hallows, K.R.; Bhalla, V. Neural precursor cell-expressed developmentally downregulated protein 4-2 (Nedd4-2) regulation by 14-3-3 protein binding at canonical serum and glucocorticoid kinase 1 (SGK1) phosphorylation sites. J. Biol. Chem. 2011, 286, 37830–37840. [Google Scholar] [CrossRef]
- Zhou, R.; Snyder, P.M. Nedd4-2 phosphorylation induces serum and glucocorticoid-regulated kinase (SGK) ubiquitination and degradation. J. Biol. Chem. 2005, 280, 4518–4523. [Google Scholar] [CrossRef]
- Kobayashi, T.; Cohen, P. Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem. J. 1999, 339, 319–328. [Google Scholar] [CrossRef]
- Lee, C.T.; Ma, Y.L.; Lee, E.H. Serum- and glucocorticoid-inducible kinase1 enhances contextual fear memory formation through downregulation of the expression of Hes5. J. Neurochem. 2007, 100, 1531–1542. [Google Scholar]
- Lee, C.T.; Tyan, S.W.; Ma, Y.L.; Tsai, M.C.; Yang, Y.C.; Lee, E.H. Serum- and glucocorticoid-inducible kinase (SGK) is a target of the MAPK/ERK signaling pathway that mediates memory formation in rats. Eur. J. Neurosci. 2006, 23, 1311–1320. [Google Scholar] [CrossRef]
- García-Martínez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef]
- Lee, I.H.; Dinudom, A.; Sanchez-Perez, A.; Kumar, S.; Cook, D.I. Akt mediates the effect of insulin on epithelial sodium channels by inhibiting Nedd4-2. J. Biol. Chem. 2007, 282, 29866–29873. [Google Scholar] [CrossRef]
- Waskiewicz, A.J.; Cooper, J.A. Mitogen and stress response pathways: MAP kinase cascades and phosphatase regulation in mammals and yeast. Curr. Opin. Cell Biol. 1995, 7, 798–805. [Google Scholar] [CrossRef]
- Gabryel, B.; Pudelko, A.; Adamczyk, J.; Fischer, I.; Malecki, A. Calcineurin and Erk1/2-signaling pathways are involved in the antiapoptotic effect of cyclosporin A on astrocytes exposed to simulated ischemia in vitro. Naunyn. Schmiedebergs Arch. Pharmacol. 2006, 374, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Leong, M.L.; Buse, P.; Maiyar, A.C.; Firestone, G.L.; Hemmings, B.A. Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulated signaling pathway. EMBO J. 1999, 18, 3024–3033. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; King, M.M.; Soderling, T.R. Regulatory interactions of calmodulin-binding proteins: Phosphorylation of calcineurin by autophosphorylated Ca2+/calmodulin-dependent protein kinase II. Proc. Natl. Acad. Sci. USA 1988, 85, 7001–7005. [Google Scholar] [CrossRef] [PubMed]
- MacDonnell, S.M.; Weisser-Thomas, J.; Kubo, H.; Hanscome, M.; Liu, Q.; Jaleel, N.; Berretta, R.; Chen, X.; Brown, J.H.; Sabri, A.K.; et al. CaMKII negatively regulates calcineurin-NFAT signaling in cardiac myocytes. Circ. Res. 2009, 105, 316–325. [Google Scholar] [CrossRef]
- Schwarz, L.A.; Patrick, G.N. Ubiquitin-dependent endocytosis, trafficking and turnover of neuronal membrane proteins. Mol. Cell. Neurosci. 2012, 49, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Hallengren, J.; Chen, P.C.; Wilson, S.M. Neuronal ubiquitin homeostasis. Cell Biochem. Biophys. 2013, 67, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.C.; Kanelis, V.; Fouladkou, F.; Debonneville, A.; Staub, O.; Rotin, D. Regulation of Nedd4-2 self-ubiquitination and stability by a PY motif located within its HECT-domain. Biochem. J. 2008, 415, 155–163. [Google Scholar] [CrossRef] [PubMed]
- García-Tardón, N.; González-González, I.M.; Martínez-Villarreal, J.; Fernández-Sánchez, E.; Giménez, C.; Zafra, F. Protein kinase C (PKC)-promoted endocytosis of glutamate transporter GLT-1 requires ubiquitin ligase Nedd4-2-dependent ubiquitination but not phosphorylation. J. Biol. Chem. 2012, 287, 19177–19187. [Google Scholar] [CrossRef]
- Snyder, P.M.; Olson, D.R.; Kabra, R.; Zhou, R.; Steines, J.C. cAMP and serum and glucocorticoid-inducible kinase (SGK) regulate the epithelial Na+ channel through convergent phosphorylation of Nedd4-2. J. Biol. Chem. 2004, 279, 45753–45758. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, C.; Zhu, Q.; Luo, J.; Xu, Y.; Huang, Y.; Zhang, X.; Wang, X. Upregulation of serum- and glucocorticoid-induced protein kinase 1 in the brain tissue of human and experimental epilepsy. Neurochem. Int. 2010, 57, 899–905. [Google Scholar] [CrossRef]
- Pérez, M.; Avila, J. The expression of casein kinase 2α’ and phosphatase 2A activity. Biochim. Biophys. Acta 1999, 1449, 150–156. [Google Scholar] [CrossRef]
- Pellegrini-Giampietro, D.E.; Gorter, J.A.; Bennett, M.V.; Zukin, R.S. The GluR2 (GluR-B) hypothesis: Ca2+-permeable AMPA receptors in neurological disorders. Trends Neurosci. 1997, 20, 464–470. [Google Scholar] [CrossRef]
- Hell, J.W. How Ca2+-permeable AMPA receptors, the kinase PKA, and the phosphatase PP2B are intertwined in synaptic LTP and LTD. Sci. Signal. 2016, 9, e2. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Sun, X.; Li, J.; Jia, R.; Yuan, F.; Wei, D.; Jiang, W. Melatonin Alleviates the Epilepsy-Associated Impairments in Hippocampal LTP and Spatial Learning Through Rescue of Surface GluR2 Expression at Hippocampal CA1 Synapses. Neurochem. Res. 2017, 42, 1438–1448. [Google Scholar] [CrossRef]
- Lorgen, J.Ø.; Egbenya, D.L.; Hammer, J.; Davanger, S. PICK1 facilitates lasting reduction in GluA2 concentration in the hippocampus during chronic epilepsy. Epilepsy Res. 2017, 137, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Taubøll, E.; Gerdts, R.; Gjerstad, L. Cyclosporin A and brain excitability studied in vitro. Epilepsia 1998, 39, 687–691. [Google Scholar] [CrossRef] [PubMed]
- Handreck, A.; Mall, E.M.; Elger, D.A.; Gey, L.; Gernert, M. Different preparations, doses, and treatment regimens of cyclosporine A cause adverse effects but no robust changes in seizure thresholds in rats. Epilepsy Res. 2015, 112, 1–17. [Google Scholar] [CrossRef]
- Huo, Y.; Khatri, N.; Hou, Q.; Gilbert, J.; Wang, G.; Man, H.Y. The deubiquitinating enzyme USP46 regulates AMPA receptor ubiquitination and trafficking. J. Neurochem. 2015, 134, 1067–1080. [Google Scholar] [CrossRef]
- Li, C.; Zhang, J.; Xu, H.; Chang, M.; Lv, C.; Xue, W.; Song, Z.; Zhang, L.; Zhang, X.; Tian, X. Retigabine ameliorates acute stress-induced impairment of spatial memory retrieval through regulating USP2 signaling pathways in hippocampal CA1 area. Neuropharmacology 2018, 135, 151–162. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Host | Manufacturer (Catalog Number) | Dilution Used |
|---|---|---|---|
| NEDD4-2 | Rabbit | Abcam (ab131167): IP Abcam (ab46521): WB | 1:100 (IP) 1:1000 (WB) |
| GRIA1 | Mouse | Synaptic systems (#182011) | 1:100 (IP) 1:1000 (WB) |
| p-NEDD4-2 S342 | Rabbit | Cell signaling (#12146) | 1:1000 (WB) |
| p-NEDD4-2 S448 | Rabbit | Abcam (ab168349) | 1:1000 (WB) |
| SGK1 | Rabbit | ST John’s Laboratory (STJ25513) | 1:100 (IP) 1:1000 (WB) |
| p-SGK1 S78 | Rabbit | Thermo (PA5-38392) | 1:1000 (WB) |
| p-SGK1 S422 | Rabbit | Abcam (ab55281) | 1:1000 (WB) |
| Ubiquitin | Rabbit | Abcam(ab7780) | 1:1000 (WB) |
| ERK1/2 | Rabbit | Biorbyt (Orb160960) | 1:1000 (WB) |
| p-ERK1/2 | Rabbit | Bioss (bs-3330R) | 1:1000 (WB) |
| PDK1 | Rabbit | Cell signaling(#3062) | 1:1000 (WB) |
| p-PDK1 S241 | Rabbit | Cell signaling (#3061) | 1:1000(WB) |
| AKT | Rabbit | Cell signaling (#9272) | 1:1000 (WB) |
| pAKT S473 | Rabbit | Cell signalling (#4060) | 1:1000 (WB) |
| PP2A | Rabbit | Cell signaling (#2038) | 1:5000 (WB) |
| p-PP2A Y308 | Rabbit | Sigma (SAB4503975) | 1:1000 (WB) |
| PP2B | Rabbit | Millipore (07-068-I) | 1:1000 (WB) |
| p-PP2B S197 | Rabbit | Badrilla (A010-80) | 1:1000 (WB) |
| β-actin | Mouse | Sigma (#A5316) | 1:5000 (WB) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-E.; Lee, D.-S.; Park, H.; Kim, T.-H.; Kang, T.-C. AMPA Receptor Antagonists Facilitate NEDD4-2-Mediated GRIA1 Ubiquitination by Regulating PP2B-ERK1/2-SGK1 Pathway in Chronic Epilepsy Rats. Biomedicines 2021, 9, 1069. https://doi.org/10.3390/biomedicines9081069
Kim J-E, Lee D-S, Park H, Kim T-H, Kang T-C. AMPA Receptor Antagonists Facilitate NEDD4-2-Mediated GRIA1 Ubiquitination by Regulating PP2B-ERK1/2-SGK1 Pathway in Chronic Epilepsy Rats. Biomedicines. 2021; 9(8):1069. https://doi.org/10.3390/biomedicines9081069
Chicago/Turabian StyleKim, Ji-Eun, Duk-Shin Lee, Hana Park, Tae-Hyun Kim, and Tae-Cheon Kang. 2021. "AMPA Receptor Antagonists Facilitate NEDD4-2-Mediated GRIA1 Ubiquitination by Regulating PP2B-ERK1/2-SGK1 Pathway in Chronic Epilepsy Rats" Biomedicines 9, no. 8: 1069. https://doi.org/10.3390/biomedicines9081069
APA StyleKim, J.-E., Lee, D.-S., Park, H., Kim, T.-H., & Kang, T.-C. (2021). AMPA Receptor Antagonists Facilitate NEDD4-2-Mediated GRIA1 Ubiquitination by Regulating PP2B-ERK1/2-SGK1 Pathway in Chronic Epilepsy Rats. Biomedicines, 9(8), 1069. https://doi.org/10.3390/biomedicines9081069