Abstract
Background: Data from published studies about the effect of HFE polymorphisms on ALS risk, phenotype, and survival are still inconclusive. We aimed at evaluating whether the p.H63D polymorphism is a modifier of phenotype and survival in SOD1-mutated patients. Methods: We included 183 SOD1-mutated ALS patients. Mutations were classified as severe or mild according to the median survival of the study population. Patients were screened for the HFE p.H63D polymorphism. Survival was calculated using the Kaplan–Meier modeling, and differences were measured by the log-rank test. Multivariable analysis was performed with the Cox proportional hazards model (stepwise backward). Results: SOD1 severe mutation carriers show more frequent familial history for ALS and shorter survival compared to mild mutation carriers. Carriers and non-carriers of the p.H63D polymorphism did not differ in terms of sex ratio, frequency of positive familial history, age at onset, and bulbar/spinal ratio. In univariate and in Cox multivariable analysis using sex, age at onset, site of onset, family history, country of origin, and mutation severity as covariates, p.H63D carriers had a longer survival (p = 0.034 and p = 0.004). Conclusions: We found that SOD1-mutated ALS patients carrying the p.H63D HFE polymorphism have a longer survival compared to non-carriers, independently of sex, age and site of onset, family history, nation of origin, and severity of mutations, suggesting a possible role as disease progression modifier for the p.H63D HFE polymorphism in SOD1-ALS.
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease affecting upper and lower motor neurons leading to death approximately three years after the onset, usually from respiratory failure. Genetic mutations are detectable in about two thirds of familial cases (fALS) and more than 10% of apparently sporadic patients, with C9ORF72, SOD1, TARDBP, and FUS being the most frequent causative genes [1]. Furthermore, different genes have been identified as modifiers of disease survival. Polymorphisms of UNC13A [2,3] and NIPA1 [4] genes and intermediate-length expansion of the ATXN2 [5] gene have been associated with reduced survival. The impact of HFE gene variants on ALS risk and phenotype has been investigated as it is involved in iron homeostasis, and the p.C282Y and p.H63D polymorphisms are associated with hereditary hemochromatosis (HH). HH is characterized by iron accumulation in the liver, adrenal glands, heart, skin, gonads, joints, and pancreas. Indeed, there are data suggesting that brain iron overload, possibly related to oxidative stress, has a role in different neurodegenerative diseases, including Alzheimer’s disease and Parkinson’s disease [6]. The possible role of the p.H63D (p.His63Asp) polymorphism of the HFE gene as a risk factor for ALS has been evaluated in several studies with conflicting results. Of note are two recent meta-analyses that do not show any detrimental effect on disease risk [7,8].
In a large series of Italian ALS patients, we previously found that this polymorphism did not influence ALS phenotype and survival, with the possible exception of patients carrying SOD1 mutations [9]. Nevertheless, the number of SOD1 patients in that series was relatively small (n = 26). The aim of the present study is to assess whether the HFE p.H63D (rs1799945) common polymorphism is a modifier of phenotype and survival in a series of Italian and French SOD1-mutated patients, in order to replicate the results of the previous study in a larger cohort.
2. Materials and Methods
2.1. Patients
We included ALS patients carrying SOD1 mutations, collected through the Italian ALS Genetic (ITALSGEN) consortium and at three French ALS centres (Montpellier, Clermont-Ferrand, Paris) in the period 1996–2015. All patients were of Caucasian ethnicity and did not carry mutations in the other most common ALS-related genes (C9ORF72, TARDBP, and FUS). In order to control for the heterogeneity of clinical course related to different SOD1 mutations, each mutation was classified either as severe or mild according to the median survival of the whole study population (i.e., 7.41 years). Accordingly, mutations whose median survival was <7.41 years were considered severe and those whose median survival was >7.41 years were considered mild. A further sensitivity analysis was performed excluding all mutations identified in only one or two patients.
2.2. Genotyping
All the coding exons and 50 bp of the flanking intron–exon boundaries of SOD1 were PCR-amplified, sequenced using the BigDye Terminator v3.1 sequencing kit (Applied Biosystems Inc.), and run on an ABIPrism 3500 genetic analyzer. The exon 2 of HFE was amplified by PCR and analyzed by denaturing high-performance liquid chromatography (DHPLC) (Transgenomic, Inc., Omaha, NE, USA). PCR products with heteroduplex profiles were sequenced on an ABI 3500 sequencer (Life Technologies, Foster City, CA, USA) with BigDye termination v.1.1 (Life Technologies) technologies according to standard protocols. Samples with homozygous profiles were coupled with a wild-type reference, denaturated and re-analyzed by DHPLC in order to also detect homozygous sequence alterations. If a mixed profile was positive, the original sample was sequenced. All sequencing products were analyzed with SeqScape Software v.3.0 (Applied Biosystems—Life Technologies). Other ALS-related genes were screened according to previously described procedures [10].
2.3. Statistical Analysis
The adherence to the Hardy–Weinberg equilibrium was tested for the HFE alleles through the appropriate equation. Comparisons between means were made with the Student’s t-test or analysis of variance; comparison between categorical variables was made with the χ2 test and Fisher’s test when applicable; Levene’s test was used to confirm the equality of variances. Survival was calculated from onset to death/tracheostomy or censoring date (30 June 2016) using the Kaplan–Meier survival modeling, and differences in survival were measured by the log-rank test. No patients were lost to follow up. Multivariable analysis was performed with the Cox proportional hazards model (stepwise backward) with a retention criterion of p < 0.1. The significance level was set at p < 0.05. Data were processed using SPSS statistical package, version 22.0 (IBM Corporation, Chicago, IL, USA).
3. Results
A total of 183 Italian and French ALS patients carrying different mutations of SOD1 gene were included (30 from France and 153 from Italy). SOD1 mutations found in our sample are reported with the relative frequency (in decreasing order) and classification (mild/severe) in Table 1.

Table 1.
List of SOD1 mutations in order of decreasing frequency in our series (n = 183).
Table 2 shows demographic and clinical characteristics of patients of the whole series and of the two groups (mild and severe mutation carriers).
Table 2.
Demographic and clinical characteristics of the whole series (n = 183) and of the two groups (mild and severe mutation carriers).
Familial history for ALS is significantly more reported in patients with severe mutations compared to those carrying mild mutations (p = 0.04), possibly due to higher penetrance of severe mutations compared to mild mutations. A significant difference is found in median survival, as expected, based on the criterion used to classify mutations as mild or severe.
Patients were assessed for the rs1799945 (p.H63D) polymorphism of the HFE gene. The following allelic frequencies were found: CC (absence of the p.H63D polymorphism), 127 cases (69.4%); CG (heterozygous for p.H63D), 51 cases (27.9%); GG (homozygous for p.H63D), 5 cases (2.7%). The frequencies of observed HFE genotypes meet the Hardy–Weinberg equilibrium (Table 3).
Table 3.
Calculation of Hardy–Weinberg equilibrium for HFE genotypes.
Patient groups corresponding to different HFE genotypes did not differ in terms of sex ratio (p = 0.165), frequency of familial history (p = 0.863), and age at onset (p = 0.904). The bulbar/spinal ratio resulted different (p < 0.01) but the difference becomes not significant considering together heterozygous and homozygous carriers of the p.H63D polymorphism.
In univariate analysis, patients carrying the p.H63D (CG + GG) polymorphism showed a longer median survival (median survival, 13.58 years, IQR 3.92-NA, vs 6.09 years, IQR 2.08-NA, p = 0.034). A stratified analysis showed that the observed effect was confirmed in each group (for mild and severe mutation carriers, see Supplemental Figures S1 and S2, respectively). The presence of the p.H63D polymorphism also remained significant in Cox multivariable analysis using sex, age at onset, site of onset, family history, country of origin, and severity of mutations as covariates (hazard ratio, 0.49, 95% CI 0.30–0.80, p = 0.004). Kaplan–Meier curves are reported in Figure 1. Similar results were obtained excluding mutations identified in only one or two patients (data not reported).
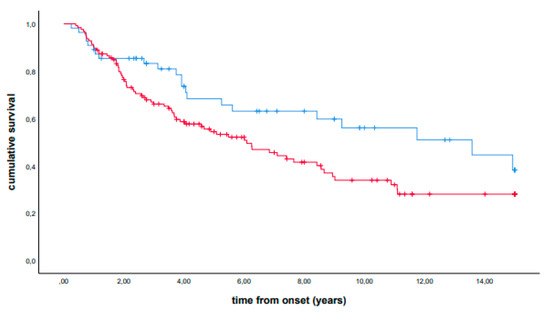
Figure 1.
Cumulative survival (Kaplan–Meier curves) of patients carrying the p.H63D (CG + GG) polymorphism (blue line) versus non-carrier patients (red line).
4. Discussion
Among 183 Italian and French ALS patients carrying SOD1 mutations, we found that the presence of the G allele of the HFE gene (i.e., p.H63D polymorphism) was significantly associated with a longer survival. Published data about HFE variants and ALS risk, phenotype, and survival are conflicting. Some studies reported a higher frequency of the p.H63D polymorphism in ALS patients compared to healthy controls, but without any influence on male/female ratio, age at and site of onset, and survival [11,12,13,14]. Sutedja et al. reported a positive association between homozygosity for the p.H63D polymorphism and ALS, suggesting that HFE is a contributing factor in the development of the disease in the Dutch population. In addition, they found that heterozygosity was associated with a higher age at ALS onset, possibly indicating that p.H63D is a risk factor for a later-onset form of ALS [15]. Other studies failed to detect any association between the p.H63D variant and ALS risk, phenotype, and survival [16,17]. Particularly, a survey collecting 3962 ALS patients and 5072 healthy controls showed no difference in the p.H63D allele frequency between patients and controls, and no effect of the p.H63D polymorphism on age at onset and survival [7]. Furthermore, a metanalysis published in 2014 did not observe any evidence for an association of the p.H63D polymorphism with ALS risk [8]. Interestingly, a study by Su and colleagues [18] reported that the p.H63D HFE polymorphism (either homozygous or heterozygous) was associated with increased disease duration and decreased muscle superoxide dismutase-1 expression in ALS patients. However, the sample size was small (22 ALS patients with wild-type HFE and 16 ALS patients either homozygous or heterozygous for the p.H63D HFE polymorphism) and a selection bias was contemplated by the authors, since many of the patients selected for muscle biopsy underwent this procedure because their clinical and/or electrodiagnostic findings were in some way atypical. In 2015, a study [9] including 1119 Italian and 232 Sardinian ALS patients and 1302 Italian and 121 Sardinian age- and gender-matched healthy controls did not find any significant difference in either population. Patients with CC, CG, and GG genotypes did not differ by age at onset and site of onset. No difference of survival was found considering both the CC/CG/GG phenotypes and the presence of a G allele in either cohorts of patients. In the 26 patients with SOD1 mutations, an increased survival was found in patients with CG or GG compared with CC genotype or in patients carrying the G allele (dominant assumption) (p = 0.04). This finding was confirmed by the multivariable Cox model, where the G allele was retained as an independent prognostic factor (p = 0.03). The conflicting findings of published studies may arise from different aspects. First, some papers are based on small, underpowered series; second, in some studies, controls are not matched by ethnic origin to cases. To overcome the limitation of the small sample size, in the present study we enrolled 183 Italian and French ALS patients carrying SOD1 mutations and showed that the presence of the G allele of the HFE gene (i.e., p.H63D polymorphism) was significantly associated with a longer survival. Conversely, in the SOD1 transgenic mouse, the p.His67Asp polymorphism of the HFE gene (analogue of the p.His63Asp human polymorphism) has been observed to negatively influence the survival of the SOD1 transgenic mouse [19]. The double transgenic mice had shorter survival and disease duration compared to SOD1 mice, but only in the female group. No further differences between the two transgenic mice were found in age at onset, motor neuron loss, and total iron concentrations in lumbar spinal cord. Such conflicting data suggest the possibility that genetic interactions in mice are biologically different compared to humans. The mechanism through which the p.His63Asp HFE polymorphism might prolong the survival of SOD1-mutated ALS patients remains to be elucidated. Although some HFE variants are related to HH, they are common in the Caucasian population, probably because they provide some advantage to asymptomatic carriers. Recent studies suggest that HFE variants, including the p.H63D, can positively influence the immune system, the general fitness, and reproductive status of carriers [20]. The increase in iron uptake may have helped humans, particularly women of reproductive age, to compensate for the reduction in iron content in the cereal grain-based diet that took the place of the paleo-diet (rich in red meat) in Europe in the Neolithic Age, when the first HFE mutation arose [21]. We can hypothesize that the p.H63D polymorphism of HFE may provide some advantage to carriers in the presence of a pathogenic SOD1 mutation. Nevertheless, such a hypothesis needs to be explored by further studies. The possible involvement of iron homeostasis dysregulation in ALS pathogenesis deserves further evaluation since it could be a therapeutic target. Currently, a phase 2–3, multicentre, parallel-group, placebo-controlled, randomised clinical trial of deferiprone, an iron chelator, as disease-modifying strategy is currently ongoing (NCT03293069). Nevertheless, the precise comprehension of the characteristics of the eventual dysregulation of iron metabolism in ALS is of outstanding importance to understand how to modulate it through therapeutic interventions.
5. Conclusions
We found that SOD1-mutated ALS patients carrying the p.H63D polymorphism of HFE have a significantly longer median survival compared to non-carriers, and the result is independent of sex, age at onset, site of onset, positive family history, nation of origin, and severity of mutations. The underlying basis of such an effect of the HFE genotype on SOD1 ALS survival needs further investigation to be clarified.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biomedicines11030704/s1: Supplemental Figure S1 [Univariate analysis in the mild mutation carrier group: Kaplan–Meier curves showing that patients carrying the p.H63D (CG + GG) polymorphism (blue line) have a longer median survival compared to non-carriers (red line)]; Supplemental Figure S2 [Univariate analysis in the severe mutation carrier group: Kaplan–Meier curves showing that patients carrying the p.H63D (CG + GG) polymorphism (blue line) have a longer median survival as compared to non-carriers (red line)]; list of the members of the ITALSGEN consortium.
Author Contributions
Study concept and design: A.C. (Antonio Canosa), W.C. and A.C. (Adriano Chiò). Acquisition of data: A.C. (Antonio Canosa), A.C. (Andrea Calvo), G.M., C.M., M.B. (Maura Brunetti), M.B. (Marco Barberis), G.B., C.C., F.T., R.S., P.V., I.L.S., F.S., F.O.L., N.R., L.T., F.G., J.M., R.T., M.R.M., P.M., F.L.C., M.Z., M.S., C.T., C.L., L.M., S.D., N.G., V.M., P.C., W.C. and A.C. (Adriano Chiò). Analysis and interpretation of data: A.C. (Antonio Canosa), A.C. (Andrea Calvo), W.C. and A.C. (Adriano Chiò). Drafting of the manuscript: A.C. (Antonio Canosa), W.C. and A.C. (Adriano Chiò). Critical revision of the manuscript for important intellectual content: A.C. (Antonio Canosa), A.C. (Andrea Calvo), G.M., C.M., M.B. (Maura Brunetti), M.B. (Marco Barberis), G.B., C.C., F.T., R.S., P.V., I.L.S., F.S., F.O.L., N.R., L.T., F.G., J.M., R.T., M.R.M., P.M., F.L.C., M.Z., M.S., C.T., C.L., L.M., S.D., N.G., V.M., P.C., W.C. and A.C. (Adriano Chiò). Obtained funding: A.C. (Adriano Chiò). Study supervision: A.C. (Antonio Canosa), W.C. and A.C. (Adriano Chiò). A.C. (Antonio Canosa) had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. All authors have approved the submitted version of the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This study was in part supported by the Italian Ministry of Health (Ministero della Salute, Ricerca Sanitaria Finalizzata, grant RF-2016-02362405), the European Commission’s Health Seventh Framework Programme (FP7/2007–2013 under grant agreement 259867), the Joint Programme—Neurodegenerative Disease Research (Strength, ALS-Care, and Brain-Mend projects), granted by Italian Ministry of Education, University and Research, and the Progetti di Ricerca di Rilevante Interesse Nazionale (PRIN, grant 2017SNW5MB), granted by the Italian Ministry of Education, University and Research. This study was performed under the Department of Excellence grant of the Italian Ministry of Education, University and Research to the “Rita Levi Montalcini” Department of Neuroscience, University of Turin, Italy. This study was partly funded by the AGING Project for Department of Excellence at the Department of Translational Medicine (DIMET), Università del Piemonte Orientale, Novara, Italy. Funding sources had no role in study design and conduct; data collection, management, analysis, and interpretation; preparation, review, approval, and decision to submit the manuscript.
Institutional Review Board Statement
The study was approved at the Coordinator Centre (ALS Centre of Turin) by the Ethical Committee of the ‘Azienda Ospedaliero-Universitaria Città della Salute e della Scienza di Torino’ (approval code 0036344).
Informed Consent Statement
Patients signed a written informed consent for genetic analysis for research purposes and deidentification of patients was performed to ensure the respect of privacy regulations. The study conforms with World Medical Association Declaration of Helsinki.
Data Availability Statement
Data are available upon request from interested researchers.
Conflicts of Interest
Antonio Canosa serves on the Editorial Board of Biomedicines and serves as Guest Editor for the Special Issues ‘Recent Advances in Amyotrophic Lateral Sclerosis Genetics and Pathophysiology’ and ‘Recent Advances in Amyotrophic Lateral Sclerosis Genetics and Pathophysiology 2.0′ of Biomedicines. Andrea Calvo received a research grant from Cytokinetics. Adriano Chiò serves on scientific advisory boards for Mitsubishi Tanabe, Roche, Biogen, Cytokinetics, Denali, and AveXis, and received a research grant from Italfarmaco. Jessica Mandrioli received research support from Pfizer. The other authors have no conflict of interest relevant for the manuscript.
References
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Diekstra, F.P.; van Vught, P.W.J.; van Rheenen, W.; Koppers, M.; Pasterkamp, R.J.; van Es, M.A.; Schelhaas, H.J.; de Visser, M.; Robberecht, W.; Van Damme, P.; et al. UNC13A is a modifier of survival in amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 630.e3–630.e8. [Google Scholar] [CrossRef]
- Chiò, A.; Mora, G.; Restagno, G.; Brunetti, M.; Ossola, I.; Barberis, M.; Ferrucci, L.; Canosa, A.; Manera, U.; Moglia, C.; et al. UNC13A influences survival in Italian amyotrophic lateral sclerosis patients: A population-based study. Neurobiol. Aging 2013, 34, 357.e1–357.e5. [Google Scholar] [CrossRef]
- Blauw, H.M.; van Rheenen, W.; Koppers, M.; Van Damme, P.; Waibel, S.; Lemmens, R.; van Vught, P.W.J.; Meyer, T.; Schulte, C.; Gasser, T.; et al. NIPA1 polyalanine repeat expansions are associated with amyotrophic lateral sclerosis. Hum. Mol. Genet. 2012, 21, 2497–2502. [Google Scholar] [CrossRef]
- Chiò, A.; Calvo, A.; Moglia, C.; Canosa, A.; Brunetti, M.; Barberis, M.; Restagno, G.; Conte, A.; Bisogni, G.; Marangi, G.; et al. ATXN2 polyQ intermediate repeats are a modifier of ALS survival. Neurology 2015, 84, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Nandar, W.; Connor, J.R. HFE gene variants affect iron in the brain. J. Nutr. 2011, 141, 729S–739S. [Google Scholar] [CrossRef]
- van Rheenen, W.; Diekstra, F.P.; van Doormaal, P.T.C.; Seelen, M.; Kenna, K.; McLaughlin, R.; Shatunov, A.; Czell, D.; van Es, M.A.; van Vught, P.W.J.; et al. H63D polymorphism in HFE is not associated with amyotrophic lateral sclerosis. Neurobiol. Aging 2013, 34, 1517.e5–1517.e7. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, L.; Wang, W.; Qi, X.L.; Tang, Z.Y.; Li, M.; Wang, L.; Wang, W.; Qi, X.L.; Tang, Z.Y. Mutations in the HFE gene and sporadic amyotrophic lateral sclerosis risk: A meta-analysis of observational studies. Braz. J. Med. Biol. Res. 2014, 47, 215–222. [Google Scholar] [CrossRef]
- Chiò, A.; Mora, G.; Sabatelli, M.; Caponnetto, C.; Lunetta, C.; Traynor, B.J.; Johnson, J.O.; Nalls, M.A.; Calvo, A.; Moglia, C.; et al. HFE p.H63D polymorphism does not influence ALS phenotype and survival. Neurobiol. Aging 2015, 36, 2906.e7–2906.e11. [Google Scholar] [CrossRef]
- Chiò, A.; Calvo, A.; Mazzini, L.; Cantello, R.; Mora, G.; Moglia, C.; Corrado, L.; D’Alfonso, S.; Majounie, E.; Renton, A.; et al. Extensive genetics of ALS: A population-based study in Italy. Neurology 2012, 79, 1983–1989. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-S.; Lee, S.; Simmons, Z.; Boyer, P.; Scott, K.; Liu, W.; Connor, J. Increased incidence of the Hfe mutation in amyotrophic lateral sclerosis and related cellular consequences. J. Neurol. Sci. 2004, 227, 27–33. [Google Scholar] [CrossRef]
- Goodall, E.F.; Greenway, M.J.; van Marion, I.; Carroll, C.B.; Hardiman, O.; Morrison, K.E. Association of the H63D polymorphism in the hemochromatosis gene with sporadic ALS. Neurology 2005, 65, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Restagno, G.; Lombardo, F.; Ghiglione, P.; Calvo, A.; Cocco, E.; Sbaiz, L.; Mutani, R.; Chiò, A. HFE H63D polymorphism is increased in patients with amyotrophic lateral sclerosis of Italian origin. J. Neurol. Neurosurg. Psychiatry 2007, 78, 327. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Lu, X.; Hu, J.; Xi, J.; Zhou, D.; Shang, H.; Liu, L.; Zhou, H.; Yan, B.; Yu, L.; et al. H63D polymorphism in the hemochromatosis gene is associated with sporadic amyotrophic lateral sclerosis in China. Eur. J. Neurol. 2011, 18, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Sutedja, N.A.; Sinke, R.J.; Van Vught, P.W.J.; Van der Linden, M.W.; Wokke, J.H.J.; Van Duijn, C.M.; Njajou, O.T.; Van der Schouw, Y.T.; Veldink, J.H.; Van den Berg, L.H. The association between H63D mutations in HFE and amyotrophic lateral sclerosis in a Dutch population. Arch. Neurol. 2007, 64, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Yen, A.A.; Simpson, E.P.; Henkel, J.S.; Beers, D.R.; Appel, S.H. HFE mutations are not strongly associated with sporadic ALS. Neurology 2004, 62, 1611–1612. [Google Scholar] [CrossRef]
- Praline, J.; Blasco, H.; Vourc’h, P.; Rat, V.; Gendrot, C.; Camu, W.; Andres, C.R. French ALS Study Group Study of the HFE gene common polymorphisms in French patients with sporadic amyotrophic lateral sclerosis. J. Neurol. Sci. 2012, 317, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Su, X.W.; Lee, S.Y.; Mitchell, R.M.; Stephens, H.E.; Simmons, Z.; Connor, J.R. H63D HFE polymorphisms are associated with increased disease duration and decreased muscle superoxide dismutase-1 expression in amyotrophic lateral sclerosis patients. Muscle Nerve 2013, 48, 242–246. [Google Scholar] [CrossRef]
- Nandar, W.; Neely, E.B.; Simmons, Z.; Connor, J.R. H63D HFE genotype accelerates disease progression in animal models of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2014, 1842, 2413–2426. [Google Scholar] [CrossRef] [PubMed]
- Hollerer, I.; Bachmann, A.; Muckenthaler, M.U. Pathophysiological consequences and benefits of HFE mutations: 20 years of research. Haematologica 2017, 102, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Naugler, C. Hemochromatosis: A Neolithic adaptation to cereal grain diets. Med. Hypotheses 2008, 70, 691–692. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).