
Cold Exposure Exacerbates Cardiac Dysfunction in a Model of Heart Failure with Preserved Ejection Fraction in Male and Female C57Bl/6J Mice

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Metabolic and Hypertensive Stress (MHS) Protocol
2.3. Echocardiography
2.4. Body Composition
2.5. Myocardial Fibrosis Evaluation
2.6. Cardiomyocyte Cross-Sectional Area
2.7. RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction
2.8. BAT Histology
2.9. UCP1 Immunohistochemistry
2.10. Statistical Analysis
3. Results
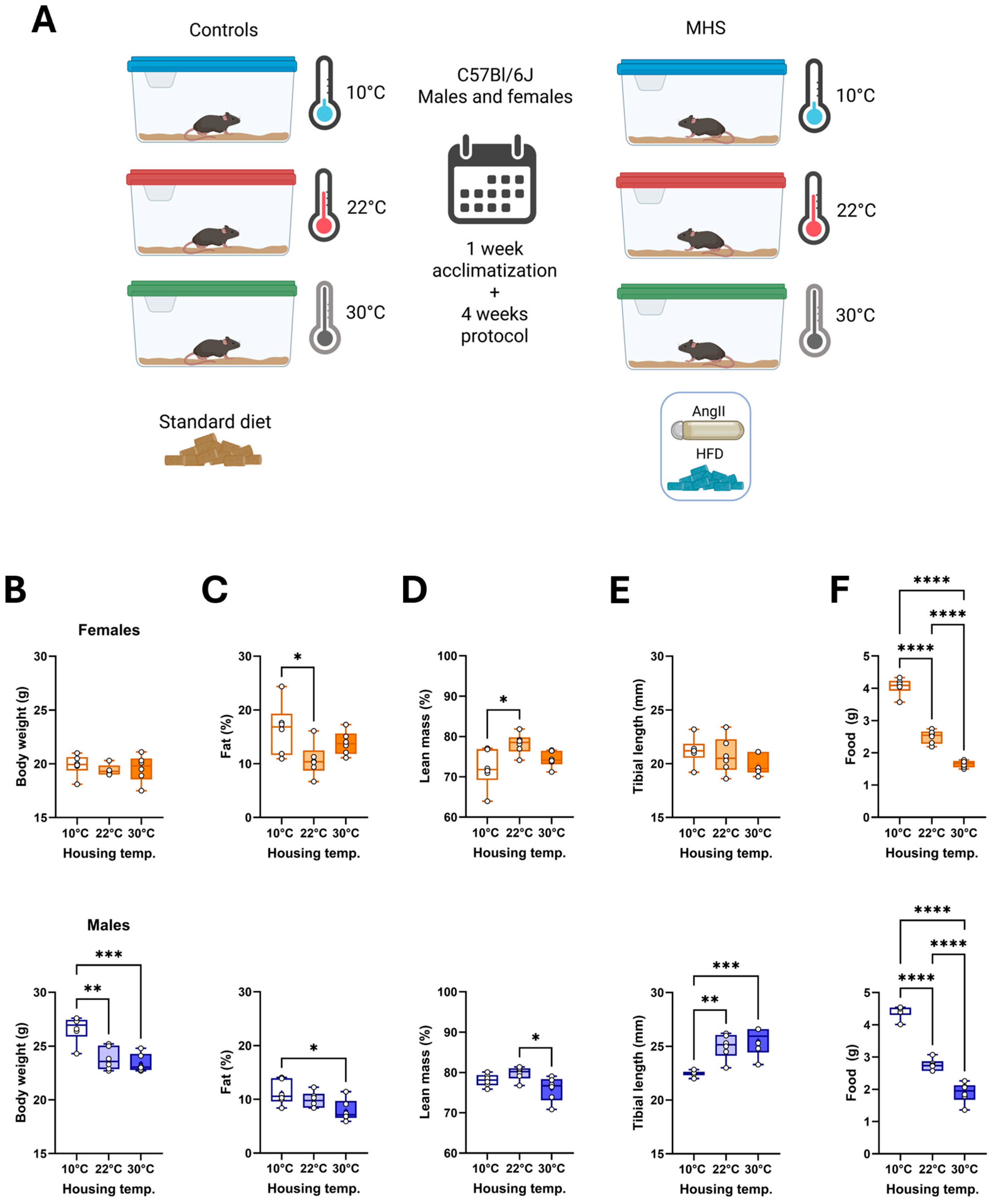
3.1. Different Housing Temperatures Result in Sex-Specific Adaptations in C57Bl6/J Mice
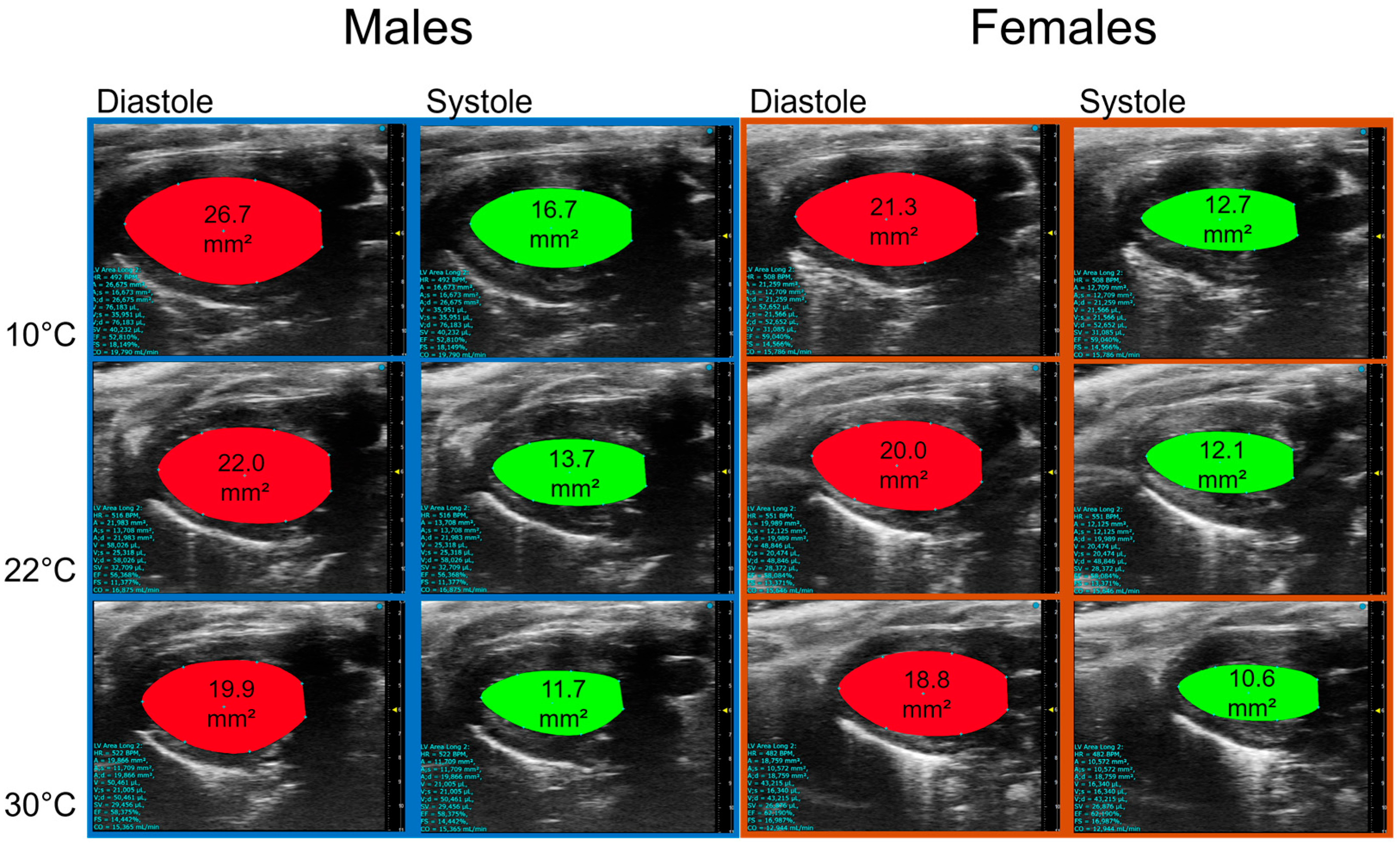
3.2. Housing Temperature Affects Cardiac Morphology and Function
3.3. Thermoneutrality (30 °C) Reduces the Cardiac Hypertrophic Response to MHS
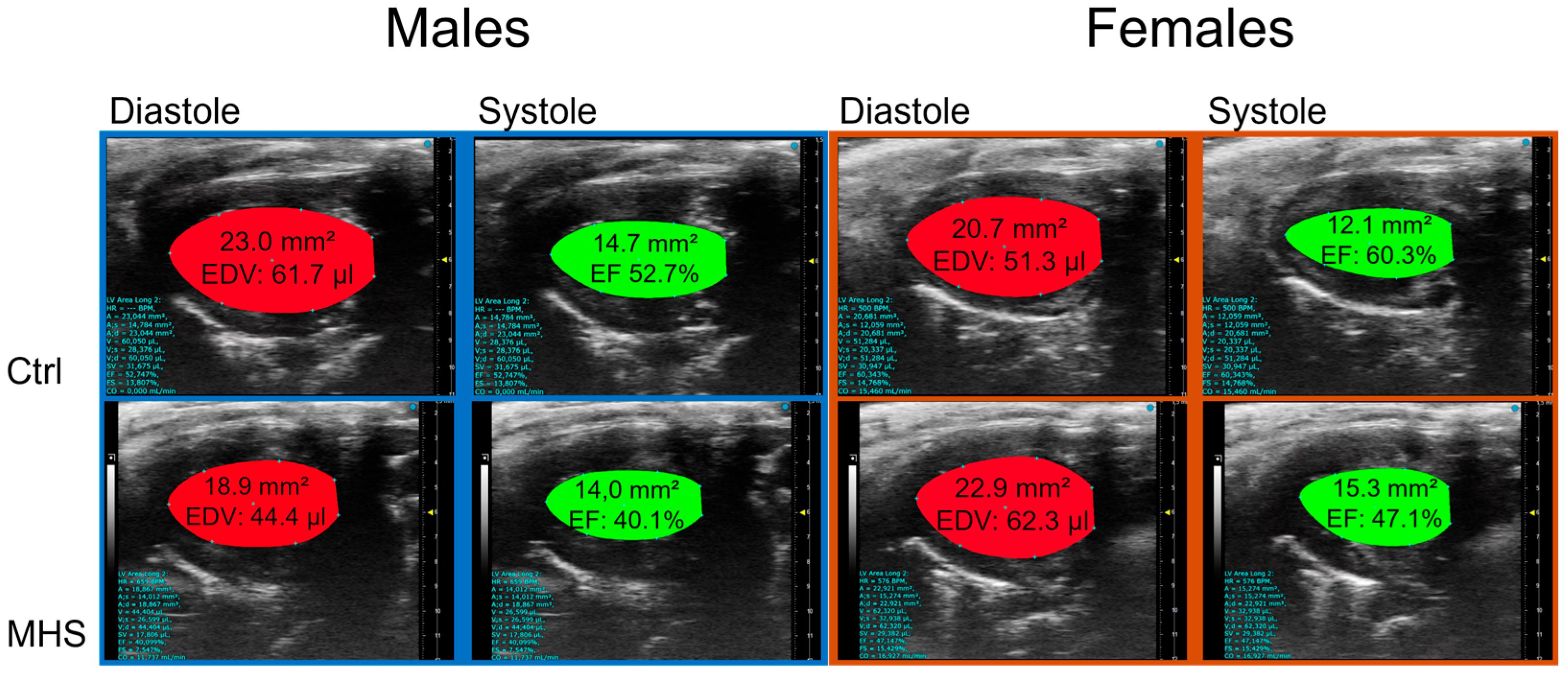
3.4. Cold Exacerbates Diastolic Dysfunction After MHS and Causes a Loss of Ejection Fraction
3.5. Housing Temperature Modulates Myocardial Hypertrophy and Fibrosis Marker Genes After MHS
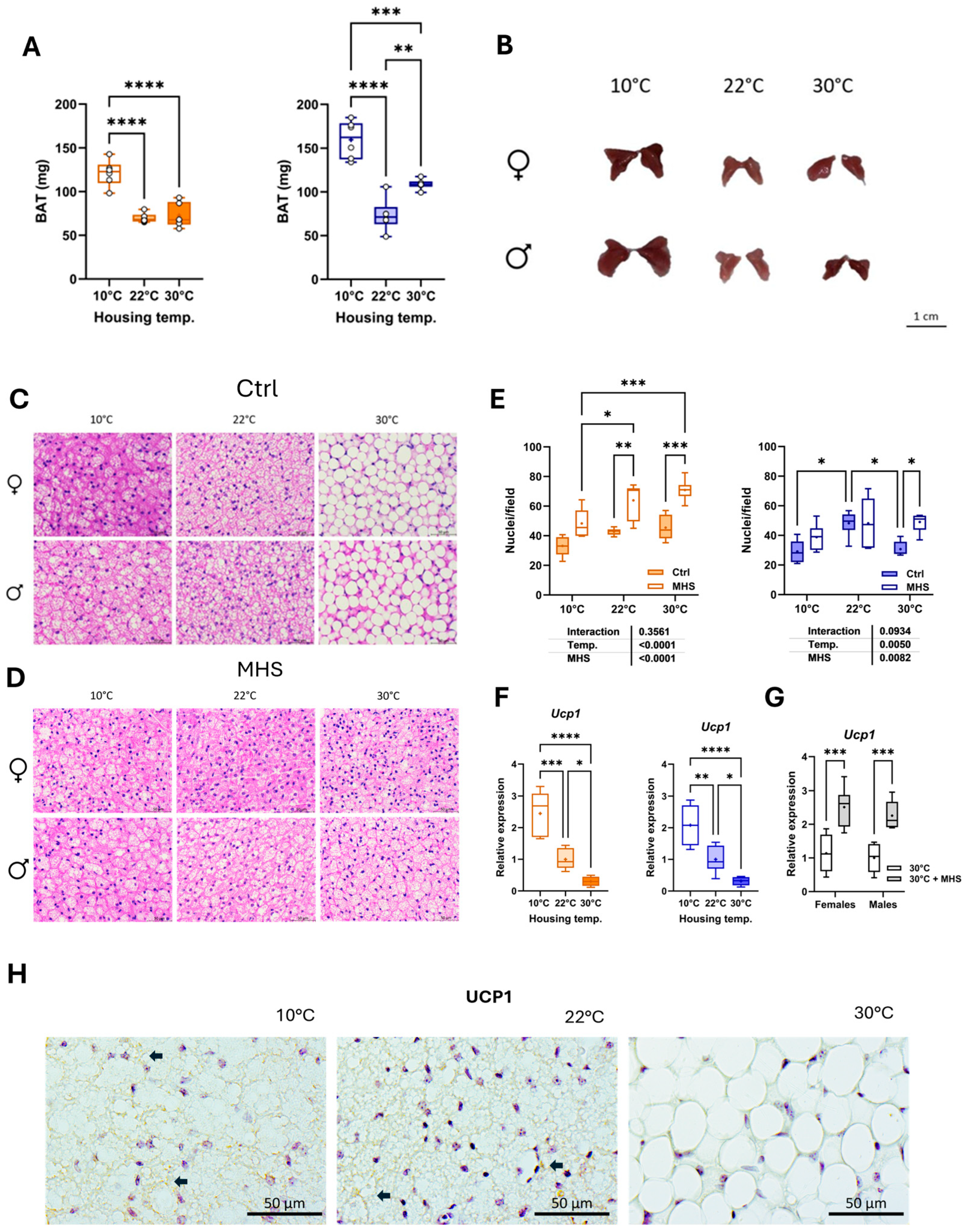
3.6. Cold Stress Activates BAT, and MHS Induces BAT Browning at Thermoneutrality
4. Discussion
5. Study Limitations
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HFpEF | Heart failure with preserved ejection fraction |
| AngII | Angiotensin II |
| MHS | Metabolic and hypertensive stress (AngII + HFD) |
| HFD | High-fat diet |
| LA | Left atrial or left atrium |
| LV | Left ventricle |
| DOCA | Deoxycorticosterone acetate |
| CIH | Cold-induced hypertension |
| CICH | Cold-induced cardiac hypertrophy |
| CH | Cardiac hypertrophy |
| EF | Ejection fraction |
| UCP1 | Uncoupled protein 1 |
| ECM | Extracellular matrix |
| EDD | End-diastolic diameter |
| ESD | End-systolic diameter |
| RWT | Relative wall thickness |
| SV | Stroke volume |
| CO | Cardiac output |
| BW | Body weight |
| CSA | Cross-sectional area |
| FGF21 | Fibroblast growth factor 21 |
| 12-HOME | 2,13-double bond of octadeca-9,12-dienoic acid |
| L-NAME | L-NG-Nitroarginine Methyl Ester |
| BP | Blood pressure |
| EDV | End-diastolic volume |
| ESV | End-systolic volume |
| Nppa | Atrial natriuretic peptide |
| Nppb | Brain natriuretic peptide |
| Col1a | Pro-collagen1 alpha |
| Col3a | Pro-collagen3 alpha |
| Postn | Periostin |
| Thbs4 | Thrombospondin 4 |
| PW | Posterior wall |
| IVSW | Interventricular septal wall |
| SEM | Standard error of the mean |
| RWT | Relative LV wall thickness |
| Ppia | Cyclophilin A |
| RPL13 | L13 ribosomal protein |
| eNOS | Nitric oxide synthase, endothelial |
| iNOS | Nitric oxide synthase, inducible |
| TAC | Transverse aortic constriction |
| AT2R | Angiotensin II receptor, type 2 |
| β3-AR | Adrenergic receptor, beta 3 |
| BAT | Brown adipose tissue |
| RAAS | Renin–angiotensin–aldosterone system |
| SNS | Sympathetic nervous system |
References
- Smith, A.N.; Altara, R.; Amin, G.; Habeichi, N.J.; Thomas, D.G.; Jun, S.; Kaplan, A.; Booz, G.W.; Zouein, F.A. Genomic, Proteomic, and Metabolic Comparisons of Small Animal Models of Heart Failure With Preserved Ejection Fraction: A Tale of Mice, Rats, and Cats. J. Am. Hear. Assoc. 2022, 11, e026071. [Google Scholar] [CrossRef] [PubMed]
- Withaar, C.; Lam, C.S.P.; Schiattarella, G.G.; A de Boer, R.; Meems, L.M.G. Heart failure with preserved ejection fraction in humans and mice: Embracing clinical complexity in mouse models. Eur. Hear. J. 2021, 42, 4420–4430. [Google Scholar] [CrossRef]
- Jasińska-Stroschein, M. Searching for Effective Treatments in HFpEF: Implications for Modeling the Disease in Rodents. Pharmaceuticals 2023, 16, 1449. [Google Scholar] [CrossRef]
- Gao, S.; Liu, X.-P.; Li, T.-T.; Chen, L.; Feng, Y.-P.; Wang, Y.-K.; Yin, Y.-J.; Little, P.J.; Wu, X.-Q.; Xu, S.-W.; et al. Animal models of heart failure with preserved ejection fraction (HFpEF): From metabolic pathobiology to drug discovery. Acta Pharmacol. Sin. 2023, 45, 23–35. [Google Scholar] [CrossRef]
- Reitman, M.L. Of mice and men–environmental temperature, body temperature, and treatment of obesity. FEBS Lett. 2018, 592, 2098–2107. [Google Scholar] [CrossRef]
- James, C.M.; Olejniczak, S.H.; Repasky, E.A. How murine models of human disease and immunity are influenced by housing temperature and mild thermal stress. Temperature 2022, 10, 166–178. [Google Scholar] [CrossRef]
- Keijer, J.; Li, M.; Speakman, J.R. What is the best housing temperature to translate mouse experiments to humans? Mol. Metab. 2019, 25, 168–176. [Google Scholar] [CrossRef]
- Hankenson, F.C.; O Marx, J.; Gordon, C.J.; David, J.M. Effects of Rodent Thermoregulation on Animal Models in the Research Environment. Comp. Med. 2018, 68, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Kowaltowski, A.J. Cold Exposure and the Metabolism of Mice, Men, and Other Wonderful Creatures. Physiology 2022, 37, 253–259. [Google Scholar] [CrossRef]
- Chaudhary, R.; Suhan, T.K.; Wu, C.; Alzamrooni, A.; Madamanchi, N.; Abdel-Latif, A. Housing temperature influences metabolic phenotype of heart failure with preserved ejection fraction in J vs N strain C57BL/6 mice. Mol. Cell. Endocrinol. 2025, 598, 112457. [Google Scholar] [CrossRef] [PubMed]
- McLeod, K.; Datta, V.; Fuller, S. Adipokines as Cardioprotective Factors: BAT Steps Up to the Plate. Biomedicines 2025, 13, 710. [Google Scholar] [CrossRef]
- Sun, Z. Cardiovascular responses to cold exposure. Front. Biosci. 2010, 2, 495–503. [Google Scholar] [CrossRef]
- Aidara, M.L.; Walsh-Wilkinson, É.; Thibodeau, S.; Labbé, E.-A.; Morin-Grandmont, A.; Gagnon, G.; Boudreau, D.K.; Arsenault, M.; Bossé, Y.; Couët, J. Cardiac reverse remodeling in a mouse model with many phenotypical features of heart failure with preserved ejection fraction: Effects of modifying lifestyle. Am. J. Physiol. Circ. Physiol. 2024, 326, H1017–H1036. [Google Scholar] [CrossRef]
- Walsh-Wilkinson, É.; Aidara, M.L.; Morin-Grandmont, A.; Thibodeau, S.; Gagnon, J.; Genest, M.; Arsenault, M.; Couet, J. Age and sex hormones modulate left ventricle regional response to angiotensin II in male and female mice. Am. J. Physiol. Circ. Physiol. 2022, 323, H643–H658. [Google Scholar] [CrossRef] [PubMed]
- Walsh-Wilkinson, E.; Arsenault, M.; Couet, J. Segmental analysis by speckle-tracking echocardiography of the left ventricle response to isoproterenol in male and female mice. Peer J. 2021, 9, e11085. [Google Scholar] [CrossRef]
- Labbé, E.-A.; Thibodeau, S.; Walsh-Wilkinson, É.; Chalifour, M.; Sirois, P.-O.; Leblanc, J.; Morin-Grandmont, A.; Arsenault, M.; Couet, J. Relative contribution of correcting the diet and voluntary exercise to myocardial recovery in a two-hit murine model of heart failure with preserved ejection fraction. Am. J. Physiol. Circ. Physiol. 2025, 329, H51–H68. [Google Scholar] [CrossRef]
- Zhao, Z.; Yang, R.; Li, M.; Bao, M.; Huo, D.; Cao, J.; Speakman, J.R. Effects of ambient temperatures between 5 and 35 °C on energy balance, body mass and body composition in mice. Mol. Metab. 2022, 64, 101551. [Google Scholar] [CrossRef]
- Richard, D.; Labrie, A.; Rivest, S. Tissue specificity of SNS response to exercise in mice exposed to low temperatures. Am. J. Physiol. Integr. Comp. Physiol. 1992, 262, R921–R925. [Google Scholar] [CrossRef] [PubMed]
- Papanek, P.E.; Wood, C.E.; Fregly, M.J. Role of the sympathetic nervous system in cold-induced hypertension in rats. J. Appl. Physiol. 1991, 71, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Dejima, Y.; Fukuda, S.; Ichijoh, Y.; Takasaka, K.; Ohtsuka, R. Cold-Induced Salt Intake in Mice and Catecholamine, Renin and Thermogenesis Mechanisms. Appetite 1996, 26, 203–220. [Google Scholar] [CrossRef]
- Sun, Z.; Fregly, M.J.; Cade, J.R. Effect of renal denervation on elevation of blood pressure in cold-exposed rats. Can. J. Physiol. Pharmacol. 1995, 73, 72–78. [Google Scholar] [CrossRef]
- Sun, Z.; Cade, R.; Zhang, Z.; Alouidor, J.; Van, H. Angiotensinogen Gene Knockout Delays and Attenuates Cold-Induced Hypertension. Hypertension 2003, 41, 322–327. [Google Scholar] [CrossRef]
- Chen, G.-F.; Sun, Z. Effects of chronic cold exposure on the endothelin system. J. Appl. Physiol. 2006, 100, 1719–1726. [Google Scholar] [CrossRef] [PubMed]
- Axsom, J.E.; Nanavati, A.P.; Rutishauser, C.A.; Bonin, J.E.; Moen, J.M.; Lakatta, E.G. Acclimation to a thermoneutral environment abolishes age-associated alterations in heart rate and heart rate variability in conscious, unrestrained mice. GeroScience 2019, 42, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Fregly, M.J. Activity of the hypothalamic-pituitary-thyroid axis during exposure to cold. Pharmacol. Ther. 1989, 41, 85–142. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.O.; Carvalho, S.D.; Schultz, J.J.; Chiellini, G.; Scanlan, T.S.; Bianco, A.C.; Brent, G.A. Thyroid hormone-sympathetic interaction and adaptive thermogenesis are thyroid hormone receptor isoform-specific. J. Clin. Investig. 2001, 108, 97–105. [Google Scholar] [CrossRef]
- Fregly, M.J.; Rossi, F.; Cade, J.R. A role for thyroid hormones in cold-induced elevation of blood pressure and cardiac hypertrophy. Can. J. Physiol. Pharmacol. 1994, 72, 1066–1074. [Google Scholar] [CrossRef]
- Maillet, M.; van Berlo, J.H.; Molkentin, J.D. Molecular basis of physiological heart growth: Fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 2012, 14, 38–48. [Google Scholar] [CrossRef]
- Wang, X.; Skelley, L.; Wang, B.; Mejia, A.; Sapozhnikov, V.; Sun, Z. AAV-Based RNAi Silencing of NADPH Oxidase gp91phox Attenuates Cold-Induced Cardiovascular Dysfunction. Hum. Gene Ther. 2012, 23, 1016–1026. [Google Scholar] [CrossRef]
- Chen, P.G.-F.; Sun, Z. AAV Delivery of Endothelin-1 shRNA Attenuates Cold-Induced Hypertension. Hum. Gene Ther. 2017, 28, 190–199. [Google Scholar] [CrossRef]
- Kong, X.; Liu, H.; He, X.; Sun, Y.; Ge, W. Unraveling the Mystery of Cold Stress-Induced Myocardial Injury. Front. Physiol. 2020, 11, 580811. [Google Scholar] [CrossRef]
- Portes, A.M.O.; Paula, A.B.R.; de Miranda, D.C.; Resende, L.T.; Coelho, B.I.C.; Teles, M.C.; Jardim, I.A.B.A.; Natali, A.J.; Castrucci, A.M.d.L.; Isoldi, M.C. A systematic review of the effects of cold exposure on pathological cardiac remodeling in mice. J. Therm. Biol. 2023, 114, 103598. [Google Scholar] [CrossRef]
- Burns, M.P.A.; Reges, C.R.; Barnhill, S.W.; Koehler, K.N.; Lewis, B.C.; Colombo, A.T.; Felter, N.J.; Schaeffer, P.J. Chronic cold exposure causes left ventricular hypertrophy that appears to be physiological. J. Exp. Biol. 2024, 227, jeb247476. [Google Scholar] [CrossRef]
- Lu, S.; Xu, D. Cold stress accentuates pressure overload-induced cardiac hypertrophy and contractile dysfunction: Role of TRPV1/AMPK-mediated autophagy. Biochem. Biophys. Res. Commun. 2013, 442, 8–15. [Google Scholar] [CrossRef]
- Teou, D.C.; Labbé, E.-A.; Thibodeau, S.; Walsh-Wilkinson, É.; Morin-Grandmont, A.; Trudeau, A.-S.; Arsenault, M.; Couet, J. The Loss of Gonadal Hormones Has a Different Impact on Aging Female and Male Mice Submitted to Heart Failure-Inducing Metabolic Hypertensive Stress. Cells 2025, 14, 870. [Google Scholar] [CrossRef] [PubMed]
- Blenck, C.L.; Harvey, P.A.; Reckelhoff, J.F.; Leinwand, L.A. The Importance of Biological Sex and Estrogen in Rodent Models of Cardiovascular Health and Disease. Circ. Res. 2016, 118, 1294–1312. [Google Scholar] [CrossRef]
- Swirski, F.K.; Nahrendorf, M. Cardioimmunology: The immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 733–744. [Google Scholar] [CrossRef]
- Rodríguez-Cuenca, S.; Pujol, E.; Justo, R.; Frontera, M.; Oliver, J.; Gianotti, M.; Roca, P. Sex-dependent Thermogenesis, Differences in Mitochondrial Morphology and Function, and Adrenergic Response in Brown Adipose Tissue. J. Biol. Chem. 2002, 277, 42958–42963. [Google Scholar] [CrossRef] [PubMed]
- Benz, V.; Bloch, M.; Wardat, S.; Böhm, C.; Maurer, L.; Mahmoodzadeh, S.; Wiedmer, P.; Spranger, J.; Foryst-Ludwig, A.; Kintscher, U.; et al. Sexual Dimorphic Regulation of Body Weight Dynamics and Adipose Tissue Lipolysis. PLoS ONE 2012, 7, e37794. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.-H.; Doria, A.; et al. Identification and Importance of Brown Adipose Tissue in Adult Humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef]
- Pfannenberg, C.; Werner, M.K.; Ripkens, S.; Stef, I.; Deckert, A.; Schmadl, M.; Reimold, M.; Häring, H.-U.; Claussen, C.D.; Stefan, N. Impact of Age on the Relationships of Brown Adipose Tissue With Sex and Adiposity in Humans. Diabetes 2010, 59, 1789–1793. [Google Scholar] [CrossRef]
- Pedersen, S.B.; Bruun, J.M.; Kristensena, K.; Richelsen, B. Regulation of UCP1, UCP2, and UCP3 mRNA Expression in Brown Adipose Tissue, White Adipose Tissue, and Skeletal Muscle in Rats by Estrogen. Biochem. Biophys. Res. Commun. 2001, 288, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Cannon, B.; Nedergaard, J. Brown Adipose Tissue: Function and Physiological Significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef]
- Lin, J.-R.; Ding, L.-L.; Xu, L.; Huang, J.; Zhang, Z.-B.; Chen, X.-H.; Cheng, Y.-W.; Ruan, C.-C.; Gao, P.-J. Brown Adipocyte ADRB3 Mediates Cardioprotection via Suppressing Exosomal iNOS. Circ. Res. 2022, 131, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Bollinger, E.; Cunio, T.; Damilano, F.; Stansfield, J.C.; Pinkus, C.A.; Kreuser, S.; Hirenallur-Shanthappa, D.; Flach, R.J.R. An assessment of thermoneutral housing conditions on murine cardiometabolic function. Am. J. Physiol. Circ. Physiol. 2022, 322, H234–H245. [Google Scholar] [CrossRef]
- Alvarez-Gallego, F.; González-Blázquez, R.; Gil-Ortega, M.; Somoza, B.; Calderón-Dominguez, M.; Moratinos, J.; Garcia-Garcia, V.; Fernández, P.; González-Moreno, D.; Viana, M.; et al. Angiotensin II type 2 receptor as a novel activator of brown adipose tissue in obesity. BioFactors 2023, 49, 1106–1120. [Google Scholar] [CrossRef] [PubMed]
- Gozalo, A.S.; Elkins, W.R. A Review of the Effects of Some Extrinsic Factors on Mice Used in Research. Comp. Med. 2023, 73, 413–431. [Google Scholar] [CrossRef]
- Balcombe, J.P. Laboratory environments and rodents’ behavioural needs: A review. Lab. Anim. 2006, 40, 217–235. [Google Scholar] [CrossRef]
- Nag, S.; Patel, S.; Mani, S.; Hussain, T. Role of angiotensin type 2 receptor in improving lipid metabolism and preventing adiposity. Mol. Cell. Biochem. 2019, 461, 195–204. [Google Scholar] [CrossRef]
- Nag, S.; Khan, M.A.; Samuel, P.; Ali, Q.; Hussain, T. Chronic angiotensin AT2R activation prevents high-fat diet-induced adiposity and obesity in female mice independent of estrogen. Metabolism 2015, 64, 814–825. [Google Scholar] [CrossRef]
- Than, A.; Xu, S.; Li, R.; Leow, M.-S.; Sun, L.; Chen, P. Angiotensin type 2 receptor activation promotes browning of white adipose tissue and brown adipogenesis. Signal Transduct. Target. Ther. 2017, 2, 17022. [Google Scholar] [CrossRef]
- Fan, J.-F.; Xiao, Y.-C.; Feng, Y.-F.; Niu, L.-Y.; Tan, X.; Sun, J.-C.; Leng, Y.-Q.; Li, W.-Y.; Wang, W.-Z.; Wang, Y.-K. A systematic review and meta-analysis of cold exposure and cardiovascular disease outcomes. Front. Cardiovasc. Med. 2023, 10, 1084611. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Description | Forward Sequence Reverse Sequence |
|---|---|---|
| Col1a1 | Collagen Type I Alpha 1 Chain | 5′-CAT TGT GTA TGC AGC TGA CTT C-3′ 5′CGC AAA CAC TCT ACA TGT CTA GG-3′ |
| Col3a1 | Collagen Type III Alpha 1 Chain | 5′-TCT CTA GAC TCA TAG GAC TGA CC-3′ 5′ TTC TTC TCA CCC TTC TTC ATC C-3′ |
| Nppa | Natriuretic Peptide B | 5′-CTC CTT GGC TGT TAT CTT CGG-3′ 5′-GGG TAG GAT TGA CAG GAT TGG-3′ |
| Nppb | Natriuretic Peptide A | 5′-AGG TGA CAC ATA TCT CAA GCT G-3′ 5′-CTT CCT ACA ACA TCA GTG C-3′ |
| Ppia | Cyclophilin A | 5′-TTC ACC TTC CCA AAG ACC AC-3′ 5′-CAA ACA CAA ACG GTT CCC AG-3′ |
| Postn | Periostin | 5′-GCT TTC GAG AAA CTG CCA CG-3′ 5′-ATG GTC TCA AAC ACG GCT CC-3′ |
| Thbs4 | Thrombospondin 4 | 5′-GAT ACT GAC GGG GAT GGG AG-3′ 5′-CGT CAC TGT CTT GGT TGG TG-3′ |
| Ucp1 | Uncoupled protein 1 | 5′-GCT TCT ACG ACT CAG TCC AA-3′ 5′-CTC TGG GCT TGC ATT CTG AC-3′ |
| Males | ||||||
|---|---|---|---|---|---|---|
| Parameters | C10 (n = 6) | M10 (n = 6) | C22 (n = 6) | M22 (n = 6) | C30 (n = 6) | M30 (n = 6) |
| PWd, mm | 0.85 ± 0.012 | 1.11 ± 0.035 d | 0.78 ± 0.017 | 1.05 ± 0.032 d | 0.76 ± 0.011 | 1.10 ± 0.038 d |
| IVSd, mm | 0.76 ± 0.010 | 0.93 ± 0.024 d | 0.71 ± 0.017 | 0.94 ± 0.043 c | 0.67 ± 0.019 | 0.89 ± 0.027 d |
| EDD, mm | 4.29 ± 0.101 | 4.12 ± 0.142 | 3.90 ± 0.057 | 3.54 ± 0.092 b | 3.73 ± 0.045 | 3.22 ± 0.091 c |
| ESD, mm | 3.25 ± 0.104 | 3.18 ± 0.128 | 2.93 ± 0.088 | 2.42 ± 0.116 b | 2.57 ± 0.051 | 2.09 ± 0.102 b |
| RWT | 0.38 ± 0.009 | 0.50 ± 0.031 b | 0.38 ± 0.012 | 0.57 ± 0.032 c | 0.38 ± 0.008 | 0.62 ± 0.023 d |
| LV mass, mg | 133 ± 5.2 | 171 ± 3.8 d | 103 ± 2.6 | 132 ± 5.0 c | 90 ± 3.1 | 115 ± 8.7 a |
| EDV, µL | 63 ± 3.2 | 53 ± 2.6 a | 51 ± 1.7 | 39 ± 2.5 b | 49 ± 1.9 | 37 ± 2.5 b |
| ESV, µL | 31 ± 1.9 | 29 ± 1.6 | 23 ± 1.4 | 17 ± 2.0 b | 19 ± 1.3 | 14 ± 1.7 a |
| SV, mm | 32 ± 1.9 | 24 ± 1.8 b | 29 ± 0.7 | 22 ± 1.3 c | 30 ± 1.1 | 23 ± 1.1 b |
| HR, bpm | 518 ± 14.5 | 579 ± 13.0 | 461 ± 16.4 | 495 ± 4.5 | 503 ± 14.1 | 532 ± 13.3 |
| EF, % | 52 ± 1.8 | 44 ± 1.8 a | 56 ± 1.6 | 58 ± 1.1 | 62 ± 1.6 | 63 ± 1.4 |
| CO, mL/min | 16.9 ± 0.93 | 13.7 ± 0.94 a | 13.9 ± 0.69 | 9.8 ± 0.75 a | 14.4 ± 0.71 | 12.4 ± 0.54 |
| Females | ||||||
| Parameters | C10 (n = 6) | M10 (n = 6) | C22(n = 6) | M22(n = 6) | C30(n = 6) | M30(n = 6) |
| PWd, mm | 0.85 ± 0.017 | 1.11 ± 0.043 c | 0.72 ± 0.009 | 0.96 ± 0.034 d | 0.72 ± 0.009 | 0.89 ± 0.018 d |
| IVSd, mm | 0.73 ± 0.097 | 0.96 ± 0.037 c | 0.70 ± 0.011 | 0.87 ± 0.034 c | 0.67 ± 0.011 | 0.88 ± 0.030 d |
| EDD, mm | 3.98 ± 0.020 | 3.89 ± 0.067 | 3.73 ± 0.037 | 3.75 ± 0.148 | 3.51 ± 0.025 | 3.27 ± 0.050 b |
| ESD, mm | 2.88 ± 0.098 | 3.02 ± 0.090 | 2.67 ± 0.054 | 2.67 ± 0.054 | 2.30 ± 0.030 | 2.15 ± 0.044 b |
| RWT | 0.40 ± 0.014 | 0.53 ± 0.024 c | 0.38 ± 0.006 | 0.50 ± 0.034 | 0.40 ± 0.004 | 0.54 ± 0.015 d |
| LV mass, mg | 115 ± 4.8 | 161 ± 8.8 b | 89 ± 2.2 | 127 ± 3.9 d | 78 ± 2.0 | 98 ± 4.1 b |
| EDV, µL | 55 ± 1.6 | 52 ± 2.4 | 43 ± 2.7 | 50 ± 3.3 | 38 ± 1.5 | 34 ± 2.25 |
| ESV, µL | 22 ± 0.8 | 27 ± 1.1 b | 18 ± 1.3 | 24 ± 2.0 a | 14 ± 0.8 | 12 ± 1.3 |
| SV, mm | 33 ± 1.9 | 26 ± 1.9 a | 26 ± 1.6 | 26 ± 1.8 | 24 ± 1.1 | 21 ± 1.1 |
| HR, bpm | 542 ± 20.1 | 531 ± 15.3 | 486 ± 12.1 | 515 ± 11.5 | 508 ± 14.8 | 513 ± 8.4 |
| EF, % | 60 ± 2.0 | 49 ± 3.2 a | 59 ± 1.3 | 59 ± 1.1 | 63 ± 1.4 | 64 ± 2.0 |
| CO, mL/min | 17.8 ± 1.12 | 13.7 ± 1.09 a | 12.5 ± 0.90 | 13.6 ± 0.96 | 12.0 ± 0.67 | 11.0 ± 0.66 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thibodeau, S.-È.; Legros, M.-L.; Labbé, E.-A.; Walsh-Wilkinson, É.; Morin-Grandmont, A.; Beji, S.; Arsenault, M.; Caron, A.; Couet, J. Cold Exposure Exacerbates Cardiac Dysfunction in a Model of Heart Failure with Preserved Ejection Fraction in Male and Female C57Bl/6J Mice. Biomedicines 2025, 13, 1900. https://doi.org/10.3390/biomedicines13081900
Thibodeau S-È, Legros M-L, Labbé E-A, Walsh-Wilkinson É, Morin-Grandmont A, Beji S, Arsenault M, Caron A, Couet J. Cold Exposure Exacerbates Cardiac Dysfunction in a Model of Heart Failure with Preserved Ejection Fraction in Male and Female C57Bl/6J Mice. Biomedicines. 2025; 13(8):1900. https://doi.org/10.3390/biomedicines13081900
Chicago/Turabian StyleThibodeau, Sara-Ève, Marie-Lune Legros, Emylie-Ann Labbé, Élisabeth Walsh-Wilkinson, Audrey Morin-Grandmont, Sarra Beji, Marie Arsenault, Alexandre Caron, and Jacques Couet. 2025. "Cold Exposure Exacerbates Cardiac Dysfunction in a Model of Heart Failure with Preserved Ejection Fraction in Male and Female C57Bl/6J Mice" Biomedicines 13, no. 8: 1900. https://doi.org/10.3390/biomedicines13081900
APA StyleThibodeau, S.-È., Legros, M.-L., Labbé, E.-A., Walsh-Wilkinson, É., Morin-Grandmont, A., Beji, S., Arsenault, M., Caron, A., & Couet, J. (2025). Cold Exposure Exacerbates Cardiac Dysfunction in a Model of Heart Failure with Preserved Ejection Fraction in Male and Female C57Bl/6J Mice. Biomedicines, 13(8), 1900. https://doi.org/10.3390/biomedicines13081900