
Irinotecan Induces Disease Remission in Xenograft Mouse Models of Pediatric MLL-Rearranged Acute Lymphoblastic Leukemia

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Drug Library Screening
2.3. In Vitro Drug Exposures
2.4. Western Blot
2.5. Animal Experiments
3. Results
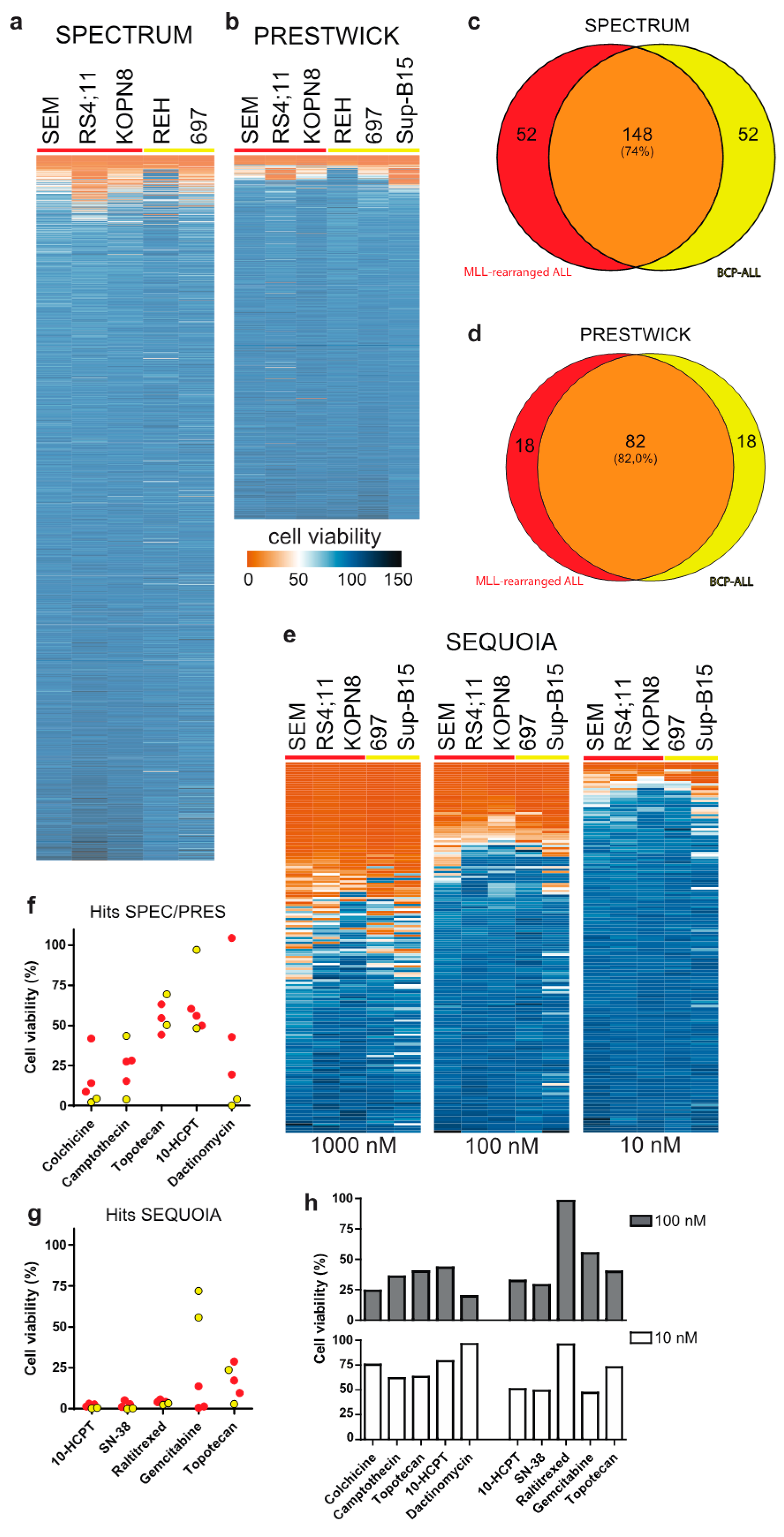
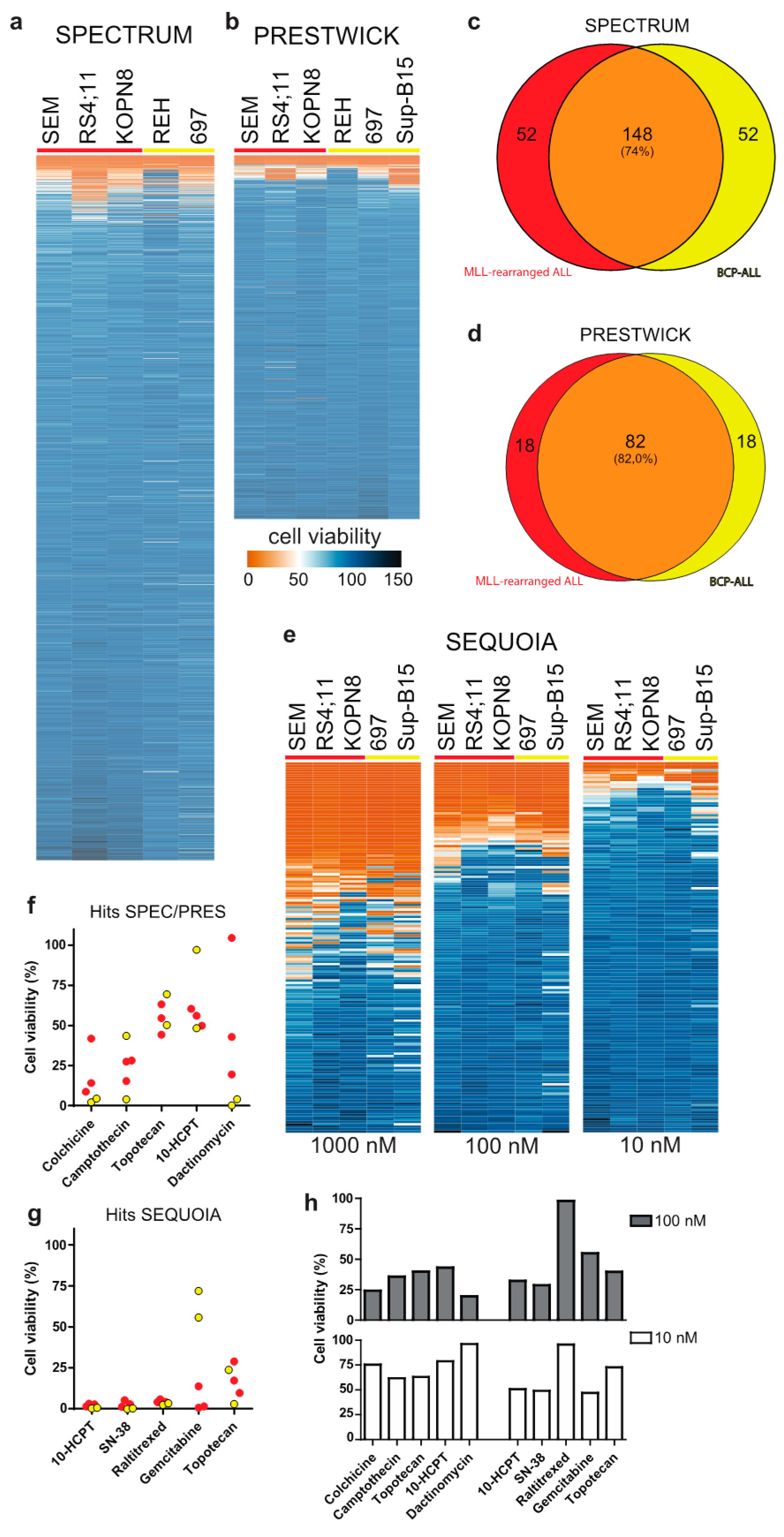
3.1. Drug Library Screening Identifies Camptothecin Derivatives as Promising Leads
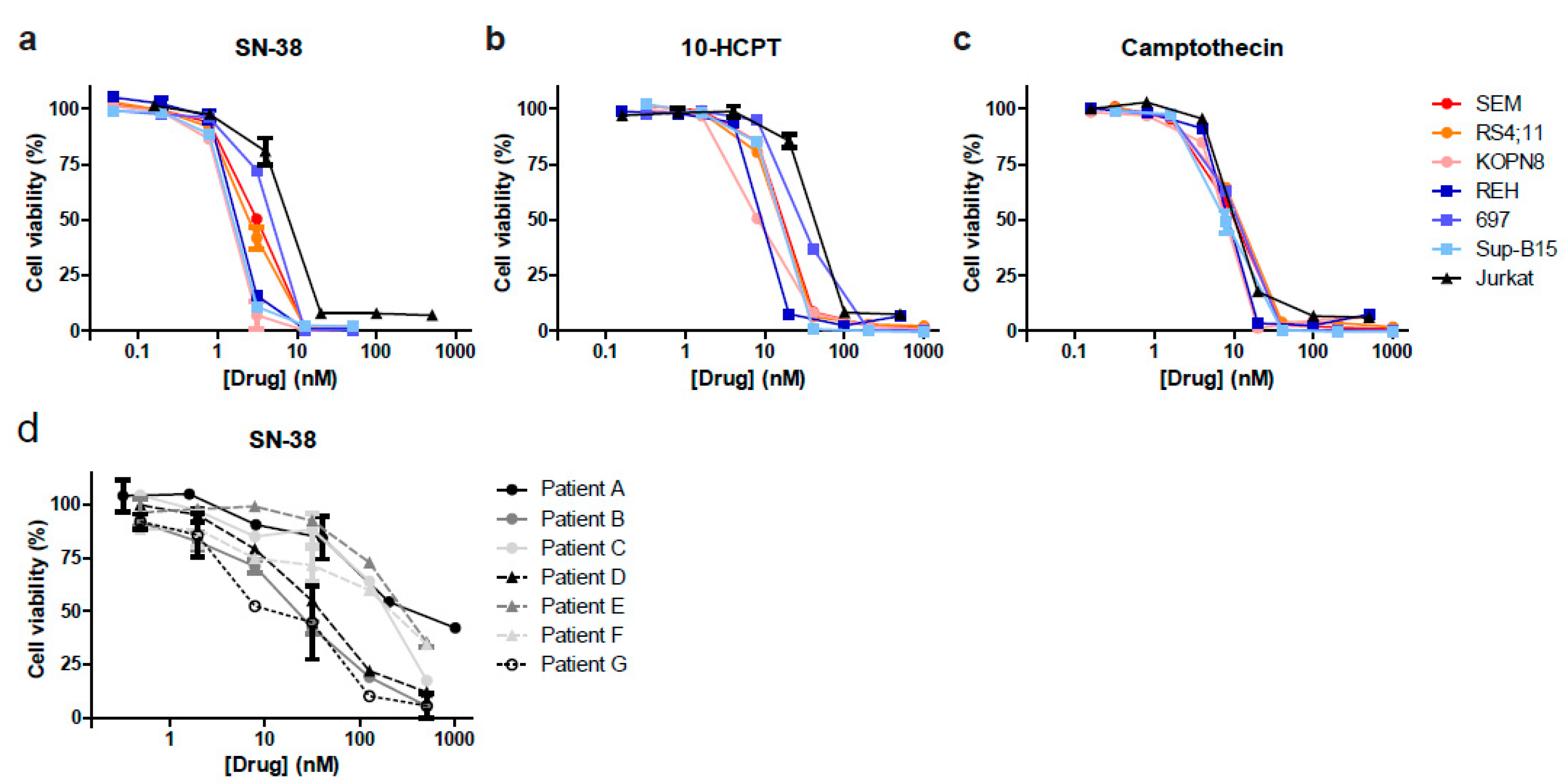
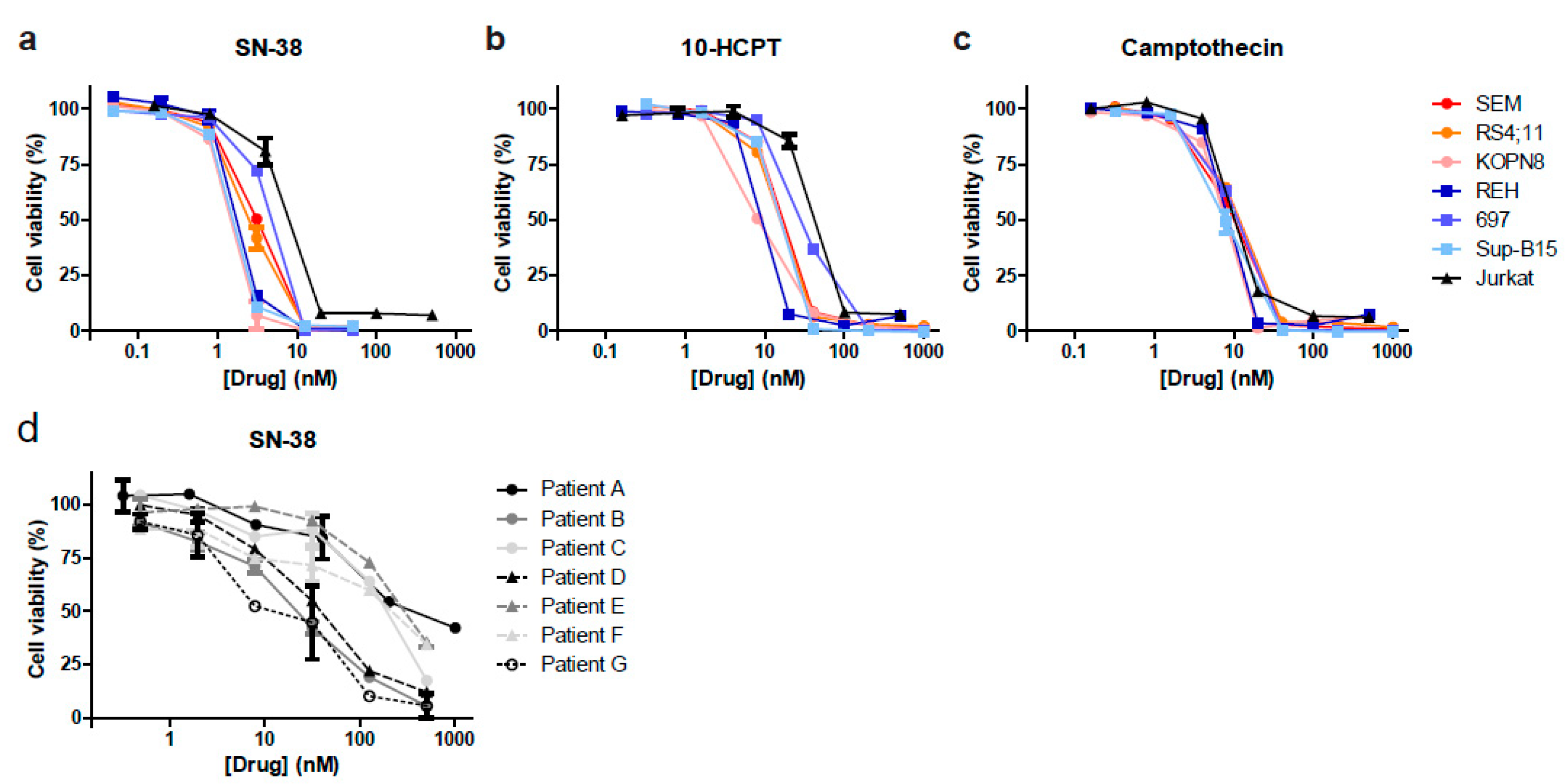
3.2. Camptothecin Derivative SN-38 Most Potently Inhibits ALL Cell Viability
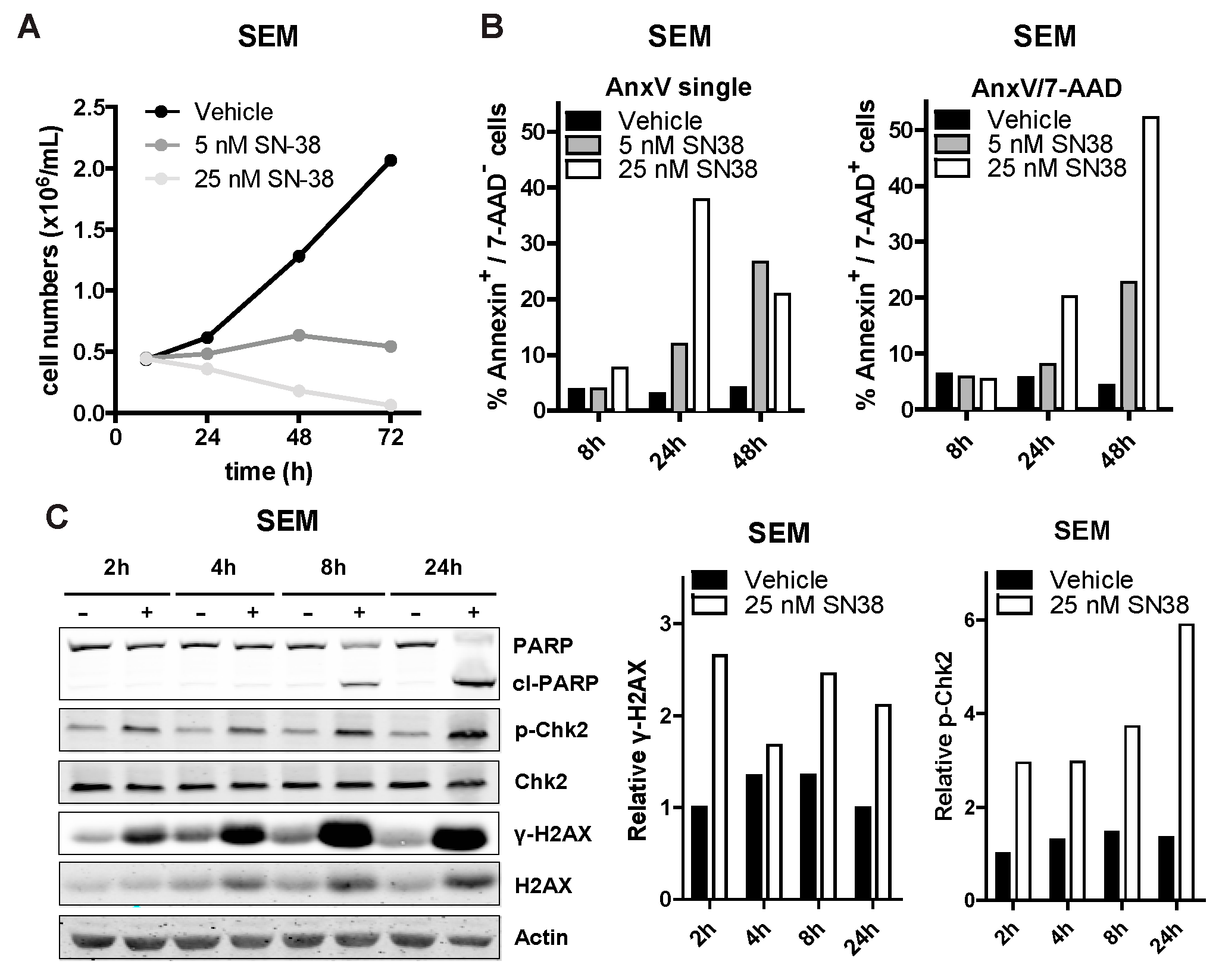
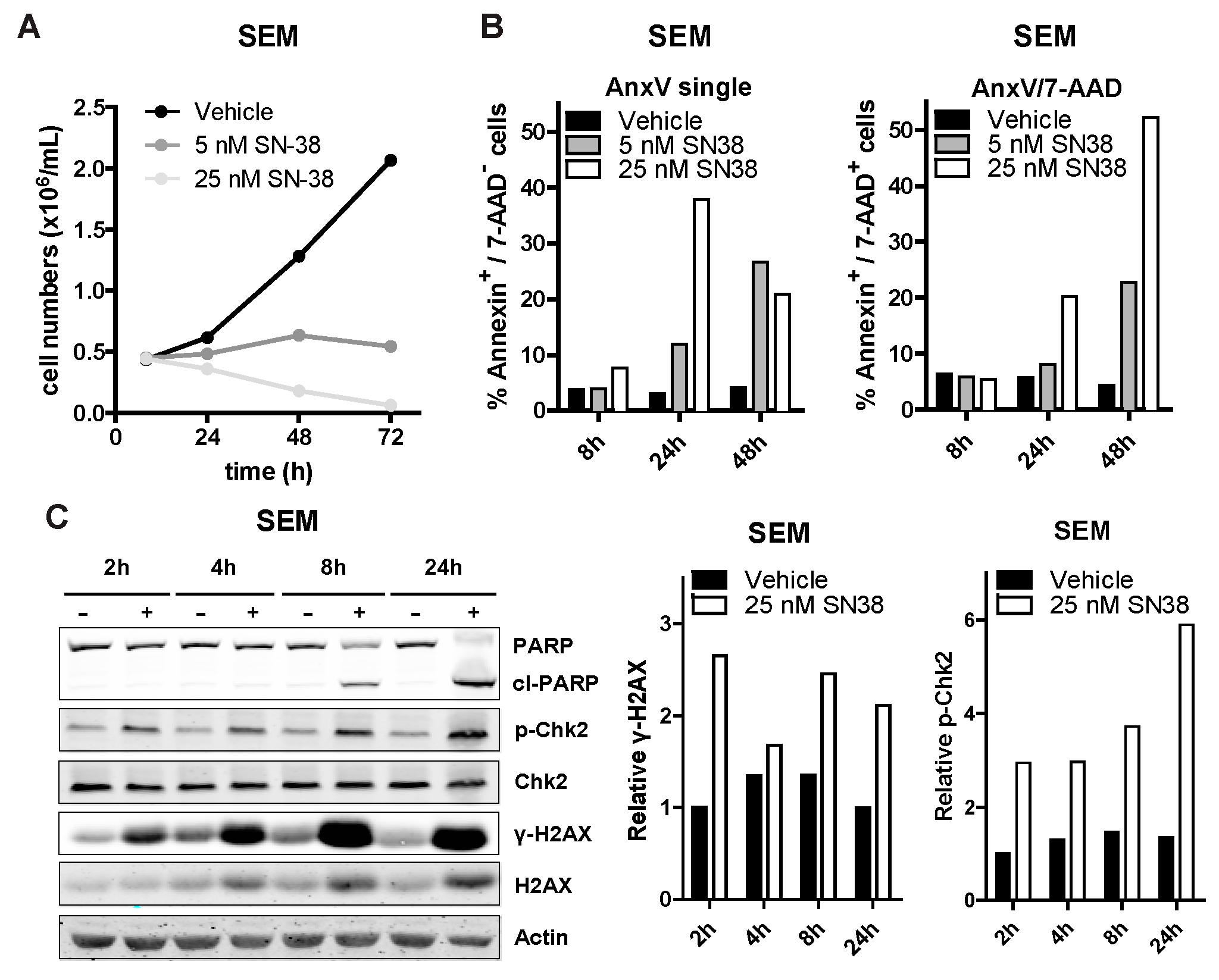
3.3. SN-38 Induces DNA Damage and Apoptotic Cell Death in ALL
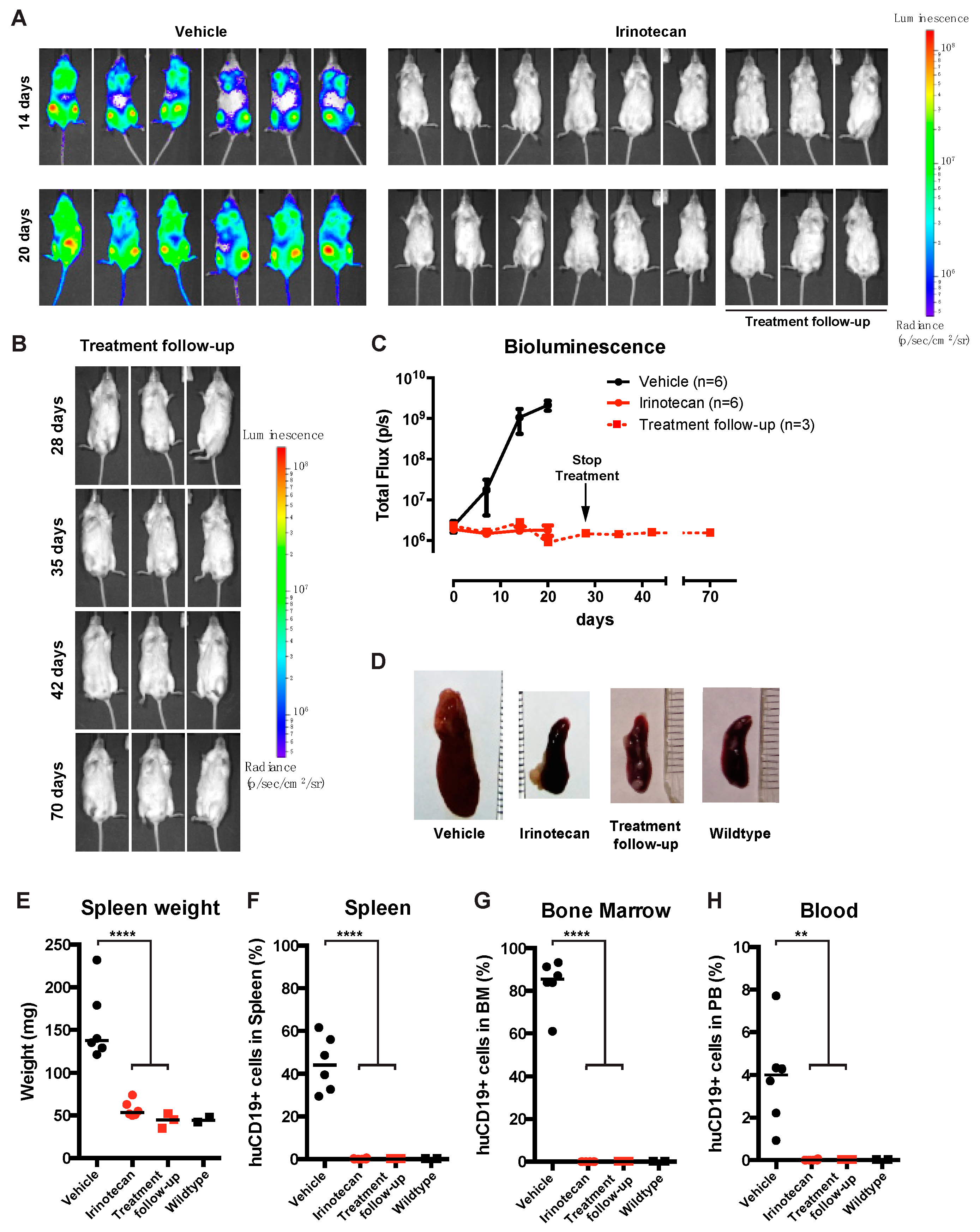
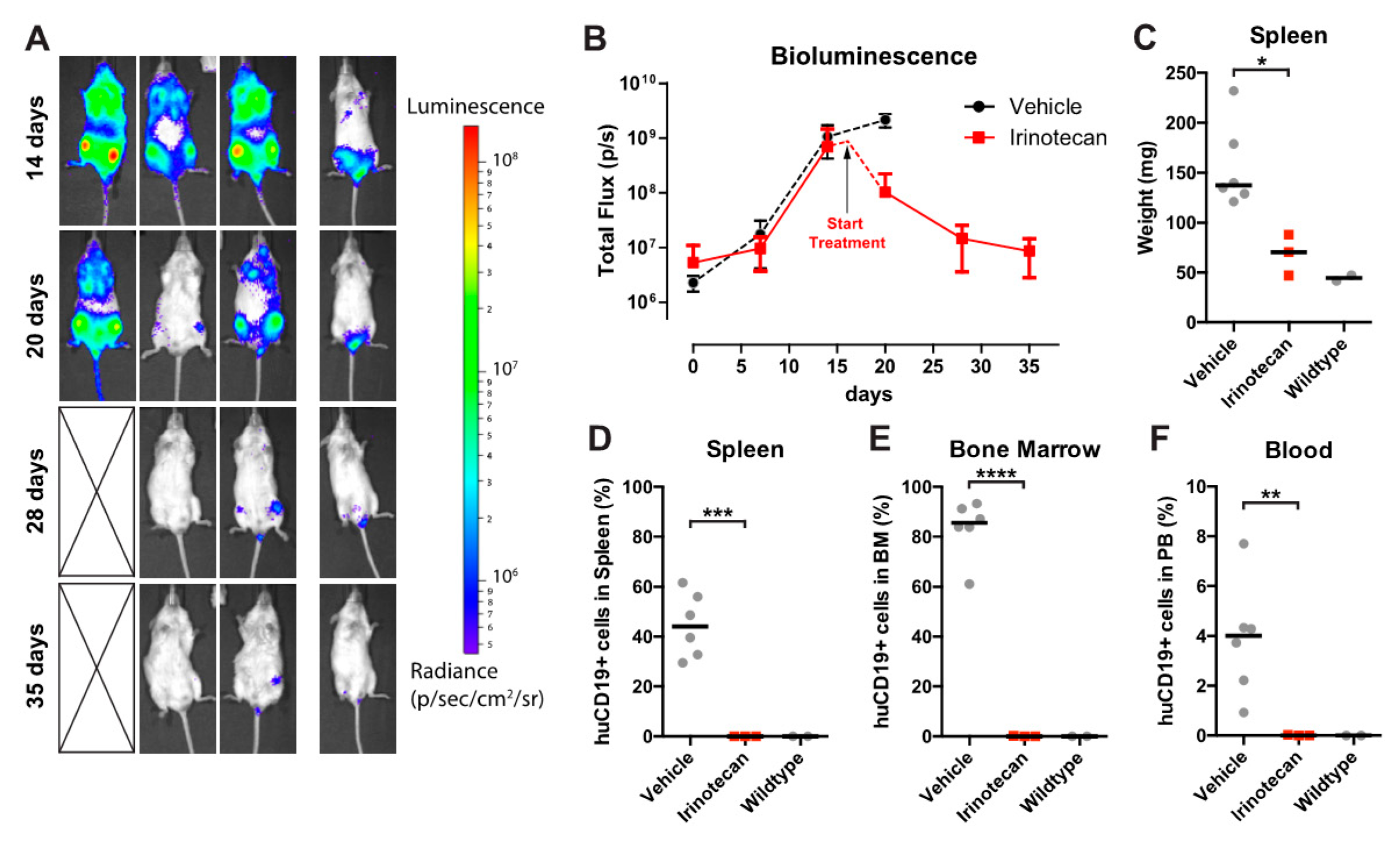
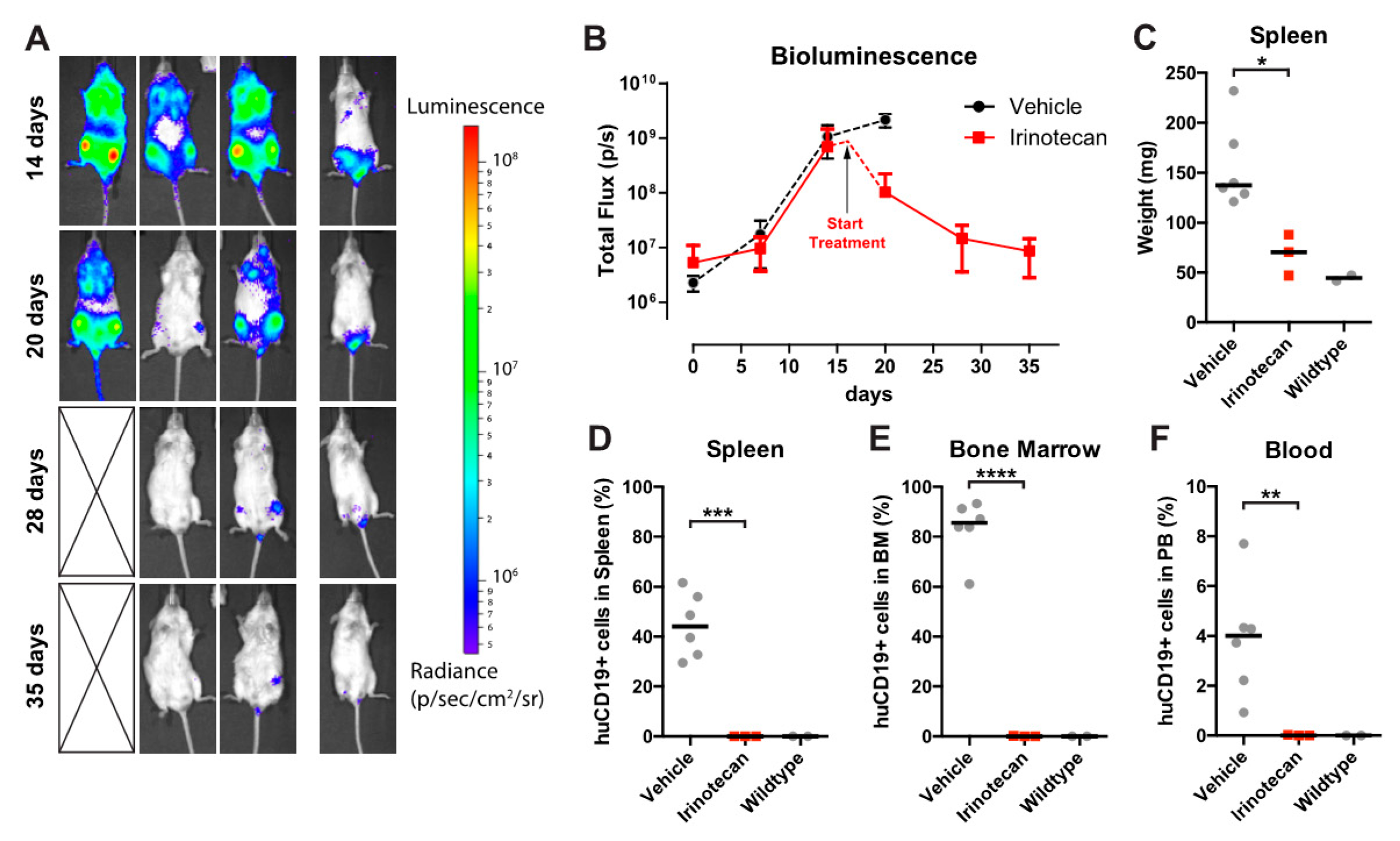
3.4. The SN-38 Pro-Drug Irinotecan Effectively Inhibits MLL-Rearranged ALL In Vivo
3.5. Irinotecan Cures Mice with Advanced MLL-Rearranged ALL
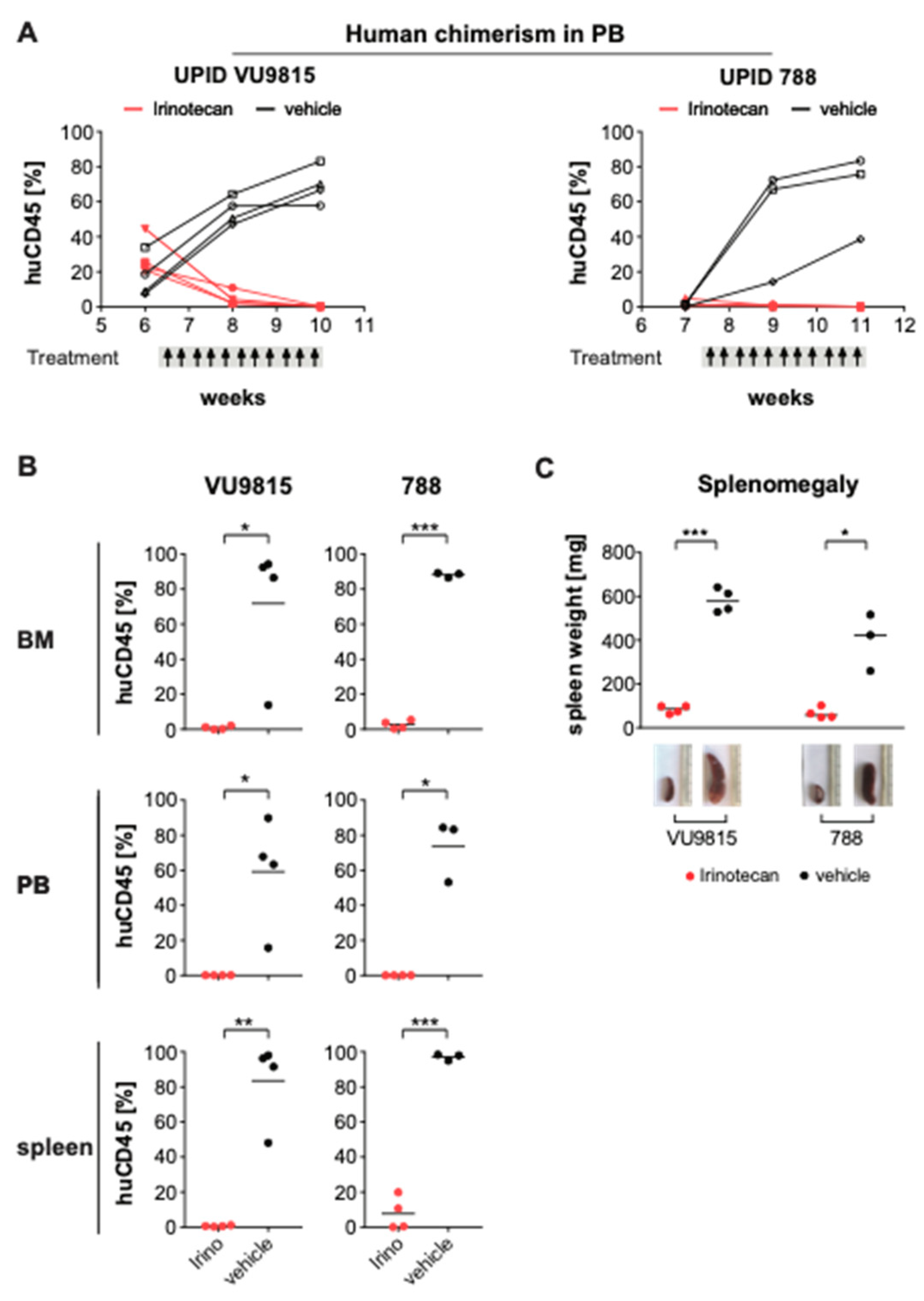
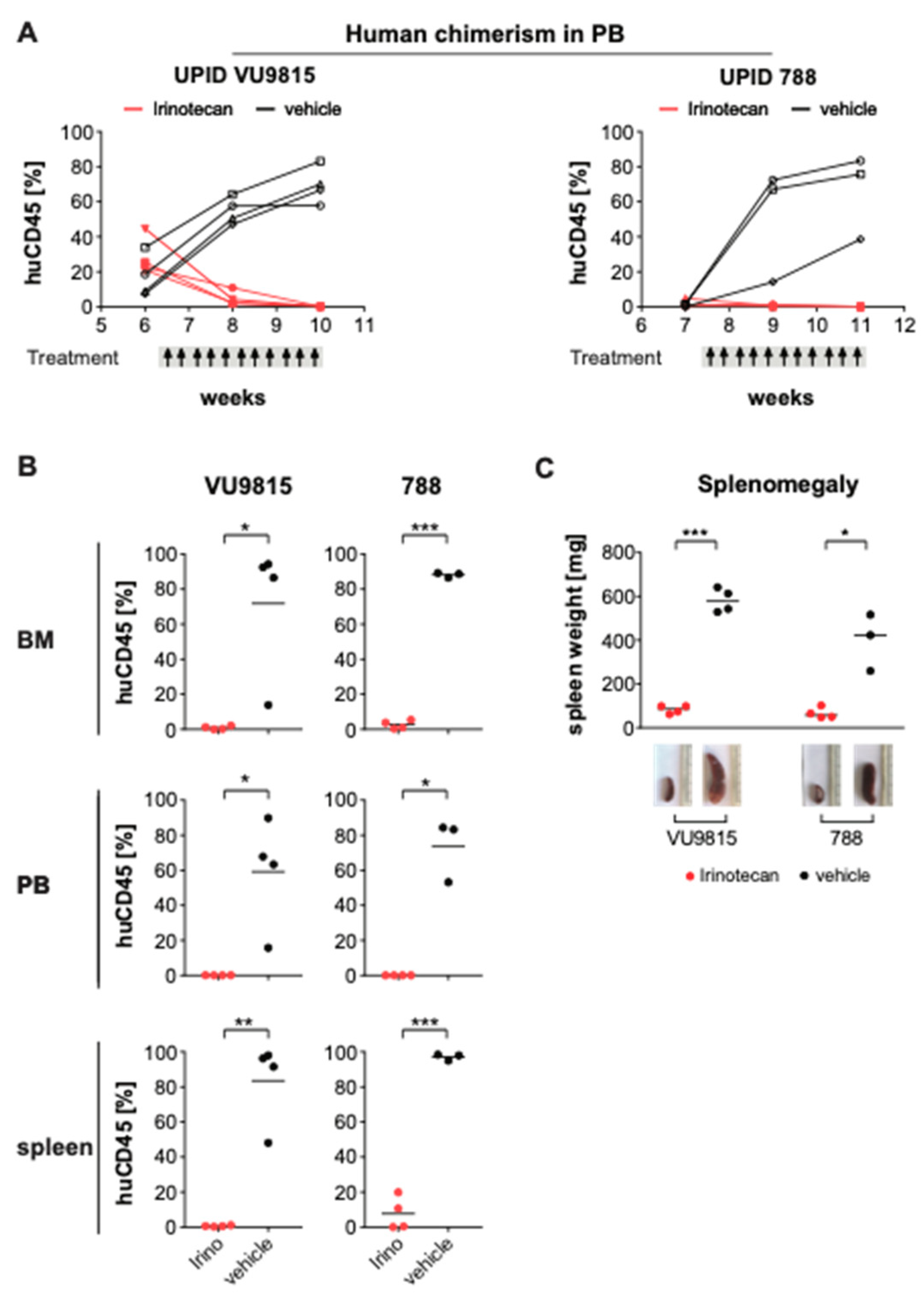
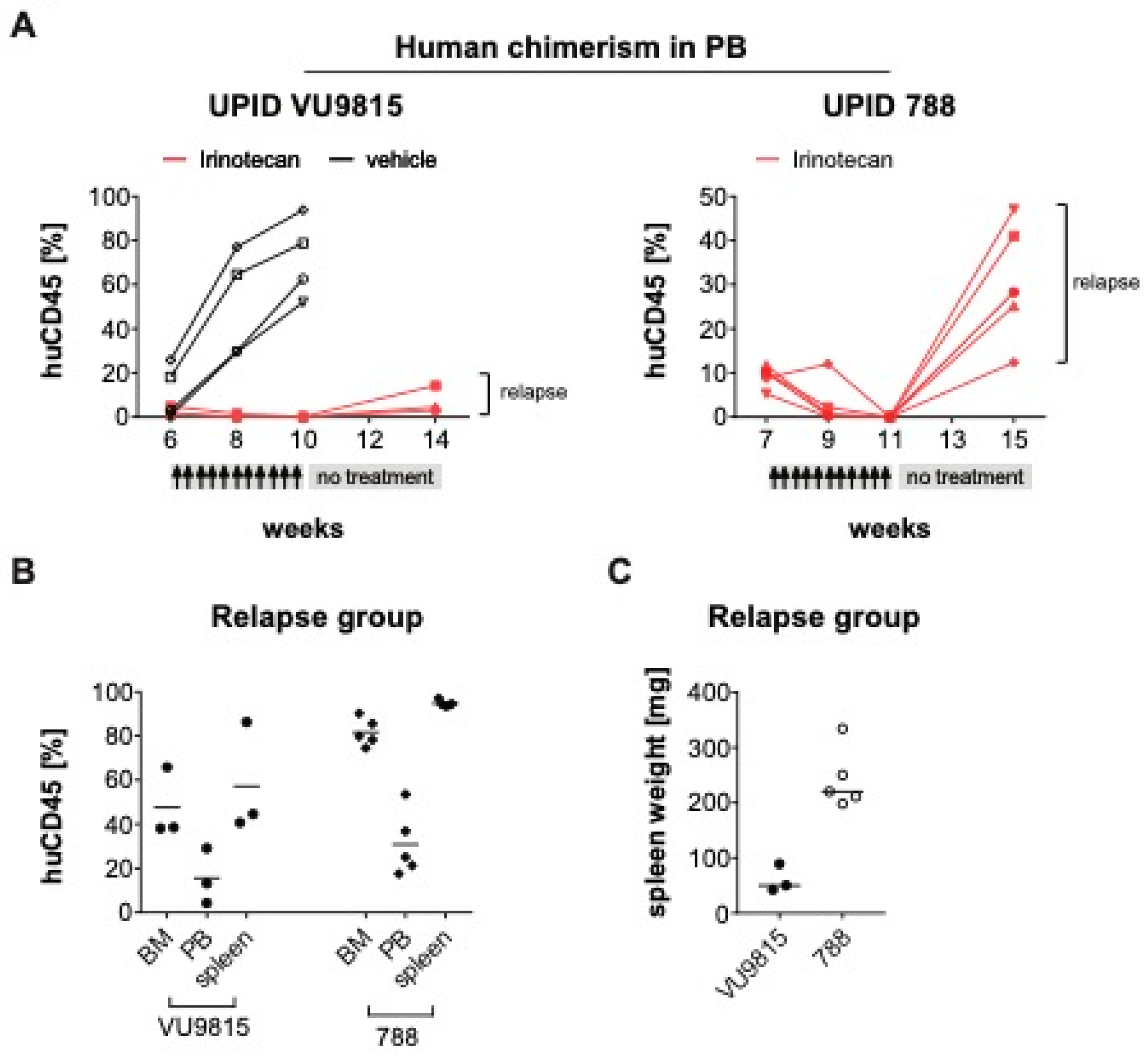
3.6. Irinotecan Shows Potent Anti-Leukemic Effects in MLL-Rearranged ALL Patient-Derived Xenograft Mouse Models
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Pieters, R.; Schrappe, M.; De Lorenzo, P.; Hann, I.; De Rossi, G.; Felice, M.; Hovi, L.; LeBlanc, T.; Szczepanski, T.; Ferster, A.; et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): An observational study and a multicentre randomised trial. Lancet 2007, 370, 240–250. [Google Scholar] [CrossRef]
- Pieters, R. Outcome of infants younger than 1 year with acute lymphoblastic leukemia treated with the inter-fant-06 protocol: Results from an international phase III randomized study. J. Clin. Oncol. 2019, 37, 2246–2256. [Google Scholar] [CrossRef]
- Tomizawa, D.; Koh, K.; Sato, T.; Kinukawa, N.; Morimoto, A.; Isoyama, K.; Kosaka, Y.; Oda, T.; Oda, M.; Hayashi, Y.; et al. Outcome of risk-based therapy for infant acute lymphoblastic leukemia with or without an MLL gene rearrangement, with emphasis on late effects: A final report of two consecutive studies, MLL96 and MLL98, of the Japan Infant Leukemia Study Group. Leukemia 2007, 21, 2258–2263. [Google Scholar] [CrossRef] [Green Version]
- Hunger, S.P.; Mullighan, C.G. Acute lymphoblastic leukemia in children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [Green Version]
- Teachey, D.T.; Pui, C.-H. Comparative features and outcomes between paediatric T-cell and B-cell acute lymphoblastic leukaemia. Lancet Oncol. 2019, 20, e142–e154. [Google Scholar] [CrossRef]
- Bernt, K.; Zhu, N.; Sinha, A.U.; Vempati, S.; Faber, J.; Krivtsov, A.V.; Feng, Z.; Punt, N.; Kedaigle, A.; Bullinger, L.; et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 2011, 20, 66–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjuan-Pla, A.; Bueno, C.; Prieto, C. Revisiting the biology of infant t(4;11)/MLL-AF4+ B-cell acute lymphoblastic leukemia. Blood 2015, 126, 2676–2685. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, S.A.; Staunton, J.E.; Silverman, L.B. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 2002, 30, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Stumpel, D.J.; Schneider, P.; van Roon, E.H. Specific promoter methylation identifies different subgroups of MLL-rearranged infant acute lymphoblastic leukemia, influences clinical outcome, and provides therapeutic options. Blood 2009, 114, 5490–5498. [Google Scholar] [CrossRef]
- Andersson, A.K.; Ma, J.; Wang, J. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Agraz-Doblas, A.; Bueno, C.; Bashford-Rogers, R.; Roy, A.; Schneider, P.; Bardini, M.; Ballerini, P.; Cazzaniga, G.; Moreno, T.; Revilla, C.; et al. Unraveling the cellular origin and clinical prognostic markers of infant B-cell acute lymphoblastic leukemia using genome-wide analysis. Haematologica 2019, 104, 1176–1188. [Google Scholar] [CrossRef] [PubMed]
- Tejedor, J.R.; Bueno, C.; Vinyoles, M.; Petazzi, P.; Agraz-Doblas, A.; Cobo, I.; Torres-Ruiz, R.; Bayón, G.F.; Pérez, R.F.; López-Tamargo, S.; et al. Integrative methylome-transcriptome analysis unravels cancer cell vulnerabilities in infant MLL-rearranged B-cell acute lymphoblastic leukemia. J. Clin. Investig. 2021. [Google Scholar] [CrossRef]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Stumpel, D.J.P.M.; Schneider, P.; Seslija, L.; Osaki, H.; Williams, O.; Pieters, R.; Stam, R.W. Connectivity mapping identifies HDAC inhibitors for the treatment of t(4;11)-positive infant acute lymphoblastic leukemia. Leukemia 2011, 26, 682–692. [Google Scholar] [CrossRef] [Green Version]
- Garrido Castro, P.; van Roon, E.H.J.; Pinhancos, S.S. The HDAC inhibitor panobinostat (LBH589) exerts in vivo an-ti-leukaemic activity against MLL-rearranged acute lymphoblastic leukaemia and involves the RNF20/RNF40/WAC-H2B ubiquitination axis. Leukemia 2017, 32, 323–331. [Google Scholar] [CrossRef]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.-I.; Robson, S.C.; Chung, C.-W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nat. Cell Biol. 2011, 478, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Bardini, M.; Trentin, L.; Rizzo, F.; Vieri, M.; Savino, A.M.; Castro, P.G.; Fazio, G.; Van Roon, E.H.; Kerstjens, M.; Smithers, N.N.; et al. Antileukemic efficacy of BET inhibitor in a preclinical mouse model of MLL-AF4+ infant all. Mol. Cancer Ther. 2018, 17, 1705–1716. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, K.A.; Roth, B.L. Finding new tricks for old drugs: An efficient route for public-sector drug discovery. Nat. Rev. Drug Discov. 2005, 4, 1005–1014. [Google Scholar] [CrossRef]
- Stam, R.W.; den Boer, M.L.; Schneider, P. Targeting FLT3 in primary MLL-gene-rearranged infant acute lymphoblastic leu-kemia. Blood 2005, 106, 2484–2490. [Google Scholar] [CrossRef]
- Stam, R.W.; den Boer, M.L.; Meijerink, J.P. Differential mRNA expression of Ara-C-metabolizing enzymes explains Ara-C sensitivity in MLL gene-rearranged infant acute lymphoblastic leukemia. Blood 2003, 101, 1270–1276. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y. Drugging topoisomerases: Lessons and challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef] [Green Version]
- Slatter, J.G.; Su, P.; Sams, J.P.; Schaaf, L.J.; Wienkers, L.C. Bioactivation of the anticancer agent CPT-11 to SN-38 by human hepatic microsomal carboxylesterases and the in vitro assessment of potential drug interactions. Drug Metab. Dispos. 1997, 25, 1157–1164. [Google Scholar]
- Kaneda, N.; Nagata, H.; Furuta, T.; Yokokura, T. Metabolism and pharmacokinetics of the camptothecin analogue CPT-11 in the mouse. Cancer Res. 1990, 50, 1715–1720. [Google Scholar]
- Kotecha, R.S.; Gottardo, N.G.; Kees, U.R.; Cole, C.H. The evolution of clinical trials for infant acute lymphoblastic leukemia. Blood Cancer J. 2014, 4, e200. [Google Scholar] [CrossRef] [Green Version]
- Corsello, S.M.; Roti, G.; Ross, K.N.; Chow, K.T.; Galinsky, I.; DeAngelo, D.J.; Stone, R.M.; Kung, A.L.; Golub, T.R.; Stegmaier, K. Identification of AML1-ETO modulators by chemical genomics. Blood 2009, 113, 6193–6205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roti, G.; Carlton, A.; Ross, K.N. Complementary genomic screens identify SERCA as a therapeutic target in NOTCH1 mutated cancer. Cancer Cell 2013, 23, 390–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiang, Y.H.; Lihou, M.G.; Liu, L.F. Arrest of replication forks by drug-stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res. 1989, 49, 5077–5082. [Google Scholar] [PubMed]
- Sakasai, R.; Teraoka, H.; Takagi, M.; Tibbetts, R. Transcription-dependent activation of ataxia telangiectasia mutated prevents DNA-dependent protein kinase-mediated cell death in response to topoisomerase I poison. J. Biol. Chem. 2010, 285, 15201–15208. [Google Scholar] [CrossRef] [Green Version]
- Furuta, T.; Takemura, H.; Liao, Z.Y. Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage com-plexes. J. Biol. Chem. 2003, 278, 20303–20312. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, S.; Huang, M.; Elledge, S.J. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998, 282, 1893–1897. [Google Scholar] [CrossRef]
- Houghton, P.J.; Cheshire, P.J.; Hallman, J.D. Efficacy of topoisomerase I inhibitors, topotecan and irinotecan, adminis-tered at low dose levels in protracted schedules to mice bearing xenografts of human tumors. Cancer Chemother. Pharmacol. 1995, 36, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, S.A.; Kung, A.L.; Mabon, M.E.; Silverman, L.B.; Stam, R.W.; Boer, M.L.D.; Pieters, R.; Kersey, J.H.; Sallan, S.E.; Fletcher, J.A.; et al. Inhibition of FLT3 in MLL: Validation of a therapeutic target identified by gene expression based classification. Cancer Cell 2003, 3, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Benito, J.M.; Godfrey, L.; Kojima, K.; Hogdal, L.; Wunderlich, M.; Geng, H.; Marzo, I.; Harutyunyan, K.G.; Golfman, L.; North, P.; et al. MLL-rearranged acute lymphoblastic leukemias activate BCL-2 through H3K79 methylation and are sensitive to the BCL-2-specific antagonist ABT-199. Cell Rep. 2015, 13, 2715–2727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borkin, D.; He, S.; Miao, H.; Kempinska, K.; Pollock, J.; Chase, J.; Purohit, T.; Malik, B.; Zhao, T.; Wang, J.; et al. Pharmacologic inhibition of the menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 2015, 27, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Richmond, J.; Carol, H.; Evans, K.; High, L.; Mendomo, A.; Robbins, A.; Meyer, C.; Venn, N.C.; Marschalek, R.; Henderson, M.; et al. Effective targeting of the P53–MDM2 axis in preclinical models of infant MLL-rearranged acute lymphoblastic leukemia. Clin. Cancer Res. 2015, 21, 1395–1405. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Yao, Y.; Zhou, C.; Chen, F.; Wu, F.; Wei, L.; Liu, W.; Dong, S.; Redell, M.; Mo, Q.; et al. Pharmacological inhibition of LSD1 for the treatment of MLL-rearranged leukemia. J. Hematol. Oncol. 2016, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Malik, B.; Borkin, D.; Miao, H.; Shukla, S.; Kempinska, K.; Purohit, T.; Wang, J.; Chen, L.; Parkin, B.; et al. Menin-MLL inhibitors block oncogenic transformation by MLL-fusion proteins in a fusion partner-independent manner. Leukemia 2015, 30, 508–513. [Google Scholar] [CrossRef] [Green Version]
- Jones, L.; Carol, H.; Evans, K.; Richmond, J.; Houghton, P.J.; Smith, M.A.; Lock, R.B. A review of new agents evaluated against pediatric acute lymphoblastic leukemia by the Pediatric Preclinical Testing Program. Leukemia 2016, 30, 2133–2141. [Google Scholar] [CrossRef] [Green Version]
- Frismantas, V.; Dobay, M.P.; Rinaldi, A.; Tchinda, J.; Dunn, S.H.; Kunz, J.; Richter-Pechanska, P.; Marovca, B.; Pail, O.; Jenni, S.; et al. Ex vivo drug response profiling detects recurrent sensitivity patterns in drug-resistant acute lymphoblastic leukemia. Blood 2017, 129, e26–e37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venditto, V.J.; Simanek, E.E. Cancer therapies utilizing the Camptothecins: A review of the in vivo literature. Mol. Pharm. 2010, 7, 307–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsh Rde, W.; Talamonti, M.S.; Katz, M.H.; Herman, J.M. Pancreatic cancer and FOLFIRINOX: A new era and new questions. Cancer Med. 2015, 4, 853–863. [Google Scholar] [CrossRef]
- Fujita, K.-I.; Kubota, Y.; Ishida, H.; Sasaki, Y. Irinotecan, a key chemotherapeutic drug for metastatic colorectal cancer. World J. Gastroenterol. 2015, 21, 12234–12248. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Beran, M.; Ellis, A. Phase I study of Topotecan, a new topoisomerase I inhibitor, in patients with refractory or relapsed acute leukemia. Blood 1993, 81, 1146–1151. [Google Scholar] [CrossRef]
- Prebet, T.; Jean, E.; Autret, A.; Charbonnier, A.; Rey, J.; Étienne, A.; D’Incan, E.; Fürst, S.; Arnoulet, C.; Blaise, D.; et al. Combination of cytarabine and topotecan in patients treated for acute myeloid leukemia with persistent disease after frontline induction. Leuk. Lymphoma 2012, 53, 2186–2191. [Google Scholar] [CrossRef] [PubMed]
- Wagner, L.M. Fifteen years of irinotecan therapy for pediatric sarcoma: Where to next? Clin. Sarcoma Res. 2015, 5, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Thompson, P.A.; Gupta, M.; Rosner, G.L.; Yu, A.; Barrett, J.; Bomgaars, L.; Bernstein, M.L.; Blaney, S.M.; Mondick, J. Pharmacokinetics of irinotecan and its metabolites in pediatric cancer patients: A report from the children’s oncology group. Cancer Chemother. Pharmacol. 2008, 62, 1027–1037. [Google Scholar] [CrossRef]
- Na, Y.-S.; Jung, K.-A.; Kim, S.-M.; Hong, Y.S.; Ryu, M.-H.; Jang, S.J.; Moon, D.H.; Cho, D.-H.; Kim, J.C.; Lee, J.S.; et al. The histone deacetylase inhibitor PXD101 increases the efficacy of irinotecan in in vitro and in vivo colon cancer models. Cancer Chemother. Pharmacol. 2010, 68, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.-S.; Kim, S.-M.; Jung, K.-A.; Yang, S.-J.; Hong, Y.S.; Ryu, M.-H.; Ro, S.; Cho, D.-H.; Kim, J.C.; Jin, D.-H.; et al. Effects of the HDAC inhibitor CG2 in combination with irinotecan, 5-fluorouracil, or oxaliplatin on HCT116 colon cancer cells and xenografts. Oncol. Rep. 2010, 24, 1509–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarcar, B.; Kahali, S.; Chinnaiyan, P. Vorinostat enhances the cytotoxic effects of the topoisomerase I inhibitor SN38 in glioblas-toma cell lines. J. Neuro-Oncol. 2010, 99, 201–207. [Google Scholar] [CrossRef]
- Sampson, V.B.; Vetter, N.S.; Kamara, D.F.; Collier, A.B.; Gresh, R.C.; Kolb, E.A. Vorinostat enhances cytotoxicity of SN-38 and Te-mozolomide in ewing sarcoma cells and activates STAT3/AKT/MAPK pathways. PLoS ONE 2015, 10, e0142704. [Google Scholar] [CrossRef]
- Bruzzese, F.; Rocco, M.; Castelli, S.; Di Gennaro, E.; Desideri, A.; Budillon, A. Synergistic antitumor effect between vorinostat and topotecan in small cell lung cancer cells is mediated by generation of reactive oxygen species and DNA damage-induced apoptosis. Mol. Cancer Ther. 2009, 8, 3075–3087. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kerstjens, M.; Garrido Castro, P.; Pinhanços, S.S.; Schneider, P.; Wander, P.; Pieters, R.; Stam, R.W. Irinotecan Induces Disease Remission in Xenograft Mouse Models of Pediatric MLL-Rearranged Acute Lymphoblastic Leukemia. Biomedicines 2021, 9, 711. https://doi.org/10.3390/biomedicines9070711
Kerstjens M, Garrido Castro P, Pinhanços SS, Schneider P, Wander P, Pieters R, Stam RW. Irinotecan Induces Disease Remission in Xenograft Mouse Models of Pediatric MLL-Rearranged Acute Lymphoblastic Leukemia. Biomedicines. 2021; 9(7):711. https://doi.org/10.3390/biomedicines9070711
Chicago/Turabian StyleKerstjens, Mark, Patricia Garrido Castro, Sandra S. Pinhanços, Pauline Schneider, Priscilla Wander, Rob Pieters, and Ronald W. Stam. 2021. "Irinotecan Induces Disease Remission in Xenograft Mouse Models of Pediatric MLL-Rearranged Acute Lymphoblastic Leukemia" Biomedicines 9, no. 7: 711. https://doi.org/10.3390/biomedicines9070711
APA StyleKerstjens, M., Garrido Castro, P., Pinhanços, S. S., Schneider, P., Wander, P., Pieters, R., & Stam, R. W. (2021). Irinotecan Induces Disease Remission in Xenograft Mouse Models of Pediatric MLL-Rearranged Acute Lymphoblastic Leukemia. Biomedicines, 9(7), 711. https://doi.org/10.3390/biomedicines9070711