


Sirtuin Inhibitor Cambinol Induces Cell Differentiation and Differently Interferes with SIRT1 and 2 at the Substrate Binding Site
 , , ,
, , ,  ,
,  , , and
, , and
Abstract
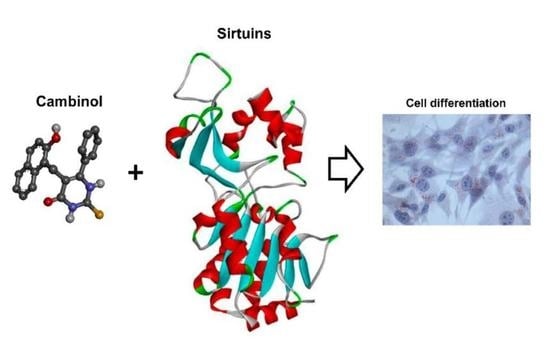
1. Introduction
2. Materials and Methods
2.1. Cell Line Culture, Treatment Conditions, and Differentiation Assay
2.2. Cell Cycle Analysis and Evaluation of Pre-G1 Phase
2.3. Western Blot Analyses
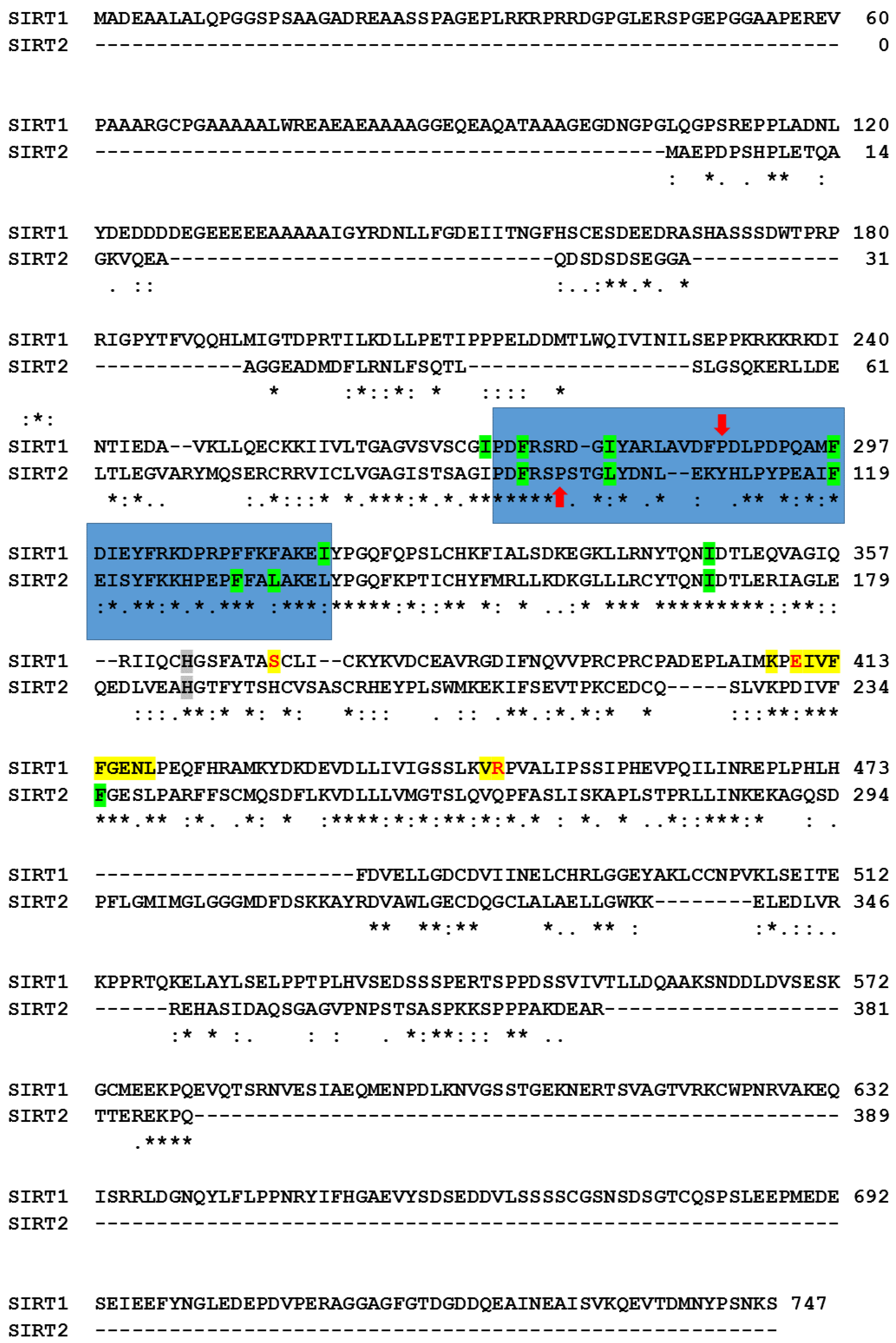
2.4. Molecular Docking Simulations and Sequence Comparison
3. Results and Discussion
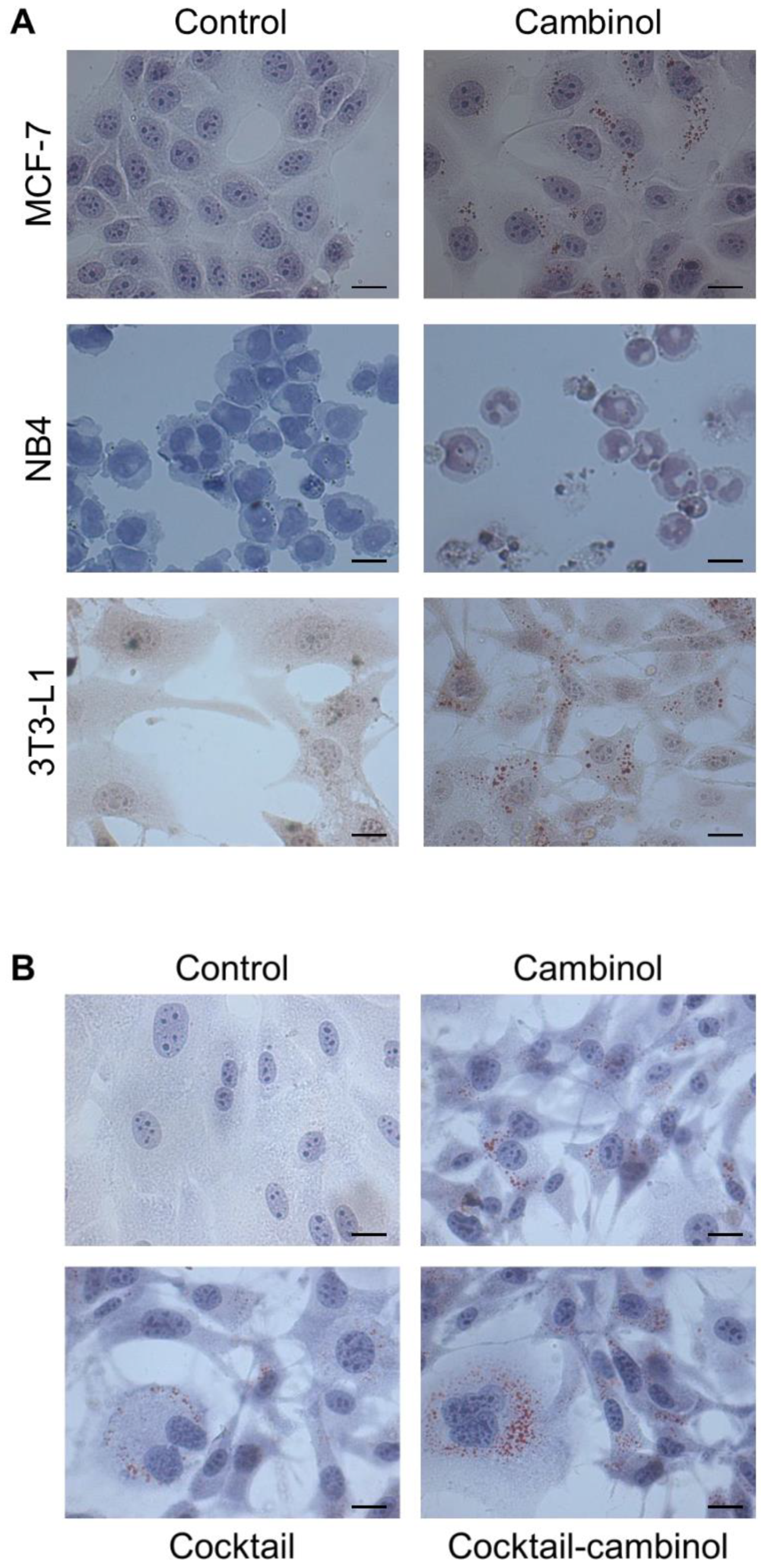
3.1. Cambinol Induces Cell Differentiation
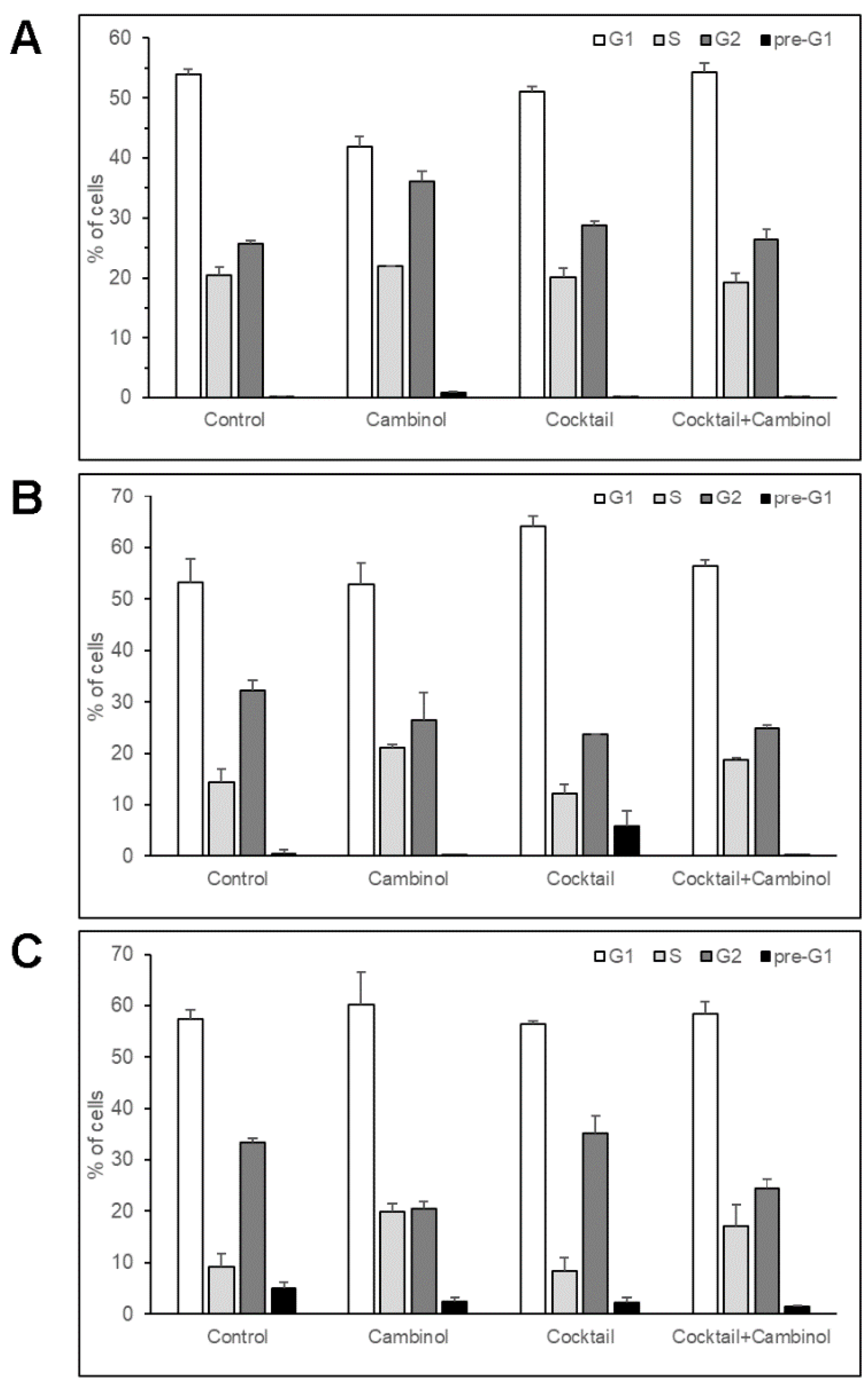
3.2. Cambinol Modulates Cell Cycle in 3T3-L1 Line
3.3. Molecular Targets Involved in the Differentiation Action of Cambinol
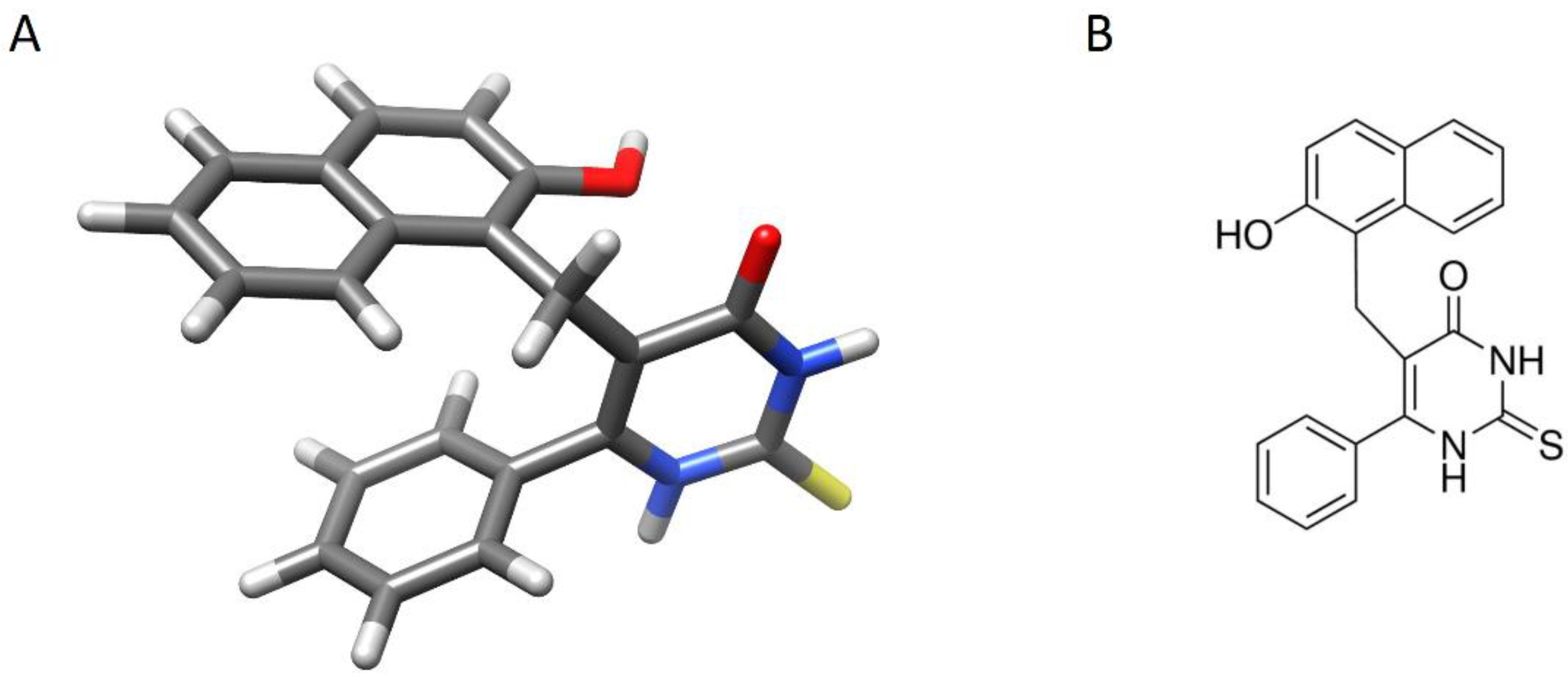
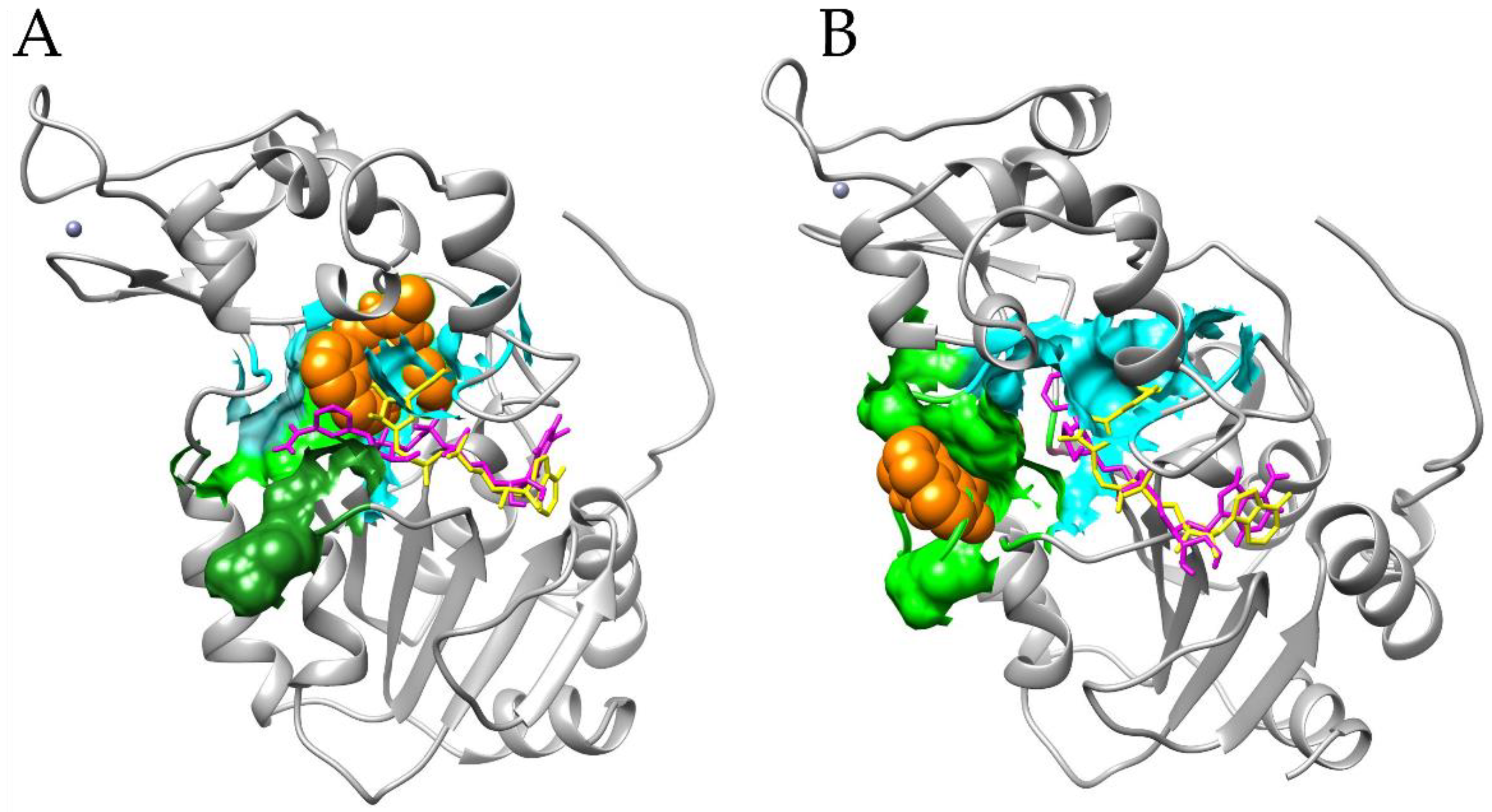
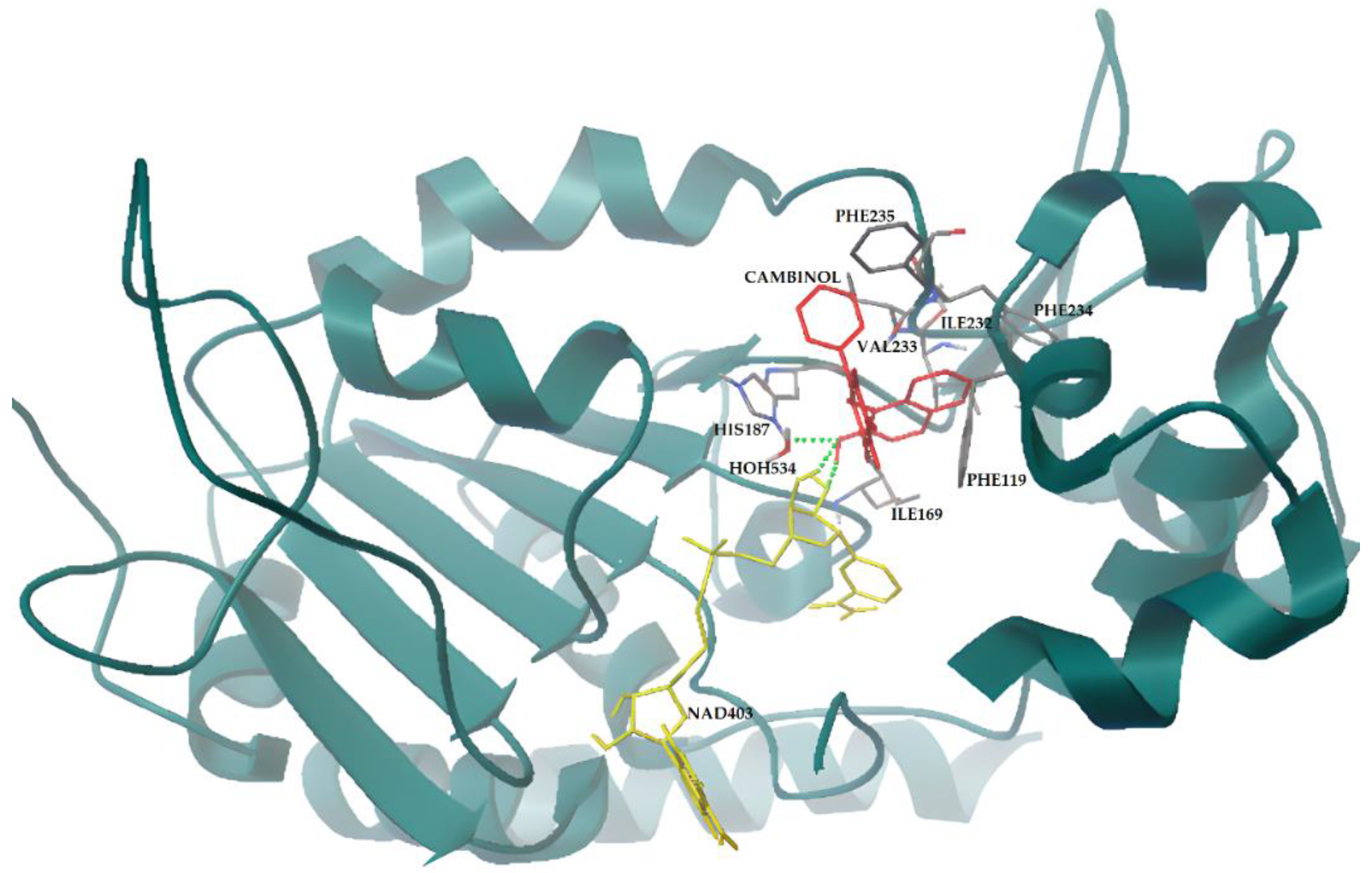
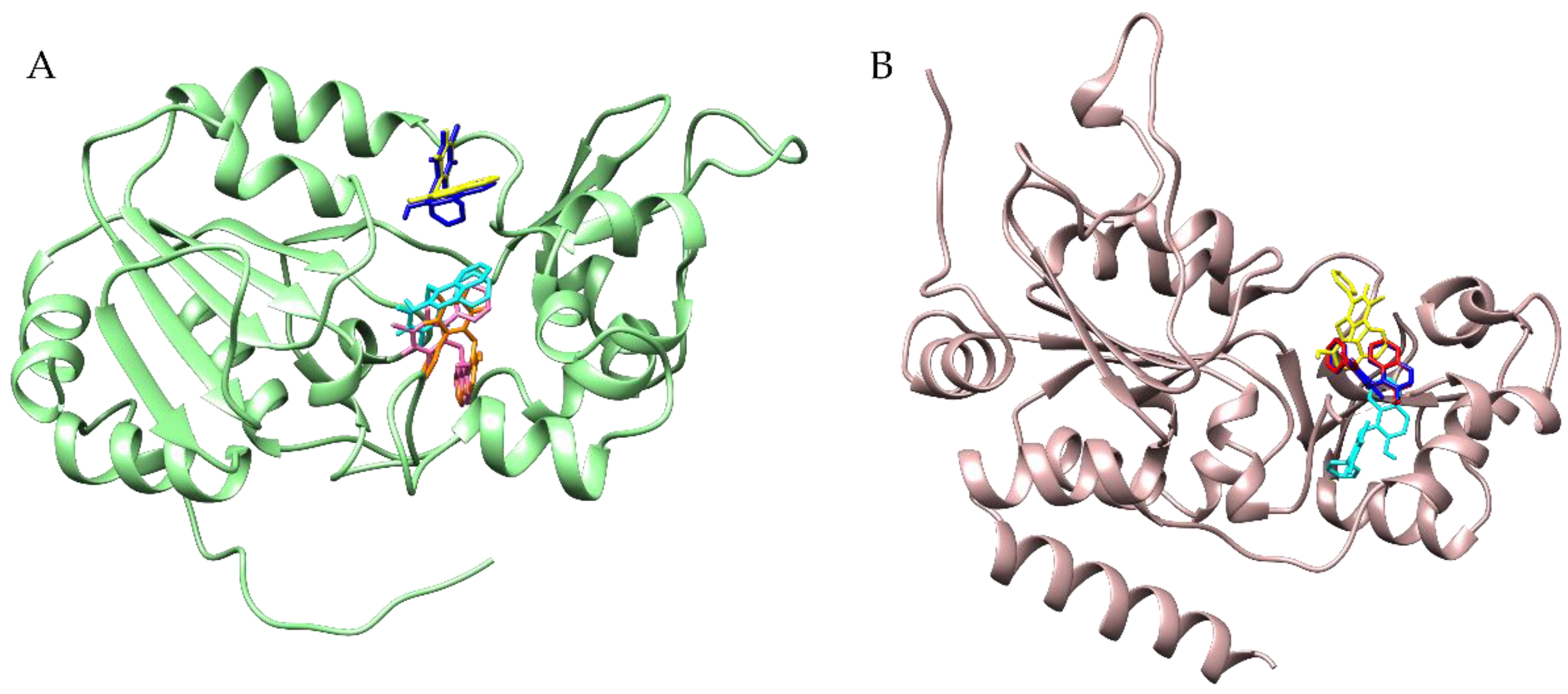
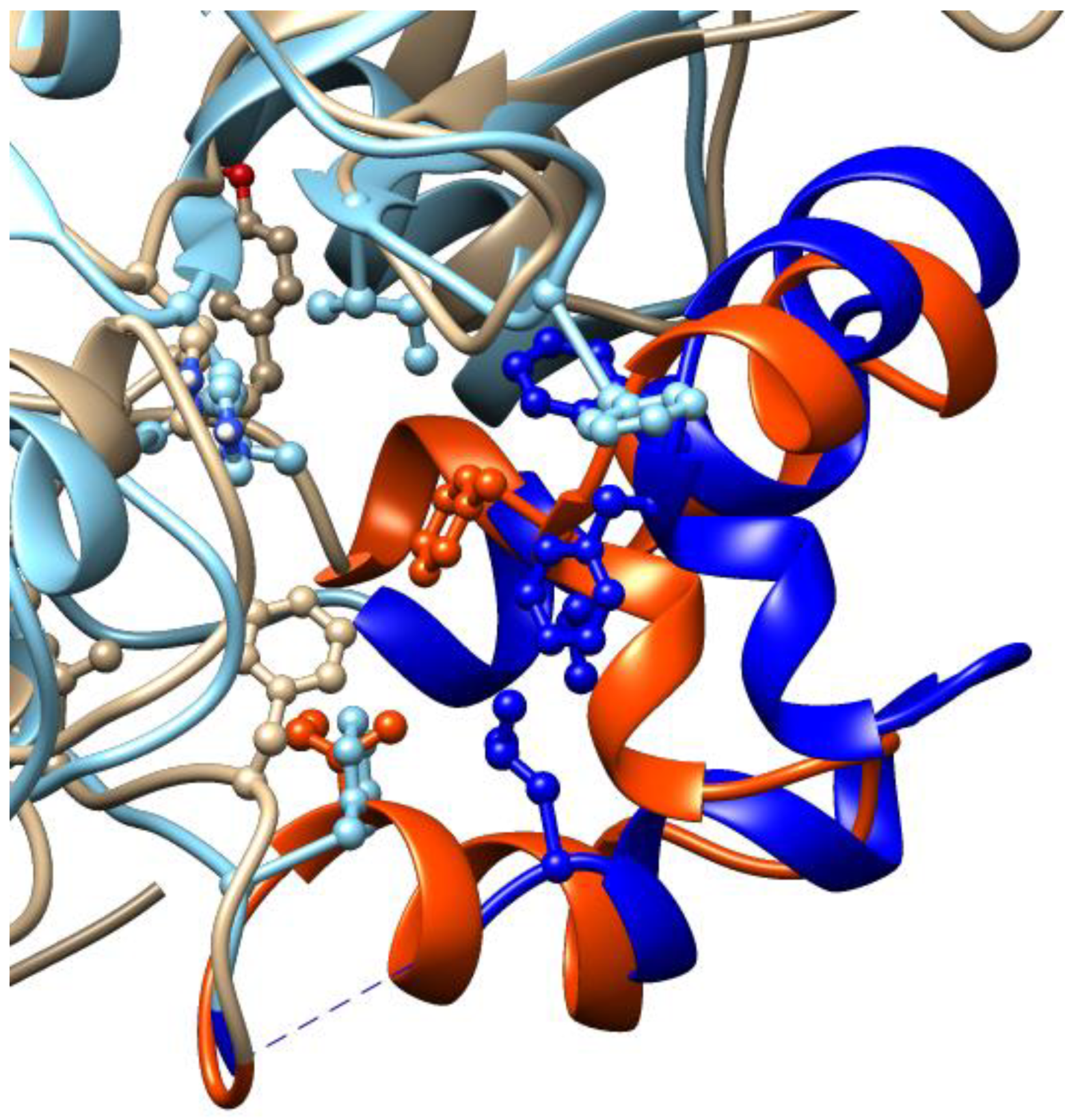
3.4. Docking Simulations of Molecular Interactions between Cambinol and the Target SIRTs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Fitz-James, M.H.; Cavalli, G. Molecular mechanisms of transgenerational epigenetic inheritance. Nat. Rev. Genet. 2022, 23, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Scafuri, B.; Bontempo, P.; Altucci, L.; De Masi, L.; Facchiano, A. Molecular Docking Simulations on Histone Deacetylases (HDAC)-1 and -2 to Investigate the Flavone Binding. Biomedicines 2020, 8, 568. [Google Scholar] [CrossRef]
- North, B.J.; Verdin, E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 2004, 5, 224. [Google Scholar] [CrossRef] [PubMed]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; De Oliveira, R.M.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef]
- Jing, E.; Gesta, S.; Kahn, C.R. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 2007, 6, 105–114. [Google Scholar] [CrossRef]
- Shi, T.; Wang, F.; Stieren, E.; Tong, Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 2005, 280, 13560–13567. [Google Scholar] [CrossRef]
- Bheda, P.; Jing, H.; Wolberger, C.; Lin, H. The substrate specificity of sirtuins. Annu. Rev. Biochem. 2016, 85, 405–429. [Google Scholar] [CrossRef]
- Sabari, B.R.; Zhang, D.; Allis, C.D.; Zhao, Y. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 2017, 18, 90–101. [Google Scholar] [CrossRef]
- Sun, Y.; Sun, D.; Li, F.; Tian, L.; Li, C.; Li, L.; Lin, R.; Wang, S. Downregulation of Sirt1 by antisense oligonucleotides induces apoptosis and enhances radiation sensitization in A549 lung cancer cells. Lung Cancer 2007, 58, 21–29. [Google Scholar] [CrossRef]
- Hiratsuka, M.; Inoue, T.; Toda, T.; Kimura, N.; Shirayoshi, Y.; Kamitani, H.; Watanabe, T.; Ohama, E.; Candice, G.T.; Kurimasa, A.; et al. Proteomics-based identification of differentially expressed genes in human gliomas: Down-regulation of SIRT2 gene. Biochem. Biophys. Res. Commun. 2003, 309, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, N.; Zino, S.; Macintyre, A.; Kingsmore, D.; Payne, A.P.; George, W.D.; Shiels, P.G. Altered sirtuin expression is associated with node-positive breast cancer. Br. J. Cancer 2006, 95, 1056–1061. [Google Scholar] [CrossRef]
- Allison, S.J.; Milner, J. SIRT3 is pro-apoptotic and participates in distinct basal apoptotic pathways. Cell Cycle 2007, 6, 2669–2677. [Google Scholar] [CrossRef] [PubMed]
- Zhao, E.; Hou, J.; Ke, X.; Abbas, M.N.; Kausar, S.; Zhang, L.; Cui, H. The roles of sirtuin family proteins in cancer progression. Cancers 2019, 11, 1949. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Villalba, J.M.; Alcaín, F.J. Sirtuin activators and inhibitors. Biofactors 2012, 38, 349–359. [Google Scholar] [CrossRef]
- Avalos, J.L.; Bever, K.M.; Wolberger, C. Mechanism of sirtuin inhibition by nicotinamide: Altering the NAD+ cosubstrate specificity of a Sir2 enzyme. Mol. Cell 2005, 17, 855–868. [Google Scholar] [CrossRef]
- Heltweg, B.; Gatbonton, T.; Schuler, A.D.; Posakony, J.; Li, H.; Goehle, S.; Kollipara, R.; De Pinho, R.A.; Gu, Y.; Simon, J.A.; et al. Antitumor activity of a small molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 2006, 66, 4368–4377. [Google Scholar] [CrossRef]
- King, J.; Patel, M.; Chandrasekaran, S. Metabolism, HDACs, and HDAC Inhibitors: A Systems Biology Perspective. Metabolites 2021, 11, 792. [Google Scholar] [CrossRef]
- Green, H.; Kehinde, O. An established preadipose cell line and its differentiation in culture II. Factors affecting the adipose conversion. Cell 1975, 5, 19–27. [Google Scholar] [CrossRef]
- Bontempo, P.; De Masi, L.; Carafa, V.; Rigano, D.; Scisciola, L.; Iside, C.; Grassi, R.; Molinari, A.M.; Aversano, R.; Nebbioso, A.; et al. Anticancer activities of anthocyanin extract from genotyped Solanum tuberosum “Vitelotte”. J. Funct. Foods 2015, 19, 584–593. [Google Scholar] [CrossRef]
- Bontempo, P.; Rigano, D.; Doto, A.; Formisano, C.; Conte, M.; Nebbioso, A.; Carafa, V.; Caserta, G.; Sica, V.; Molinari, A.M.; et al. Genista sessilifolia DC. extracts induce apoptosis across a range of cancer cell lines. Cell Proliferat. 2013, 46, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Bontempo, P.; Stiuso, P.; Lama, S.; Napolitano, A.; Piacente, S.; Altucci, L.; Molinari, A.M.; De Masi, L.; Rigano, D. Metabolite Profile and In Vitro Beneficial Effects of Black Garlic (Allium sativum L.) Polar Extract. Nutrients 2021, 13, 2771. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Zhao, X.; Allison, D.; Condon, B.; Zhang, F.; Gheyi, T.; Zhang, A.; Ashok, S.; Russell, M.; MacEwan, I.; Qian, Y.; et al. The 2.5 Å crystal structure of the SIRT1 catalytic domain bound to nicotinamide adenine dinucleotide (NAD+) and an indole (EX527 analogue) reveals a novel mechanism of histone deacetylase. J. Med. Chem. 2013, 56, 963–969. [Google Scholar] [CrossRef]
- Schiedel, M.; Rumpf, T.; Karaman, B.; Lehotzky, A.; Oláh, J.; Gerhardt, S.; Ovádi, J.; Sippl, W.; Einsle, O.; Jung, M. Aminothiazoles as potent and selective Sirt2 inhibitors: A structure-activity relationship study. J. Med. Chem. 2016, 59, 1599–1612. [Google Scholar] [CrossRef]
- Nguyen, G.T.; Schaefer, S.; Gertz, M.; Weyand, M.; Steegborn, C. Structures of human sirtuin 3 complexes with ADP-ribose and with carba-NAD+ and SRT1720: Binding details and inhibition mechanism. Acta Cryst. 2013, D69, 1423–1432. [Google Scholar] [CrossRef] [PubMed]
- Giordano, D.; Facchiano, A.; D’Auria, S.; Loreto, F. A hypothesis on the capacity of plant odorant-binding proteins to bind volatile isoprenoids based on in silico evidences. eLife 2021, 10, e66741. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Nagashima, I.; Hikita, M.; Bujo, H.; Takahashi, K.; Otabe, M.; Morisaki, N.; Saito, Y. Effect of troglitazone on plasma lipid metabolism and lipoprotein lipase. Br. J. Clin. Pharmacol. 1999, 47, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.; Bates, S.; Peters, G. Evidence for different modes of action of cyclin-dependent kinase inhibitors: p15 and p16 bind to kinases, p21 and p27 bind to cyclins. Oncogene 1995, 11, 1581–1588. [Google Scholar] [PubMed]
- Le Maire, A.; Teyssier, C.; Balaguer, P.; Bourguet, W.; Germain, P. Regulation of RXR-RAR heterodimers by RXR-and RAR-specific ligands and their combinations. Cells 2019, 8, 1392. [Google Scholar] [CrossRef]
- Audano, M.; Pedretti, S.; Caruso, D.; Crestani, M.; De Fabiani, E.; Mitro, N. Regulatory mechanisms of the early phase of white adipocyte differentiation: An overview. Cell. Mol. Life Sci. 2022, 79, 139. [Google Scholar] [CrossRef]
- Ma, X.; Wang, D.; Zhao, W.; Xu, L. Deciphering the roles of PPARγ in adipocytes via dynamic change of transcription complex. Front. Endocrinol. 2018, 9, 473. [Google Scholar] [CrossRef]
- Legutko, A.; Marichal, T.; Fiévez, L.; Bedoret, D.; Mayer, A.; De Vries, H.; Bureau, F. Sirtuin 1 promotes Th2 responses and airway allergy by repressing peroxisome proliferator-activated receptor-γ activity in dendritic cells. J. Immunol. 2011, 187, 4517–4529. [Google Scholar] [CrossRef]
- Davies, G.F.; McFie, P.J.; Khandelwal, R.L.; Roesler, W.J. Unique ability of troglitazone to up-regulate peroxisome proliferator-activated receptor-gamma expression in hepatocytes. J. Pharmacol. Exp. Ther. 2002, 300, 72–77. [Google Scholar] [CrossRef]
- Guilherme, A.; Tesz, G.J.; Guntur, K.V.P.; Czech, M.P. Tumor necrosis factor-alpha induces caspase-mediated cleavage of peroxisome proliferator-activated receptor gamma in adipocytes. J. Biol. Chem. 2009, 284, 17082–17091. [Google Scholar] [CrossRef]
- Niu, Z.; Shi, Q.; Zhang, W.; Shu, Y.; Yang, N.; Chen, B.; Wang, Q.; Zhao, X.; Chen, J.; Cheng, N.; et al. Caspase-1 cleaves PPARγ for potentiating the pro-tumor action of TAMs. Nat. Commun. 2017, 8, 766. [Google Scholar] [CrossRef]
- Du, W.; Searle, J.S. The rb pathway and cancer therapeutics. Curr. Drug Targets 2009, 10, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Macaluso, M.; Montanari, M.; Giordano, A. Rb family proteins as modulators of gene expression and new aspects regarding the interaction with chromatin remodeling enzymes. Oncogene 2006, 25, 5263–5267. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Narayanan, C.; Bian, J.; Sambo, D.; Brickler, T.; Zhang, W.; Chetty, S. A transient DMSO treatment increases the differentiation potential of human pluripotent stem cells through the Rb family. PLoS ONE 2018, 13, e0208110. [Google Scholar] [CrossRef]
- Qiang, L.; Wang, L.; Kon, N.; Zhao, W.; Lee, S.; Zhang, Y.; Rosenbaum, M.; Zhao, Y.; Gu, W.; Farmer, S.R.; et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Pparγ. Cell 2012, 150, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Wang, C.; Hagen, F.K.; Gormley, M.; Addya, S.; Soccio, R.; Casimiro, M.C.; Zhou, J.; Powell, M.J.; Xu, P.; et al. Acetylation-defective mutant of Pparγ is associated with decreased lipid synthesis in breast cancer cells. Oncotarget 2014, 5, 7303–7315. [Google Scholar] [CrossRef]
- Medda, F.; Russell, R.J.; Higgins, M.; McCarthy, A.R.; Campbell, J.; Slawin, A.M.; Lane, D.P.; Lain, S.; Westwood, N.J. Novel cambinol analogs as sirtuin inhibitors: Synthesis, biological evaluation, and rationalization of activity. J. Med. Chem. 2009, 52, 2673–2682. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor a | Docking Procedure | Binding Energy (Kcal/mol) | Interaction Region on the Receptor b | |
|---|---|---|---|---|
| SIRT1 | Blind | −7.66 c | Near the binding site (Ser370, Lys408, Glu410, Ile411, Val412, Glu416, Asn417, and Leu418) | |
| −7.57 c | Binding site (NAD, His363, Val412, Phe413, Phe414, Gly415, Glu416, Asn417, Leu418, and Arg446) | |||
| Focused | Rigid NAD+ | −7.96 d | Near the binding site (Ser370, Lys408, Glu410, Ile411, Val412, Glu416, Asn417, and Leu418) | |
| −7.91 d | Binding site (NAD, His363, Phe413, Phe414, Gly415, Glu416, Lys444, Val445, and Arg446) | |||
| Flexible NAD+ | −7.29 e | Binding site (NAD, His363, Val412, Phe413, Phe414, Gly415, Glu416, Leu418, Val445, and Arg446) | ||
| −6.35 e | Inhibition site (NAD, Ile270, Phe273, Ile279, Phe297, Ile316, Ile347, His363, HOH717, and HOH702) | |||
| SIRT2 | Blind | −9.52 | Inhibition site (NAD, Phe96, Leu103, Phe119, Phe131, Leu134, Ile169, His187, Ile232, and Phe234) | |
| Focused | Rigid NAD+ | −9.71 | Inhibition site (NAD, Phe96, Leu103, Phe119, Phe131, Leu134, Ile169, His187, Ile232, and Phe234) | |
| Flexible NAD+ | −9.35 | Inhibition site partially occluding the binding site (NAD, Phe119, Ile169, His187, Ile232, Val233, Phe234, Phe235, and HOH534) | ||
| SIRT3 | Blind | −7.06 | Protein surface (Phe157, Arg158, Leu168, Gln169, Gln171, Asp172, Leu173, Tyr175, and Pro176) | |
| Focused | Rigid CNA | −7.36 | Binding site (Phe157, Glu177, Phe180, Val292, Phe293, and Phe294) | |
| Flexible CNA | −8.12 | Binding site (NAD, Glu177, Gln228, Ile230, His248, Phe180, Ile291, Val292, Phe294, and Val324) | ||
| Receptor/Inhibitor | Docking Procedure | Binding Energy (Kcal/mol) | Interaction Region on the Receptor |
|---|---|---|---|
| SIRT1/EX527 analog a | Blind | −11.07 b | Inhibition site |
| −6.44 b | Binding site | ||
| Focused | −11.25 | Inhibition site | |
| SIRT2/SirReal a | Blind | −10.92 | Inhibition site |
| Focused | −12.39 | Inhibition site | |
| SIRT3/SRT1720 a | Blind | −13.66 | Inhibition site |
| Focused | −13.59 | Inhibition site |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giordano, D.; Scafuri, B.; De Masi, L.; Capasso, L.; Maresca, V.; Altucci, L.; Nebbioso, A.; Facchiano, A.; Bontempo, P. Sirtuin Inhibitor Cambinol Induces Cell Differentiation and Differently Interferes with SIRT1 and 2 at the Substrate Binding Site. Biomedicines 2023, 11, 1624. https://doi.org/10.3390/biomedicines11061624
Giordano D, Scafuri B, De Masi L, Capasso L, Maresca V, Altucci L, Nebbioso A, Facchiano A, Bontempo P. Sirtuin Inhibitor Cambinol Induces Cell Differentiation and Differently Interferes with SIRT1 and 2 at the Substrate Binding Site. Biomedicines. 2023; 11(6):1624. https://doi.org/10.3390/biomedicines11061624
Chicago/Turabian StyleGiordano, Deborah, Bernardina Scafuri, Luigi De Masi, Lucia Capasso, Viviana Maresca, Lucia Altucci, Angela Nebbioso, Angelo Facchiano, and Paola Bontempo. 2023. "Sirtuin Inhibitor Cambinol Induces Cell Differentiation and Differently Interferes with SIRT1 and 2 at the Substrate Binding Site" Biomedicines 11, no. 6: 1624. https://doi.org/10.3390/biomedicines11061624
APA StyleGiordano, D., Scafuri, B., De Masi, L., Capasso, L., Maresca, V., Altucci, L., Nebbioso, A., Facchiano, A., & Bontempo, P. (2023). Sirtuin Inhibitor Cambinol Induces Cell Differentiation and Differently Interferes with SIRT1 and 2 at the Substrate Binding Site. Biomedicines, 11(6), 1624. https://doi.org/10.3390/biomedicines11061624