
The Enigmatic Genetic Landscape of Hereditary Hearing Loss: A Multistep Diagnostic Strategy in the Italian Population
 , , , , , , , and
, , , , , , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Statement
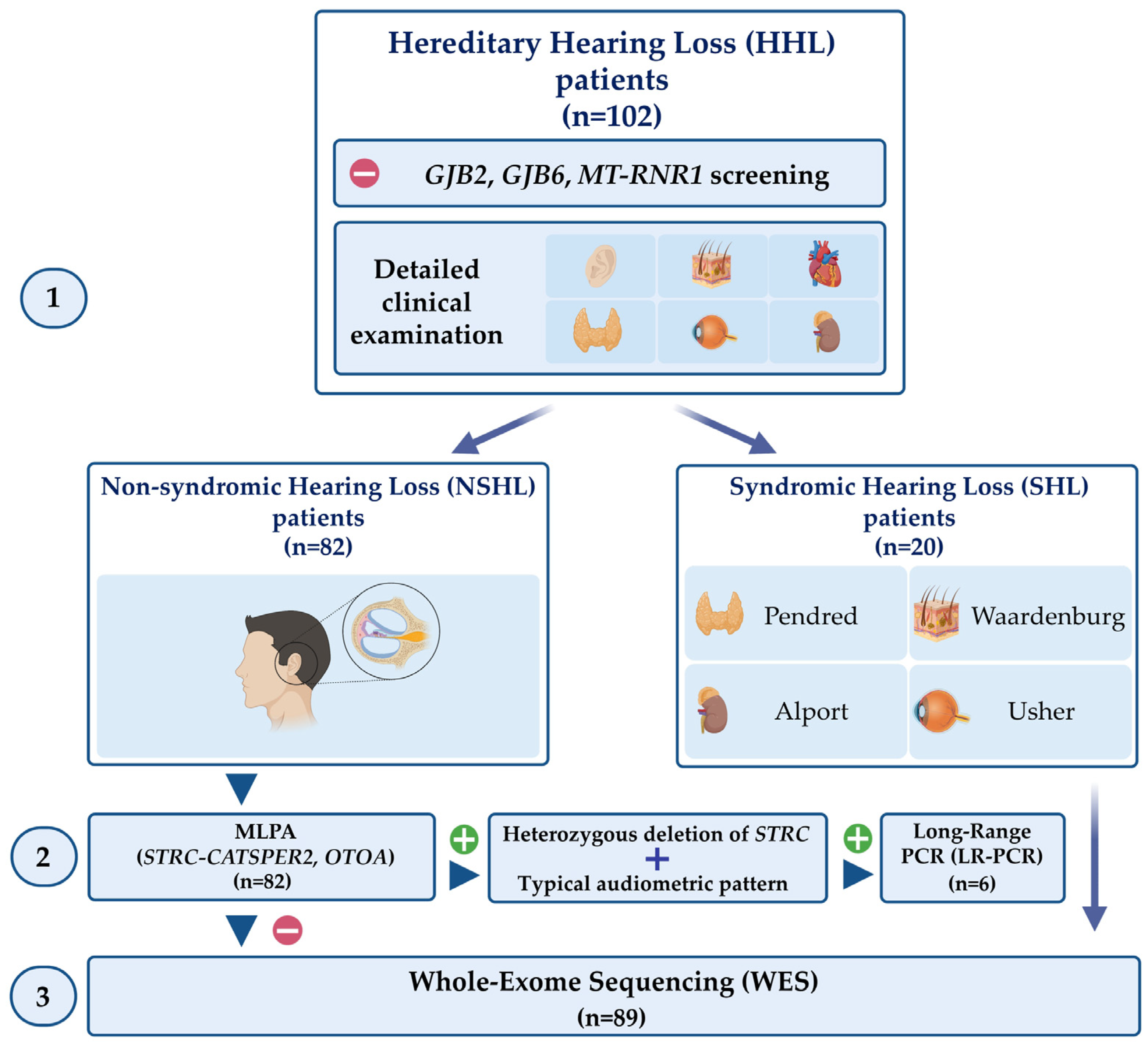
2.2. Study Design
2.3. DNA Extraction and Quality Control
2.4. MLPA
2.5. LR-PCR and STRC Sequencing
2.6. WES and Data Analysis
2.7. Prevalence of USH2A and ADGRV1 Pathogenic Variant Carriers in Italian Cohorts
3. Results
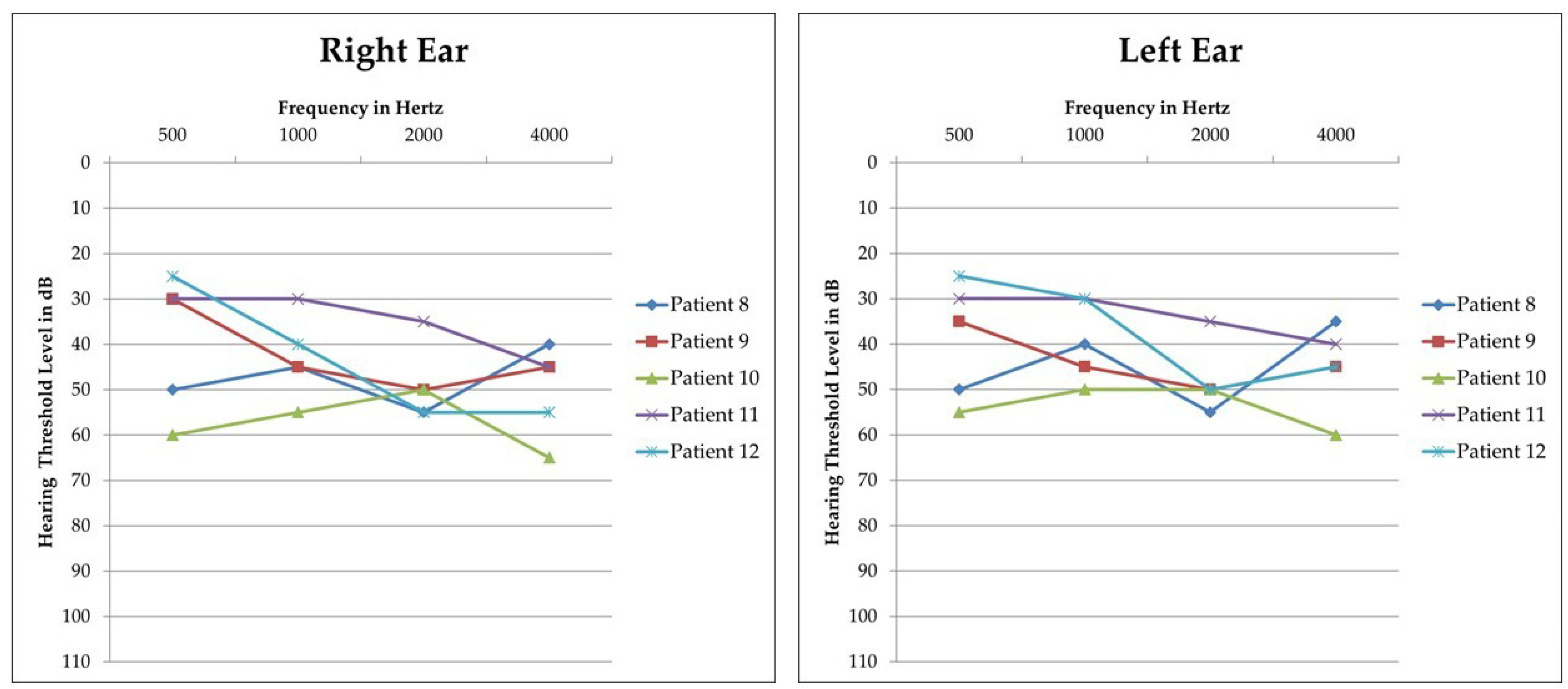
3.1. STRC Analysis
3.2. WES Analysis in NSHL Patients
3.3. WES Analysis in SHL Patients
3.4. Prevalence of USH2A and ADGRV1 Pathogenic Variant Carriers in Italian Cohorts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Posey, J.E.; O’Donnell-Luria, A.H.; Chong, J.X.; Harel, T.; Jhangiani, S.N.; Coban Akdemir, Z.H.; Buyske, S.; Pehlivan, D.; Carvalho, C.M.B.; Baxter, S.; et al. Insights into Genetics, Human Biology and Disease Gleaned from Family Based Genomic Studies. Genet. Med. 2019, 21, 798–812. [Google Scholar] [CrossRef] [PubMed]
- Korver, A.M.H.; Smith, R.J.H.; Van Camp, G.; Schleiss, M.R.; Bitner-Glindzicz, M.A.K.; Lustig, L.R.; Usami, S.-I.; Boudewyns, A.N. Congenital Hearing Loss. Nat. Rev. Dis. Prim. 2017, 3, 16094. [Google Scholar] [CrossRef] [PubMed]
- Bademci, G.; Cengiz, F.B.; Foster, J.; Duman, D.; Sennaroglu, L.; Diaz-Horta, O.; Atik, T.; Kirazli, T.; Olgun, L.; Alper, H.; et al. Variations in Multiple Syndromic Deafness Genes Mimic Non-Syndromic Hearing Loss. Sci. Rep. 2016, 6, 31622. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Kawashima, Y.; Fujikawa, T.; Honda, K.; Makabe, A.; Kitamura, K.; Tsutsumi, T. Rapid Screening of Copy Number Variations in STRC by Droplet Digital PCR in Patients with Mild-to-Moderate Hearing Loss. Hum. Genome Var. 2019, 6, 41. [Google Scholar] [CrossRef]
- Ideura, M.; Nishio, S.; Moteki, H.; Takumi, Y.; Miyagawa, M.; Sato, T.; Kobayashi, Y.; Ohyama, K.; Oda, K.; Matsui, T.; et al. Comprehensive Analysis of Syndromic Hearing Loss Patients in Japan. Sci. Rep. 2019, 9, 11976. [Google Scholar] [CrossRef]
- Gettelfinger, J.; Dahl, J. Syndromic Hearing Loss: A Brief Review of Common Presentations and Genetics. J. Pediatr. Genet. 2018, 07, 001–008. [Google Scholar] [CrossRef]
- Faridi, R.; Rea, A.; Fenollar-Ferrer, C.; O’Keefe, R.T.; Gu, S.; Munir, Z.; Khan, A.A.; Riazuddin, S.; Hoa, M.; Naz, S.; et al. New Insights into Perrault Syndrome, a Clinically and Genetically Heterogeneous Disorder. Hum. Genet. 2022, 141, 805–819. [Google Scholar] [CrossRef]
- Barakat, A.J.; Raygada, M.; Rennert, O.M. Barakat Syndrome Revisited. Am. J. Med. Genet. Part A 2018, 176, 1341–1348. [Google Scholar] [CrossRef]
- Morgan, A.; Faletra, F.; Severi, G.; La Bianca, M.; Licchetta, L.; Gasparini, P.; Graziano, C.; Girotto, G. There Is More Than Meets the Eye: Identification of Dual Molecular Diagnosis in Patients Affected by Hearing Loss. Biomedicines 2022, 10, 12. [Google Scholar] [CrossRef]
- Gooch, C.; Rudy, N.; Smith, R.J.; Robin, N.H. Genetic Testing Hearing Loss: The Challenge of Non Syndromic Mimics. Int. J. Pediatr. Otorhinolaryngol. 2021, 150, 110872. [Google Scholar] [CrossRef]
- Clark, J.G. Uses and Abuses of Hearing Loss Classification. ASHA 1981, 23, 493–500. [Google Scholar] [PubMed]
- Vona, B.; Hofrichter, M.A.H.; Neuner, C.; Schröder, J.; Gehrig, A.; Hennermann, J.B.; Kraus, F.; Shehata-Dieler, W.; Klopocki, E.; Nanda, I.; et al. DFNB16 Is a Frequent Cause of Congenital Hearing Impairment: Implementation of STRC Mutation Analysis in Routine Diagnostics. Clin. Genet. 2015, 87, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 7, 20. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Limongelli, I.; Marini, S.; Bellazzi, R. PaPI: Pseudo Amino Acid Composition to Score Human Protein-Coding Variants. BMC Bioinform. 2015, 16, 123. [Google Scholar] [CrossRef]
- Quang, D.; Chen, Y.; Xie, X. DANN: A Deep Learning Approach for Annotating the Pathogenicity of Genetic Variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef]
- Jian, X.; Boerwinkle, E.; Liu, X. In Silico Prediction of Splice-Altering Single Nucleotide Variants in the Human Genome. Nucleic Acids Res. 2014, 42, 13534–13544. [Google Scholar] [CrossRef]
- Cocca, M.; Barbieri, C.; Concas, M.P.; Robino, A.; Brumat, M.; Gandin, I.; Trudu, M.; Sala, C.F.; Vuckovic, D.; Girotto, G.; et al. A Bird’s-Eye View of Italian Genomic Variation through Whole-Genome Sequencing. Eur. J. Hum. Genet. 2020, 28, 435–444. [Google Scholar] [CrossRef]
- Spedicati, B.; Cocca, M.; Palmisano, R.; Faletra, F.; Barbieri, C.; Francescatto, M.; Mezzavilla, M.; Morgan, A.; Pelliccione, G.; Gasparini, P.; et al. Natural Human Knockouts and Mendelian Disorders: Deep Phenotyping in Italian Isolates. Eur. J. Hum. Genet. 2021, 29, 1272–1281. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Imtiaz, A. ARNSHL Gene Identification: Past, Present and Future. Mol. Genet. Genom. 2022, 297, 1185–1193. [Google Scholar] [CrossRef]
- Duman, D.; Tekin, M. Autosomal Recessive Nonsyndromic Deafness Genes: A Review. Front. Biosci. Landmark Ed. 2012, 17, 2213–2236. [Google Scholar] [CrossRef] [PubMed]
- Pingault, V.; Ente, D.; Dastot-Le Moal, F.; Goossens, M.; Marlin, S.; Bondurand, N. Review and Update of Mutations Causing Waardenburg Syndrome. Hum. Mutat. 2010, 31, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Timberlake, A.T.; Griffin, C.; Heike, C.L.; Hing, A.V.; Cunningham, M.L.; Chitayat, D.; Davis, M.R.; Doust, S.J.; Drake, A.F.; Duenas-Roque, M.M.; et al. Haploinsufficiency of SF3B2 Causes Craniofacial Microsomia. Nat. Commun. 2021, 12, 4680. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.J.; Alkhunaizi, E.; Kruszka, P.; Pyle, L.C.; Grange, D.K.; Berger, S.I.; Payne, K.K.; Masser-Frye, D.; Hu, T.; Christie, M.R.; et al. Loss-of-Function Variants in PPP1R12A: From Isolated Sex Reversal to Holoprosencephaly Spectrum and Urogenital Malformations. Am. J. Hum. Genet. 2020, 106, 121–128. [Google Scholar] [CrossRef]
- Chan, D.K.; Chang, K.W. GJB2-Associated Hearing Loss: Systematic Review of Worldwide Prevalence, Genotype, and Auditory Phenotype. Laryngoscope 2014, 124, E34–E53. [Google Scholar] [CrossRef]
- Marshall, B.A.; Permutt, M.A.; Paciorkowski, A.R.; Hoekel, J.; Karzon, R.; Wasson, J.; Viehover, A.; White, N.H.; Shimony, J.S.; Manwaring, L.; et al. Phenotypic Characteristics of Early Wolfram Syndrome. Orphanet J. Rare Dis. 2013, 8, 64. [Google Scholar] [CrossRef]
- Miksch, S.; Lumsden, A.; Guenther, U.P.; Foernzler, D.; Christen-Zäch, S.; Daugherty, C.; Ramesar, R.K.S.; Lebwohl, M.; Hohl, D.; Neldner, K.H.; et al. Molecular Genetics of Pseudoxanthoma Elasticum: Type and Frequency of Mutations in ABCC6. Hum. Mutat. 2005, 26, 235–248. [Google Scholar] [CrossRef]
- Koenekoop, R.; Arriaga, M.; Trzupek, K.M.; Lentz, J. Usher Syndrome Type II. 1999 Dec 10 [Updated 2020 Oct 22]. In GeneReviews® [Internet]; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; National Organization for Rare Disorders: Seattle, WA, USA, 1993–2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1341/ (accessed on 25 January 2023).
- Morgan, A.; Lenarduzzi, S.; Spedicati, B.; Cattaruzzi, E.; Murru, F.M.; Pelliccione, G.; Mazzà, D.; Zollino, M.; Graziano, C.; Ambrosetti, U.; et al. Lights and Shadows in the Genetics of Syndromic and Non-Syndromic Hearing Loss in the Italian Population. Genes 2020, 11, 1237. [Google Scholar] [CrossRef]
- Sang, S.; Ling, J.; Liu, X.; Mei, L.; Cai, X.; Li, T.; Li, W.; Li, M.; Wen, J.; Liu, X.; et al. Proband Whole-Exome Sequencing Identified Genes Responsible for Autosomal Recessive Non-Syndromic Hearing Loss in 33 Chinese Nuclear Families. Front. Genet. 2019, 10, 639. [Google Scholar] [CrossRef]
- Van Heurck, R.; Carminho-Rodrigues, M.T.; Ranza, E.; Stafuzza, C.; Quteineh, L.; Gehrig, C.; Hammar, E.; Guipponi, M.; Abramowicz, M.; Senn, P.; et al. Benefits of Exome Sequencing in Children with Suspected Isolated Hearing Loss. Genes 2021, 12, 1277. [Google Scholar] [CrossRef]
- Elander, J.; Ullmark, T.; Ehrencrona, H.; Jonson, T.; Piccinelli, P.; Samuelsson, S.; Löwgren, K.; Falkenius-Schmidt, K.; Ehinger, J.; Stenfeldt, K.; et al. Extended Genetic Diagnostics for Children with Profound Sensorineural Hearing Loss by Implementing Massive Parallel Sequencing. Diagnostic Outcome, Family Experience and Clinical Implementation. Int. J. Pediatr. Otorhinolaryngol. 2022, 159, 111218. [Google Scholar] [CrossRef]
- Yokota, Y.; Moteki, H.; Nishio, S.Y.; Yamaguchi, T.; Wakui, K.; Kobayashi, Y.; Ohyama, K.; Miyazaki, H.; Matsuoka, R.; Abe, S.; et al. Frequency and Clinical Features of Hearing Loss Caused by STRC Deletions. Sci. Rep. 2019, 9, 4408. [Google Scholar] [CrossRef] [PubMed]
- Markova, T.G.; Alekseeva, N.N.; Mironovich, O.L.; Galeeva, N.M.; Lalayants, M.R.; Bliznetz, E.A.; Chibisova, S.S.; Polyakov, A.V.; Tavartkiladze, G.A. Clinical Features of Hearing Loss Caused by STRC Gene Deletions/Mutations in Russian Population. Int. J. Pediatr. Otorhinolaryngol. 2020, 138, 110247. [Google Scholar] [CrossRef] [PubMed]
- Simi, A.; Perry, J.; Schindler, E.; Oza, A.; Luo, M.; Hartman, T.; Krantz, I.D.; Germiller, J.A.; Kawai, K.; Kenna, M. Audiologic Phenotype and Progression in Pediatric STRC-Related Autosomal Recessive Hearing Loss. Laryngoscope 2021, 131, E2897–E2903. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Oh, D.-Y.; Han, J.H.; Oh, J.; Kim, M.Y.; Park, H.-R.; Seok, J.; Cho, S.-D.; Lee, S.-Y.; Kim, Y.; et al. Significant Mendelian Genetic Contribution to Pediatric Mild-to-Moderate Hearing Loss and Its Comprehensive Diagnostic Approach. Genet. Med. Off. J. Am. Coll. Med. Genet. 2020, 22, 1119–1128. [Google Scholar] [CrossRef]
- Usami, S.; Isaka, Y.; Miyagawa, M.; Nishio, S. Variants in CDH23 Cause a Broad Spectrum of Hearing Loss: From Non-Syndromic to Syndromic Hearing Loss as Well as from Congenital to Age-Related Hearing Loss. Hum. Genet. 2022, 141, 903–914. [Google Scholar] [CrossRef]
- Davies, C.; Bergman, J.; Misztal, C.; Ramchandran, R.; Mittal, J.; Bulut, E.; Shah, V.; Mittal, R.; Eshraghi, A.A. The Outcomes of Cochlear Implantation in Usher Syndrome: A Systematic Review. J. Clin. Med. 2021, 10, 2915. [Google Scholar] [CrossRef]
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 2016, 376, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Song, J.; He, C.; Cai, X.; Yuan, K.; Mei, L.; Feng, Y. Genetic Insights, Disease Mechanisms, and Biological Therapeutics for Waardenburg Syndrome. Gene Ther. 2022, 29, 479–497. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Patient ID | Gene | Type of Deletion | Genotype | Inheritance |
|---|---|---|---|---|
| 1 | STRC | Entire gene | Homozygous | Paternal and Maternal |
| 2 | STRC | Entire gene | Homozygous | Paternal and Maternal |
| 3 | STRC STRC | Entire gene | Compound heterozygous | Paternal |
| Exon 19 | Maternal | |||
| 4 | STRC | Entire gene | Homozygous | Paternal and Maternal |
| 5 | STRC | Entire gene | Homozygous | Paternal and Maternal |
| 6 | STRC | Entire gene | Homozygous | Paternal and Maternal |
| 7 | STRC | Entire gene | Homozygous | Paternal and Maternal |
| Patient ID | Gene | cDNA Change | Protein Change | Genotype | Inheritance | ACMG/AMP Classification | References |
|---|---|---|---|---|---|---|---|
| 8 | STRC (NM_153700.2) | Entire-gene deletion | NA | Compound heterozygous | Paternal | NA | PMID: 21686705 |
| c.1873C>T | p.Arg625Cys | Maternal | Uncertain significance | PMID: 22147502 | |||
| 9 | STRC (NM_153700.2) | Entire-gene deletion | NA | Compound heterozygous | Paternal | NA | PMID: 21686705 |
| c.1873C>T | p.Arg625Cys | Maternal | Uncertain significance | PMID: 22147502 | |||
| 10 | STRC (NM_153700.2) | Entire-gene deletion | NA | Compound heterozygous | Paternal | NA | PMID: 21686705 |
| c.4510delG | p.(Glu1504Argfs * 32) | Maternal | Pathogenic | PMID: 27068579 | |||
| 11 | STRC (NM_153700.2) | Entire-gene deletion | NA | Compound heterozygous | Paternal | NA | PMID: 21686705 |
| c.4917_4918delACinsCT | p.(Leu1640Phe) | Maternal | Uncertain significance | PMID: 36086952 | |||
| 12 | STRC (NM_153700.2) | c.2405_2407delTGT | p.(Leu802_Ser803 delinsPro) | Compound heterozygous | Paternal | Uncertain significance | NA |
| Entire-gene deletion | NA | Maternal | NA | PMID: 21686705 | |||
| 13 | STRC (NM_153700.2) | c.4402C>T | p.(Arg1468 *) | Compound heterozygous | Paternal | Pathogenic | PMID: 26011646NA |
| Entire-gene deletion | NA | Maternal | NA | PMID: 21686705 |
| Patient ID | Gene | Associated Disease | Transmission Pattern | cDNA Change | Protein Change | Genotype | Inheritance | ACMG/AMP Classification | References |
|---|---|---|---|---|---|---|---|---|---|
| 14 | ATP2B2 (NM_00168.3) | Deafness, autosomal dominant 82 (MIM:# 619804) | AD | c.1924 T>G | p.(Trp642Gly) | Heterozygous | Paternal | Uncertain significance | NA |
| EDN3 (NM_207034.1) | Waardenburg syndrome, type 4B (MIM: # 613265) | AD | c.167_190dupAGACTGTGGCTGGCCCTGGCGAGG | p.(Glu56_Glu63dup) | Heterozygous | Paternal | Uncertain significance | NA | |
| 15 | GATA3 (NM_001002295.1) | Hypoparathyroidism, sensorineural deafness, and renal dysplasia (MIM: # 146255) | AD | c.812C>T | p.(Ser271Leu) | Heterozygous | De novo | Likely pathogenic | NA |
| 16 | TRIOBP (NM_001039141.2) | Deafness, autosomal recessive 28 (MIM: # 609823) | AR | c.2827C>T | p.(Gln943 *) | Compound heterozygous | Paternal | Pathogenic | NA |
| c.5014G>T | p.(Gly1672 *) | Maternal | Pathogenic | PMID: 29197352 | |||||
| 17 | HARS2 (NM_012208.3) | Perrault syndrome 2 (MIM: # 614926) | AR | c.1273C>T | p.(Arg425Trp) | Compound heterozygous | Paternal | Uncertain significance | NA |
| c.389A>G | p.(Leu1640Phe) | Maternal | Uncertain significance | NA | |||||
| 18 | OTOF (NM_194248.2) | Auditory neuropathy, autosomal recessive, 1 (MIM: # 601071) | AR | c.4981G>A | p.(Glu1661Lys) | Compound heterozygous | Paternal | Uncertain significance | PMID: 36672845 |
| c.5533+13G>T | NA | De novo | Uncertain significance | NA | |||||
| 19 | TMPRSS3 (NM_024022.4) | Deafness, autosomal recessive 8/10 (MIM: # 601072) | AR | c.1224delA | p.(Glu409Argfs * 7) | Compound heterozygous | Paternal | Likely pathogenic | NA |
| c.646C>T | p.(Arg216Cys) | Maternal | Pathogenic | PMID: 34440452 | |||||
| 20 | LOXHD1 (NM_144612.6) | Deafness, autosomal recessive 77 (MIM: # 613079) | AR | c.4480C>T | p.(Arg1494 *) | Compound heterozygous | Paternal | Pathogenic | PMID: 25792669 |
| c.5085+970T>C | NA | Maternal | Uncertain significance | NA | |||||
| 21 | LRTOMT (NM_001145309.3) | Deafness, autosomal recessive 63 (MIM: # 611451) | AR | c.358+4A>C | NA | Homozygous | Paternal and Maternal | Likely pathogenic | PMID: 18953341 |
| 22 | CEACAM16 (NM_001039213.2) | Deafness, autosomal dominant 4B (MIM: # 614614) | AD | c.505G>A | p.(Gly169Arg) | Heterozygous | NA | Uncertain significance | PMID: 25589040 |
| 23 | OTOF (NM_194248.2) | Auditory neuropathy, autosomal recessive, 1 (MIM: # 601071) | AR | c.1699delG | p.(Ala567fs) | Compound heterozygous | Paternal | Pathogenic | NA |
| c.1601delC | p.(Pro534fs) | Maternal | Pathogenic | PMID: 18381613 | |||||
| 24 | OTOGL (NM_173591.3) | Deafness, autosomal recessive 84B (MIM: # 614944) | AR | c.6754+4A>C | NA | Compound heterozygous | Paternal | Uncertain significance | NA |
| c.448C>T | p.(Arg150Trp) | Maternal | Uncertain significance | NA | |||||
| 25 | MYH14 (NM_001145809.2) | Deafness, autosomal dominant 4A (MIM: # 600652) | AD | c.4088G>A | p.(Arg1363His) | Heterozygous | Maternal | Uncertain significance | NA |
| 26 | COL11A1 (NM_001854.4) | Deafness, autosomal dominant 37 (MIM: # 618533) | AD | c.611C>A | p.(Thr204Lys) | Heterozygous | Paternal | Uncertain significance | NA |
| PAX3 (NM_000438.6) | Waardenburg syndrome, type 1 (MIM: # 193500) | AD | c.599G>A | p.(Arg200His) | Heterozygous | Paternal | Uncertain significance | NA | |
| 27 | CLDN14 (NM_001146079.2) | Deafness, autosomal recessive 29 (MIM: # 614035) | AR | c.664delG | p.(Ala222fs) | Compound heterozygous | Paternal | Pathogenic | PMID: 32747562 |
| c.467T>C | p.(Met156Thr) | Maternal | Uncertain significance | NA | |||||
| 28 | KCNQ4 (NM_004700.4) | Deafness, autosomal dominant 2A (MIM: # 600101) | AD | c.845C>A | p.(Thr282Lys) | Heterozygous | Paternal | Likely pathogenic | NA |
| 29 | MYO15A (NM_016239.4) | Deafness, autosomal recessive 3 (MIM: # 600316) | AR | c.1390delG | p.(Asp464Ilefs * 22) | Compound heterozygous | Paternal | Pathogenic | NA |
| c.4777G>A | p.(Glu1593Lys) | Maternal | Uncertain significance | PMID: 32860223 | |||||
| 30 | ADGRV1 (NM_032119.4) | Usher syndrome, type 2C (MIM: # 605472) | AR | c.9447+1G>A | NA | Compound heterozygous | Paternal | Pathogenic | NA |
| c.13655dupT | p.(Asn4553Glufs * 18) | Maternal | Pathogenic | PMID: 33105617 | |||||
| 31 | PLS1 (NM_001145319.2) | Deafness, autosomal dominant 76 (MIM: # 618787) | AD | c.542C>A | p.(Ala181Asp) | Heterozygous | Maternal | Uncertain significance | NA |
| 32 | GJB2 (NM_004004.5) | Deafness, autosomal recessive 1A (MIM: # 220290) | AR | c.-22-2A>C | NA | Compound heterozygous | Paternal | Pathogenic | PMID: 34062854 |
| c.269T>C | p.(Leu90Pro) | Maternal | Likely pathogenic | PMID: 29293505 | |||||
| 33 | ADGRV1 (NM_032119.4) | Usher syndrome, type 2C (MIM: # 605472) | AR | c.18907G>A | p.(Asp6303Asn) | Compound heterozygous | Paternal | Uncertain significance | NA |
| c.17951A>C | p.(Gln5984Pro) | Maternal | Uncertain significance | NA | |||||
| 34 | MITF (NM_001354604.2) | Waardenburg syndrome, type 2A (MIM: # 193510) | AD | c.1031+1G>A | NA | Heterozygous | De novo | Pathogenic | PMID: 32013026 |
| 35 | USH2A (NM_206933.4) | Usher syndrome, type 2A (MIM: # 276901) | AR | c.9270C>A | p.(Cys3090 *) | Compound heterozygous | Paternal | Pathogenic | PMID: 35266249 |
| c.1876C>T | p.(Arg626 *) | Maternal | Pathogenic | PMID: 27460420 | |||||
| 36 | USH2A (NM_206933.4) | Usher syndrome, type 2A (MIM: # 276901) | AR | c.2099_2120delGGACAGTGGATGGAGATATTAC | p.(Gly700Alafs * 49) | Compound heterozygous | Paternal | Pathogenic | NA |
| c.8167C>T | p.(Arg2723 *) | Maternal | Pathogenic | PMID: 19683999 | |||||
| 37 | USH2A (NM_206933.4) | Usher syndrome, type 2A (MIM: # 276901) | AR | c.6778T>C | p.(Ser2260Pro) | Compound heterozygous | Paternal | Uncertain significance | PMID: 20507924 |
| c.12067-2A>G | NA | Maternal | Pathogenic | PMID: 27460420 | |||||
| 38 | KARS1 (NM_001130089.1) | Deafness, autosomal recessive 89 (MIM: # 613916) | AR | c.346A>G | p.(Lys116Glu) | Compound heterozygous | Paternal | Uncertain significance | NA |
| c.1124A>G | p.(Tyr375Cys) | Maternal | Uncertain significance | PMID: 29615062 | |||||
| 39 | STRC (NM_153700.2) | Deafness, autosomal recessive 16 (MIM: # 603720) | AR | c.3511T>C | p.(Trp1171Arg) | Compound heterozygous | Paternal | Uncertain significance | NA |
| c.4917_4918delACinsCT | p.(Leu1640Phe) | Maternal | Uncertain significance | PMID: 36086952 |
| Patient ID | Gene | Associated Disease | Transmission Pattern | cDNA Change | Protein Change | Genotype | Inheritance | ACMG/AMP Classification | References |
|---|---|---|---|---|---|---|---|---|---|
| 40 | CHD7 (NM_017780.3) | CHARGE syndrome (MIM: # 214800) | AD | c.550C>T | p.(Gln184 *) | Heterozygous | De novo | Pathogenic | PMID: 16615981 |
| 41 | USH2A (NM_206933.2) | Usher syndrome, type 2A (MIM: # 276901) | AR | c.11713C>T | p.(Arg3905Cys) | Compound heterozygous | Paternal | Pathogenic | PMID: 15015129 |
| c.11864G>A | p.(Trp3955 *) | Maternal | Pathogenic | PMID: 31877679 | |||||
| 42 | SF3B2 (NM_006842.2) | Craniofacial microsomia (MIM: # 164210) | AD | c.1660C>T | p.(Arg554 *) | Heterozygous | Maternal | Likely pathogenic | NA |
| 43 | EYA1 (NM_000503.4) | Branchiootorenal syndrome 1 (MIM: # 113650) | AD | c.880C>T | p.(Arg294 *) | Heterozygous | De novo | Pathogenic | PMID: 21280147 |
| 44 | CDH23 (NM_022124.6) | Usher syndrome, type 1D (MIM: # 601067) | AR | c.9433C>T | p.(Gln3145 *) | Compound heterozygous | Paternal | Pathogenic | NA |
| c.5712G>A | p.(Thr1904Thr) | Maternal | Likely pathogenic | PMID: 21738395 | |||||
| 45 | ADGRV1 (NM_032119.4) | Usher syndrome, type 2C (MIM: # 605472) | AR | c.12226_12227del ATinsGTAGAT GAGAGTAGATG | p.(Ile4076_Leu6306delinsValAspGluSerArg) | Compound heterozygous | Paternal | Pathogenic | NA |
| c.11410C>T | p.(Arg3804 *) | Maternal | Pathogenic | PMID: 28944237 | |||||
| 46 | GJB2 (NM_04004.5) | Deafness, autosomal recessive 1A (MIM: # 220290) | AR | c.35delG | p.(Gly12Valfs * 2) | Homozygous | Paternal and Maternal | Pathogenic | PMID: 9620796 |
| RPGR (NM_000328.3) | Retinitis pigmentosa 3 (MIM: # 300029) | XL | c.591_592delTG | p.(Cys197Trpfs * 13) | Heterozygous | De novo | Pathogenic | NA | |
| 47 | PPP1R12A (NM_002480.3) | Genitourinary and/or/brain malformation syndrome (MIM: # 618820) | AD | c.792+3A>C | NA | Heterozygous | De novo | Uncertain significance | NA |
| 48 | PCDH15 (NM_001384140.1) | Usher syndrome, type 1F (MIM: # 602083) | AR | c.1737C>G | p.(Tyr579 *) | Compound heterozygous | NA | Pathogenic | PMID: 22815625 |
| Gene deletion | NA | Pathogenic | NA | ||||||
| 49 | CDH23 (NM_022124.6) | Usher syndrome, type 1D (MIM: # 601067) | AR | c.5985C>A | p.(Tyr1995 *) | Homozygous | Paternal and Maternal | Pathogenic | PMID: 20513143 |
| 50 | WFS1 (NM_006005.3) | Deafness, autosomal dominant 6/14/38 (MIM: # 600965) | AD | c.2339G>C | p.(Gly780Ala) | Heterozygous | Maternal | Likely pathogenic | PMID: 23981289 |
| ABCC6 (NM_001171.6) | Pseudoxanthoma elasticum (MIM: # 264800) | AR | c.3491G>A | p.(Arg1164Gln) | Homozygous | Paternal and Maternal | Likely pathogenic | PMID: 16086317 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spedicati, B.; Santin, A.; Nardone, G.G.; Rubinato, E.; Lenarduzzi, S.; Graziano, C.; Garavelli, L.; Miccoli, S.; Bigoni, S.; Morgan, A.; et al. The Enigmatic Genetic Landscape of Hereditary Hearing Loss: A Multistep Diagnostic Strategy in the Italian Population. Biomedicines 2023, 11, 703. https://doi.org/10.3390/biomedicines11030703
Spedicati B, Santin A, Nardone GG, Rubinato E, Lenarduzzi S, Graziano C, Garavelli L, Miccoli S, Bigoni S, Morgan A, et al. The Enigmatic Genetic Landscape of Hereditary Hearing Loss: A Multistep Diagnostic Strategy in the Italian Population. Biomedicines. 2023; 11(3):703. https://doi.org/10.3390/biomedicines11030703
Chicago/Turabian StyleSpedicati, Beatrice, Aurora Santin, Giuseppe Giovanni Nardone, Elisa Rubinato, Stefania Lenarduzzi, Claudio Graziano, Livia Garavelli, Sara Miccoli, Stefania Bigoni, Anna Morgan, and et al. 2023. "The Enigmatic Genetic Landscape of Hereditary Hearing Loss: A Multistep Diagnostic Strategy in the Italian Population" Biomedicines 11, no. 3: 703. https://doi.org/10.3390/biomedicines11030703
APA StyleSpedicati, B., Santin, A., Nardone, G. G., Rubinato, E., Lenarduzzi, S., Graziano, C., Garavelli, L., Miccoli, S., Bigoni, S., Morgan, A., & Girotto, G. (2023). The Enigmatic Genetic Landscape of Hereditary Hearing Loss: A Multistep Diagnostic Strategy in the Italian Population. Biomedicines, 11(3), 703. https://doi.org/10.3390/biomedicines11030703