
Augmented Ouabain-Induced Vascular Response Reduces Cardiac Efficiency in Mice with Migraine-Associated Mutation in the Na+, K+-ATPase α2-Isoform
 , ,
, ,
Abstract
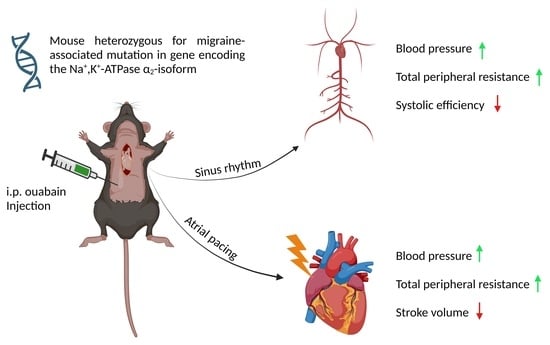
1. Introduction
2. Materials and Methods
2.1. Experimental Animals
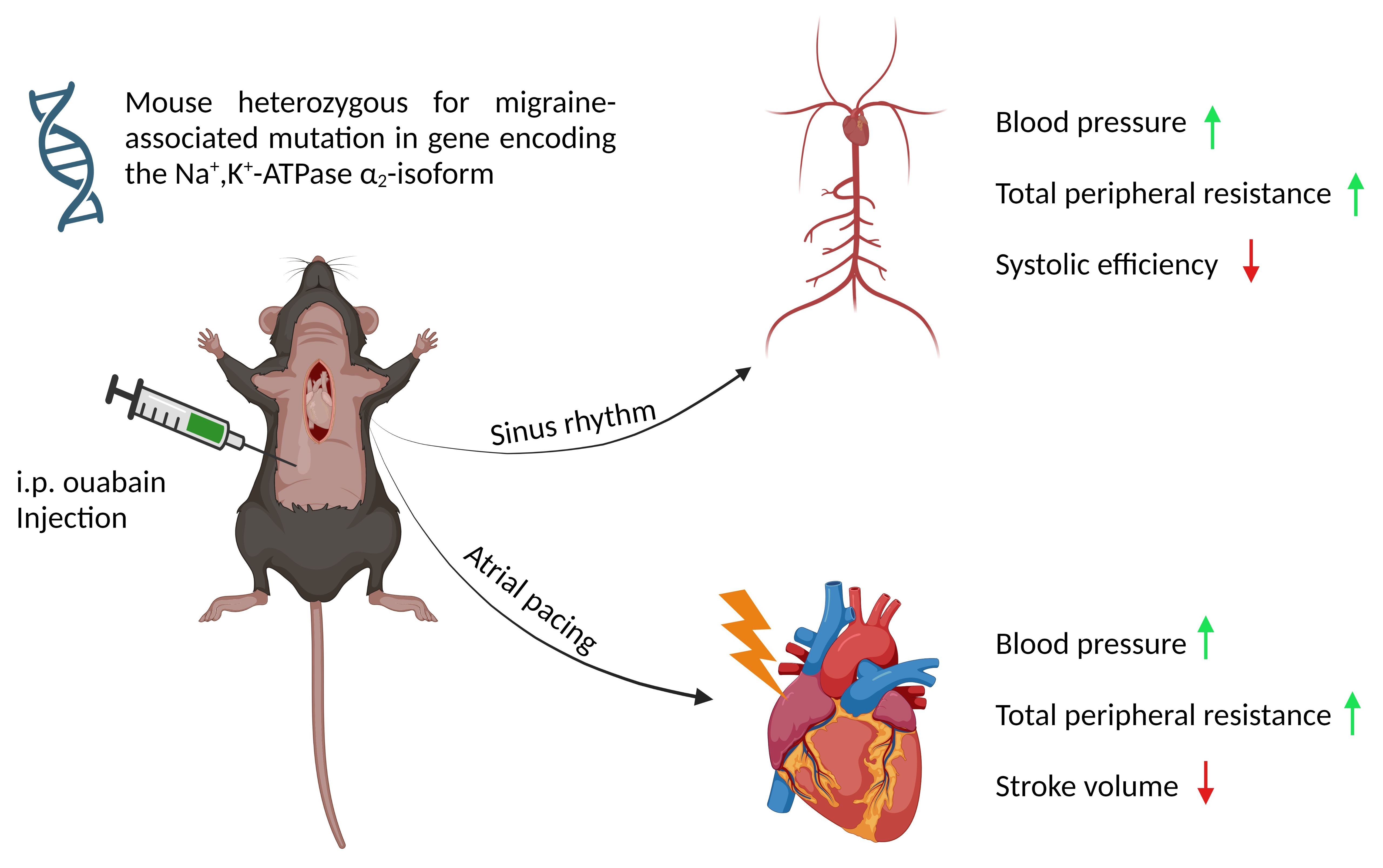
2.2. In Vivo Cardiovascular Study
2.3. Measurements of Plasma Ouabain Concentration
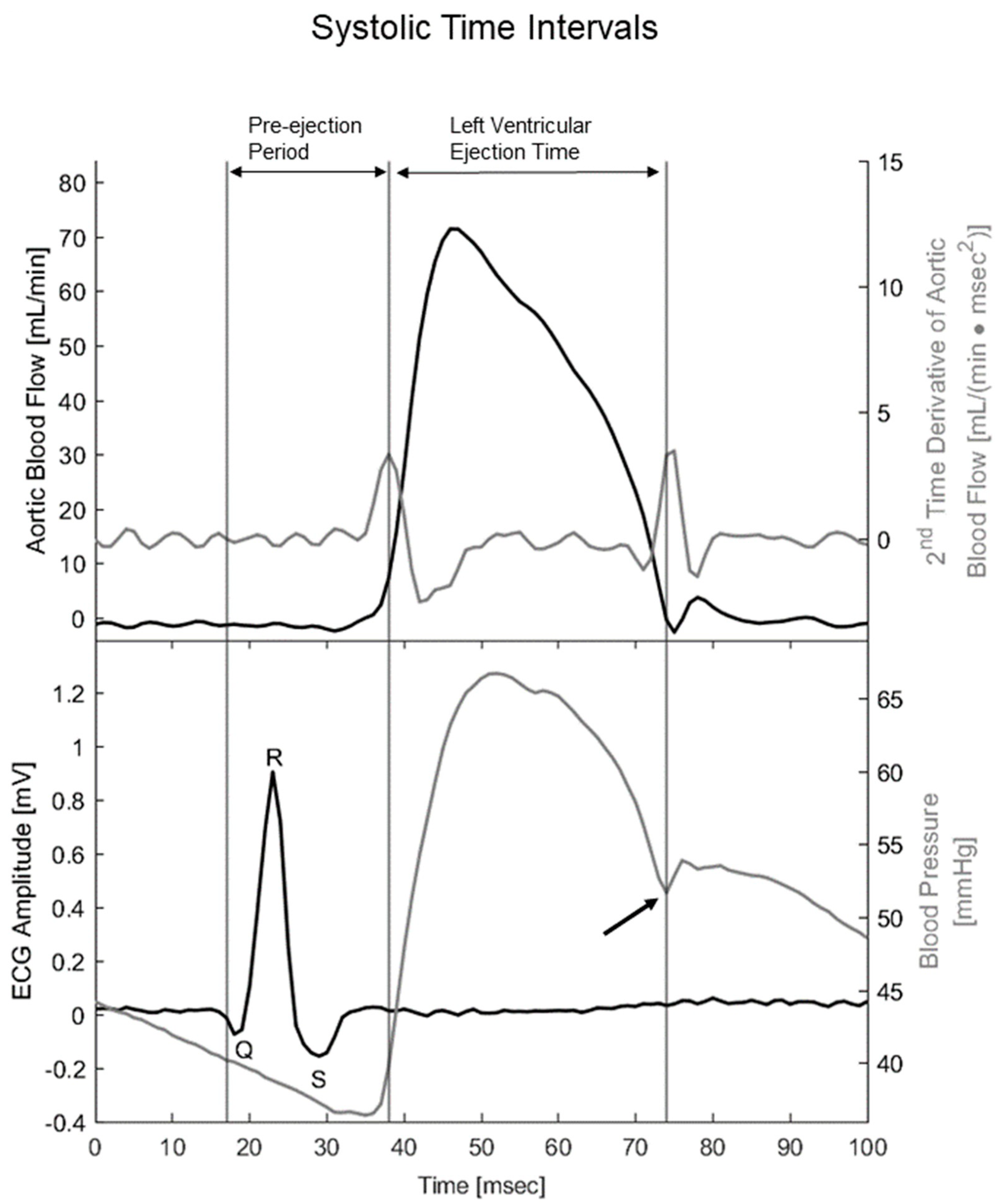
2.4. Data Acquisition and Calculations
2.5. Statistical Analysis
3. Results
3.1. Baseline Physiological Variables Were Similar between WT and α2+/G301R Mice
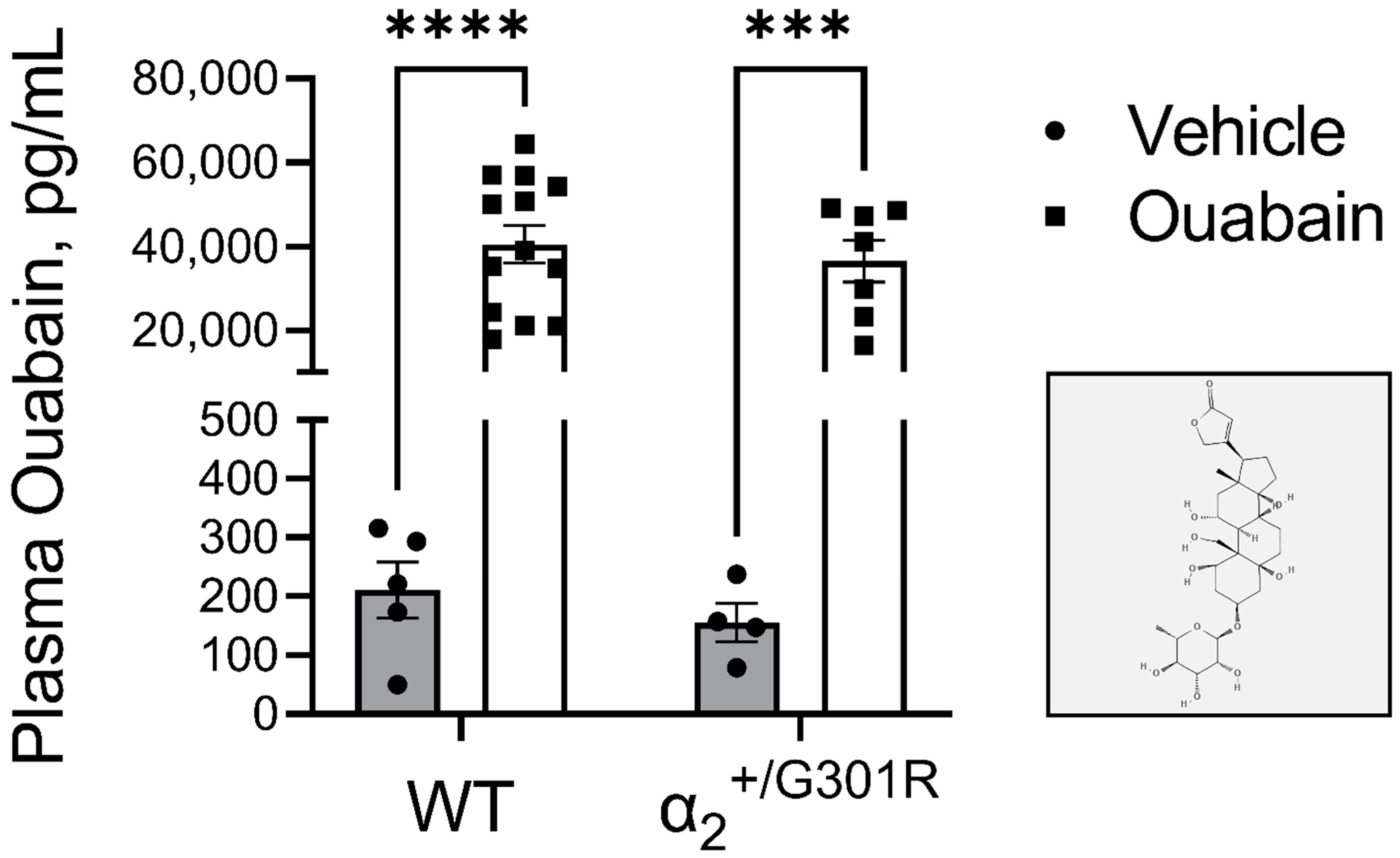
3.2. Administration of Ouabain Similarly Elevates the Plasma Ouabain Concentrations in WT and α2+/G301R Mice
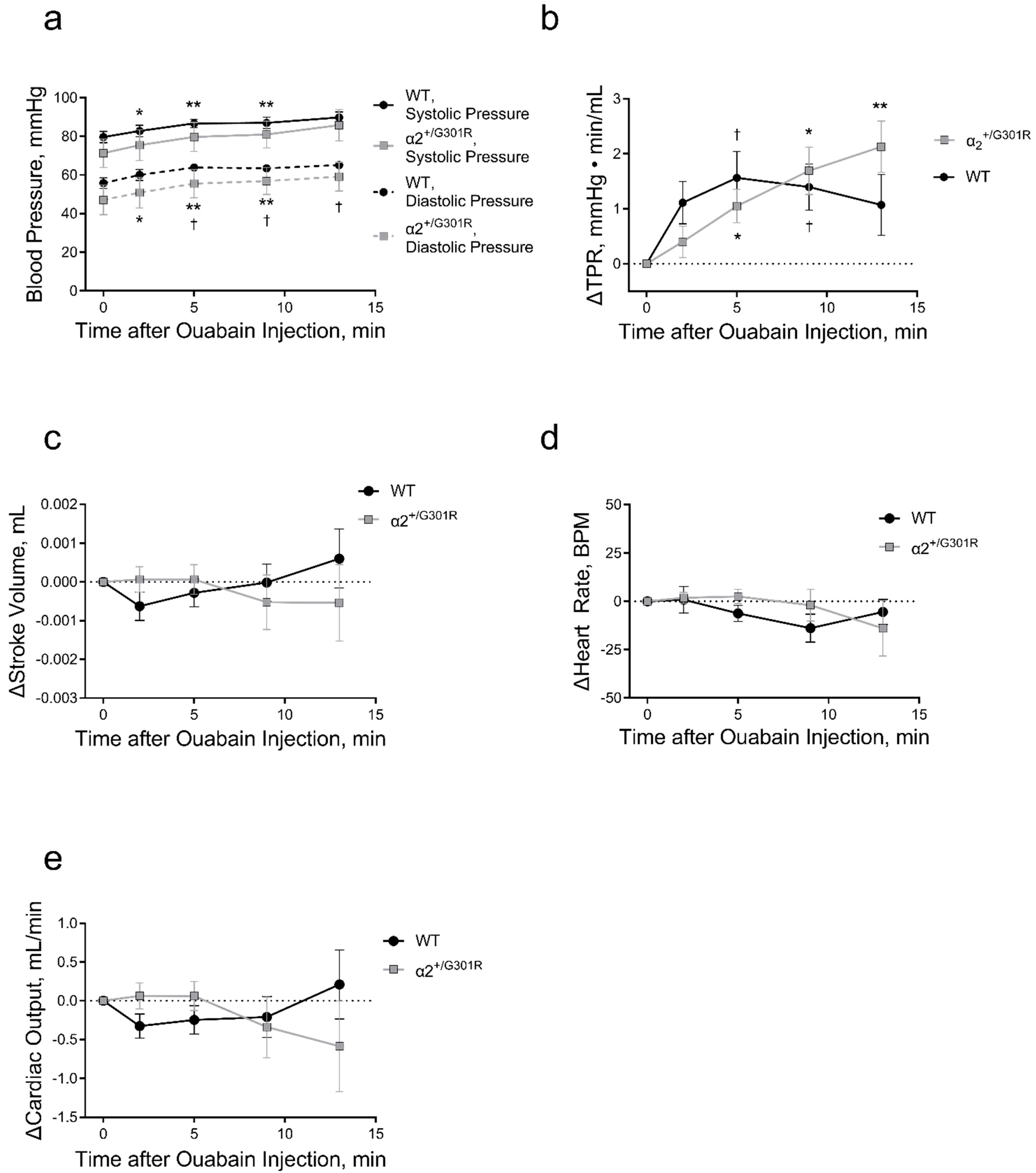
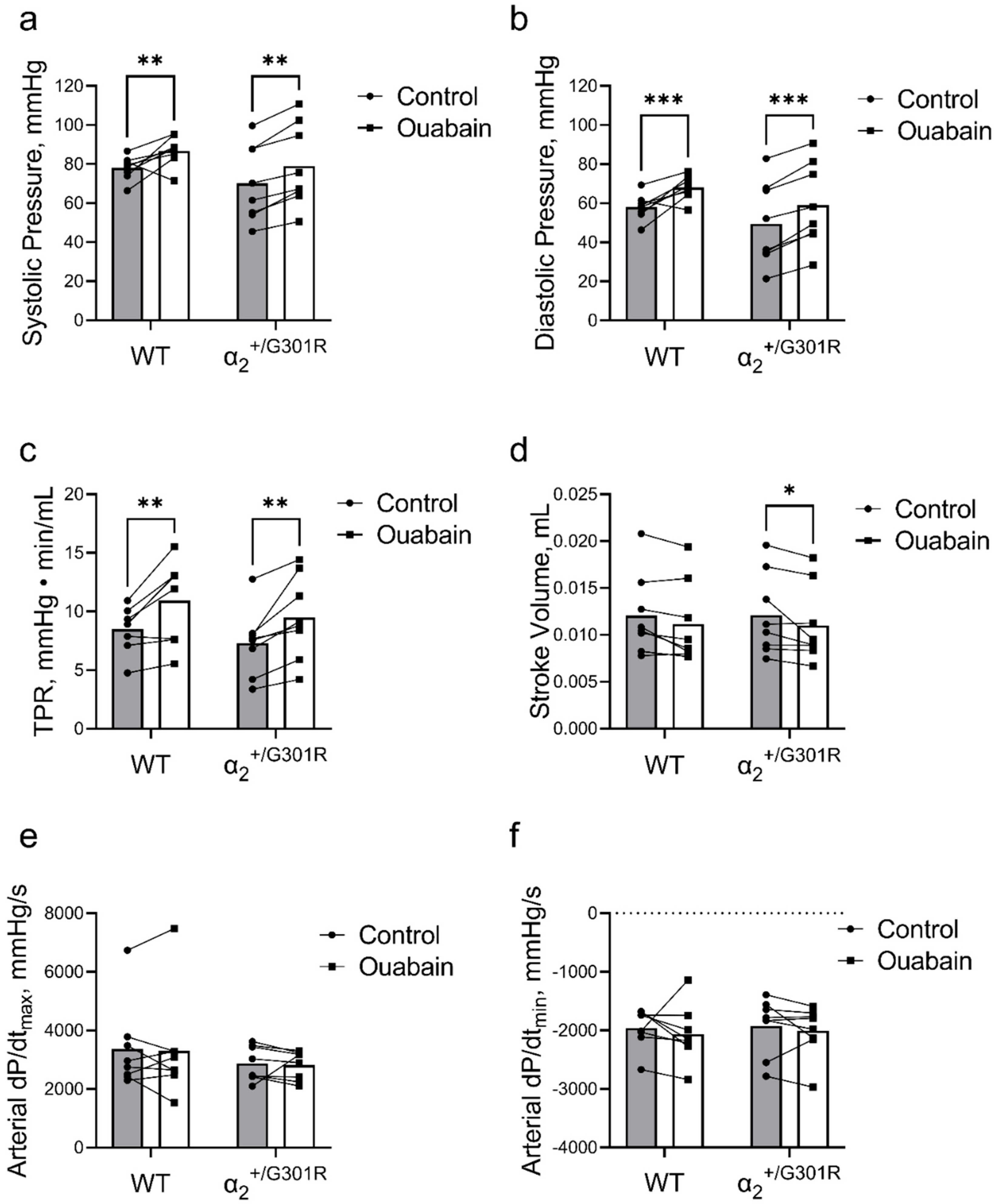
3.3. Ouabain Elevated Peripheral Vascular Resistance and Blood Pressure While Cardiac Variables Were Unchanged in Both WT and α2+/G301R Mice
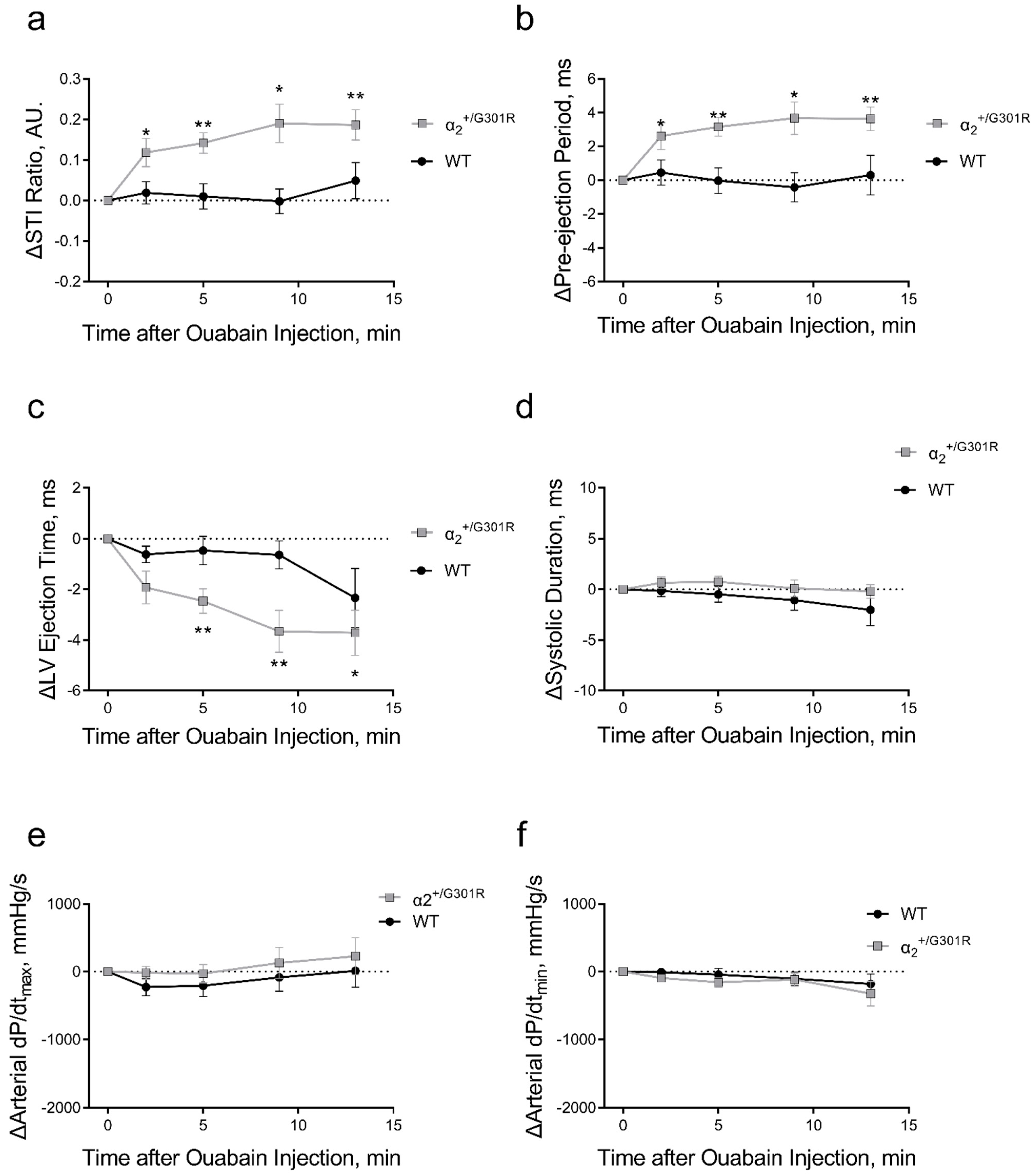
3.4. When Heart Rate Was Controlled, Ouabain Administration Reduced Stroke Volume in α2+/G301R Mice
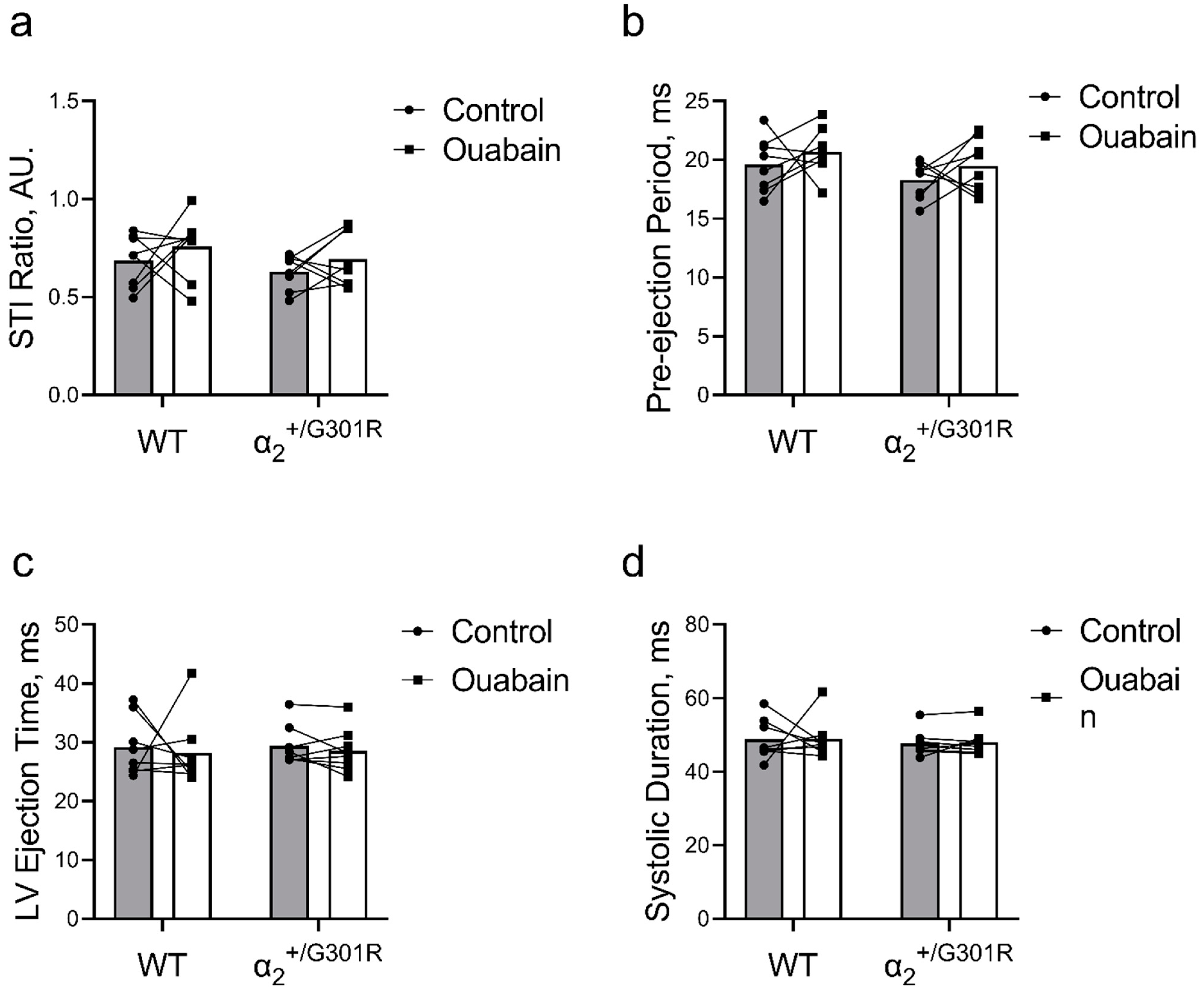
3.5. When Heart Rate Was Controlled at 11.3 Hz, Ouabain-Induced Changes to Systolic Time Intervals in α2+/G301R Mice Were Abolished
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skou, J.C. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim. Biophys. Acta Bioenerg. 1957, 23, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Clausen, M.V.; Hilbers, F.; Poulsen, H. The Structure and Function of the Na,K-ATPase Isoforms in Health and Disease. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef]
- Friedrich, T.; Tavraz, N.N.; Junghans, C. ATP1A2 Mutations in Migraine: Seeing through the Facets of an Ion Pump onto the Neurobiology of Disease. Front. Physiol. 2016, 7. [Google Scholar] [CrossRef]
- Li, Z.; Langhans, S.A. Transcriptional regulators of Na,K-ATPase subunits. Front. Cell Dev. Biol. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Huang, L.; Xie, Z.; Huang, W.H.; Askari, A. Partial inhibition of Na+/K+-ATPase by ouabain induces the Ca2+-dependent expressions of early-response genes in cardiac myocytes. J. Biol. Chem. 1996, 271, 10372–10378. [Google Scholar] [CrossRef] [PubMed]
- Pratt, R.D.; Brickman, C.R.; Cottrill, C.L.; Shapiro, J.I.; Liu, J. The Na/K-ATPase Signaling: From Specific Ligands to General Reactive Oxygen Species. Int. J. Mol. Sci. 2018, 19, 2600. [Google Scholar] [CrossRef]
- Mobasheri, A.; Avila, J.; Cózar-Castellano, I.; Brownleader, M.D.; Trevan, M.; Francis, M.J.; Lamb, J.F.; Martín-Vasallo, P. Na+, K+-ATPase isozyme diversity; comparative biochemistry and physiological implications of novel functional interactions. Biosci. Rep. 2000, 20, 51–91. [Google Scholar] [CrossRef]
- Blanco, G.; Mercer, R.W. Isozymes of the Na-K-ATPase: Heterogeneity in structure, diversity in function. Am. J. Physiol. Renal Physiol. 1998, 275, F633–F650. [Google Scholar] [CrossRef]
- Staehr, C.; Hangaard, L.; Bouzinova, E.V.; Kim, S.; Rajanathan, R.; Boegh Jessen, P.; Luque, N.; Xie, Z.; Lykke-Hartmann, K.; Sandow, S.L.; et al. Smooth muscle Ca2+ sensitization causes hypercontractility of middle cerebral arteries in mice bearing the familial hemiplegic migraine type 2 associated mutation. J. Cereb. Blood Flow Metab. 2019, 39, 1570–1587. [Google Scholar] [CrossRef]
- Staehr, C.; Rajanathan, R.; Postnov, D.D.; Hangaard, L.; Bouzinova, E.V.; Lykke-Hartmann, K.; Bach, F.W.; Sandow, S.L.; Aalkjaer, C.; Matchkov, V.V. Abnormal neurovascular coupling as a cause of excess cerebral vasodilation in familial migraine. Cardiovasc. Res. 2019, 116, 2009–2020. [Google Scholar] [CrossRef]
- Staehr, C.; Rohde, P.D.; Krarup, N.T.; Ringgaard, S.; Laustsen, C.; Johnsen, J.; Nielsen, R.; Beck, H.C.; Morth, J.P.; Lykke-Hartmann, K.; et al. Migraine Associated Mutation in the Na,K-ATPase Leads to Disturbances in Cardiac Metabolism and Reduced Cardiac Function. J. Am. Heart Assoc. 2022, 11, e021814. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, D.E.; Miller, R.B.; Thiesfeldt, S.; Lakhani, H.V.; Shapiro, J.I.; Sodhi, K. The Role of Na/K-ATPase Signaling in Oxidative Stress Related to Aging: Implications in Obesity and Cardiovascular Disease. Int. J. Mol. Sci. 2018, 19, 2139. [Google Scholar] [CrossRef]
- Rindler, T.N.; Dostanic, I.; Lasko, V.M.; Nieman, M.L.; Neumann, J.C.; Lorenz, J.N.; Lingrel, J.B. Knockout of the Na,K-ATPase α2-isoform in the cardiovascular system does not alter basal blood pressure but prevents ACTH-induced hypertension. Am. J. Physiol.-Heart Circ. Physiol. 2011, 301, H1396–H1404. [Google Scholar] [CrossRef]
- Rindler, T.N.; Lasko, V.M.; Nieman, M.L.; Okada, M.; Lorenz, J.N.; Lingrel, J.B. Knockout of the Na,K-ATPase α2-isoform in cardiac myocytes delays pressure overload-induced cardiac dysfunction. Am. J. Physiol.-Heart Circ. Physiol. 2013, 304, H1147–H1158. [Google Scholar] [CrossRef]
- Correll, R.N.; Eder, P.; Burr, A.R.; Despa, S.; Davis, J.; Bers, D.M.; Molkentin, J.D. Overexpression of the Na+/K+ ATPase α2 but not α1 isoform attenuates pathological cardiac hypertrophy and remodeling. Circ. Res. 2014, 114, 249–256. [Google Scholar] [CrossRef]
- Shattock, M.J.; Ottolia, M.; Bers, D.M.; Blaustein, M.P.; Boguslavskyi, A.; Bossuyt, J.; Bridge, J.H.B.; Chen-Izu, Y.; Clancy, C.E.; Edwards, A.; et al. Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. Physiol. J. 2015, 593, 1361–1382. [Google Scholar] [CrossRef]
- Skogestad, J.; Aronsen, J.M. Regulation of Cardiac Contractility by the Alpha 2 Subunit of the Na+/K+-ATPase. Front. Physiol. 2022, 13. [Google Scholar] [CrossRef]
- Pritchard, T.J.; Bowman, P.S.; Jefferson, A.; Tosun, M.; Lynch, R.M.; Paul, R.J. Na+-K+-ATPase and Ca2+ clearance proteins in smooth muscle: A functional unit. Am. J. Physiol.-Heart Circ. Physiol. 2010, 299, H548–H556. [Google Scholar] [CrossRef]
- Pietrobon, D. Familial hemiplegic migraine. Neurotherapeutics 2007, 4, 274–284. [Google Scholar] [CrossRef]
- Spadaro, M.; Ursu, S.; Lehmann-Horn, F.; Liana, V.; Giovanni, A.; Paola, G.; Frontali, M.; Jurkat-Rott, K. A G301R Na+/K+-ATPase mutation causes familial hemiplegic migraine type 2 with cerebellar signs. Neurogenetics 2004, 5, 177–185. [Google Scholar] [CrossRef]
- Bøttger, P.; Glerup, S.; Gesslein, B.; Illarionova, N.B.; Isaksen, T.J.; Heuck, A.; Clausen, B.H.; Füchtbauer, E.M.; Gramsbergen, J.B.; Gunnarson, E.; et al. Glutamate-system defects behind psychiatric manifestations in a familial hemiplegic migraine type 2 disease-mutation mouse model. Sci. Rep. 2016, 6. [Google Scholar]
- James, P.F.; Grupp, I.L.; Grupp, G.; Woo, A.L.; Askew, G.R.; Croyle, M.L.; Walsh, R.A.; Lingrel, J.B. Identification of a specific role for the Na,K-ATPase alpha 2 isoform as a regulator of calcium in the heart. Mol. Cell. 1999, 3, 555–563. [Google Scholar] [CrossRef]
- Botelho, A.F.M.; Pierezan, F.; Soto-Blanco, B.; Melo, M.M. A review of cardiac glycosides: Structure, toxicokinetics, clinical signs, diagnosis and antineoplastic potential. Toxicon 2019, 158, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Pulgar, V.M.; Jeffers, A.B.; Rashad, H.M.; Diz, D.I.; Aileru, A.A. Increased constrictor tone induced by ouabain treatment in rats. J. Cardiovasc. Pharmacol. 2013, 62, 174–183. [Google Scholar] [CrossRef]
- Li, M.; Martin, A.; Wen, C.; Turner, S.W.; Lewis, L.K.; Whitworth, J.A. Long-term ouabain administration does not alter blood pressure in conscious Sprague-Dawley rats. Clin. Exp. Pharmacol. Physiol. 1995, 22, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Cargnelli, G.; Trevisi, L.; Debetto, P.; Luciani, S.; Bova, S. Effect of long-term ouabain treatment on contractile responses of rat aortae. J. Cardiovasc. Pharmacol. 2000, 35, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Altamirano, J.; Li, Y.; DeSantiago, J.; Piacentino, V., 3rd; Houser, S.R.; Bers, D.M. The inotropic effect of cardioactive glycosides in ventricular myocytes requires Na+-Ca2+ exchanger function. J. Physiol. 2006, 575, 845–854. [Google Scholar] [CrossRef]
- Obrien, W.J.; Lingrel, J.B.; Wallick, E.T. Ouabain Binding Kinetics of the Rat Alpha Two and Alpha Three Isoforms of the Sodium-Potassium Adenosine Triphosphate. Arch. Biochem. 1994, 310, 32–39. [Google Scholar] [CrossRef]
- Yuan, C.M.; Manunta, P.; Hamlyn, J.M.; Chen, S.; Bohen, E.; Yeun, J.; Haddy, F.J.; Pamnani, M.B. Long-term ouabain administration produces hypertension in rats. Hypertension. 1993, 22, 178–187. [Google Scholar] [CrossRef]
- Xavier, F.E.; Rossoni, L.V.; Alonso, M.J.; Balfagón, G.; Vassallo, D.V.; Salaices, M. Ouabain-induced hypertension alters the participation of endothelial factors in α-adrenergic responses differently in rat resistance and conductance mesenteric arteries. Br. J. Pharmacol. 2004, 143, 215–225. [Google Scholar] [CrossRef]
- Shimoni, Y.; Gotsman, M.; Deutsch, J.; Kachalsky, S.; Lichtstein, D. Endogenous ouabain-like compound increases heart muscle contractility. Nature 1984, 307, 369–371. [Google Scholar] [CrossRef]
- Simaan, J.; Fawaz, G.; Jarawan, S. The effect of ouabain-induced contractility on myocardial oxygen consumption. Naunyn Schmiedebergs Arch. Pharmacol. 1971, 271, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Rajanathan, R.; Pedersen, T.M.; Thomsen, M.B.; Botker, H.E.; Matchkov, V.V. Phenylephrine-Induced Cardiovascular Changes in the Anesthetized Mouse: An Integrated Assessment of in vivo Hemodynamics Under Conditions of Controlled Heart Rate. Front. Physiol. 2022, 13, 831724. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Onaka, T.; Yamakado, M.; Nakai, J.; Ishikawa, T.-o.; Taketo, M.M.; Kawakami, K. Degeneration of the Amygdala/Piriform Cortex and Enhanced Fear/Anxiety Behaviors in Sodium Pump α2 Subunit (Atp1a2)-Deficient Mice. J. Neurosci. 2003, 23, 4667–4676. [Google Scholar] [CrossRef]
- Ikeda, K.; Onimaru, H.; Yamada, J.; Inoue, K.; Ueno, S.; Onaka, T.; Toyoda, H.; Arata, A.; Ishikawa, T.-o.; Taketo, M.M.; et al. Malfunction of Respiratory-Related Neuronal Activity in Na+, K+-ATPase α2 Subunit-Deficient Mice Is Attributable to Abnormal Cl− Homeostasis in Brainstem Neurons. J. Neurosci. 2004, 24, 10693–10701. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; Emerson, M.; et al. Reporting animal research: Explanation and elaboration for the ARRIVE guidelines 2.0. PLoS Biol. 2020, 18, e3000411. [Google Scholar] [CrossRef] [PubMed]
- Hauber, H.P.; Karp, D.; Goldmann, T.; Vollmer, E.; Zabel, P. Effect of low tidal volume ventilation on lung function and inflammation in mice. BMC Pulm. Med. 2010, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.B.; Staehr, C.; Rohde, P.D.; Homilius, C.; Kim, S.; Nyegaard, M.; Matchkov, V.V.; Boedtkjer, E. PTPRG is an ischemia risk locus essential for HCO3−-dependent regulation of endothelial function and tissue perfusion. eLife 2020, 9. [Google Scholar] [CrossRef]
- Janssen, B.; Debets, J.; Leenders, P.; Smits, J. Chronic measurement of cardiac output in conscious mice. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2002, 282, R928–R935. [Google Scholar] [CrossRef]
- Hill, L.K.; Sollers Iii, J.J.; Thayer, J.F. Resistance reconstructed estimation of total peripheral resistance from computationally derived cardiac output - biomed 2013. Biomed. Sci. Instrum. 2013, 49, 216–223. [Google Scholar]
- Monge Garcia, M.I.; Jian, Z.; Settels, J.J.; Hunley, C.; Cecconi, M.; Hatib, F.; Pinsky, M.R. Performance comparison of ventricular and arterial dP/dtmax for assessing left ventricular systolic function during different experimental loading and contractile conditions. Crit. Care 2018, 22, 325. [Google Scholar] [CrossRef] [PubMed]
- Cohn, P.F.; Liedtke, A.J.; Serur, J.; Sonnenblick, E.H.; Urschel, C.W. Maximal rate of pressure fall (peak negative dP/dt) during ventricular relaxation. Cardiovasc. Res. 1972, 6, 263–267. [Google Scholar] [CrossRef]
- Alhakak, A.S.; Teerlink, J.R.; Lindenfeld, J.; Böhm, M.; Rosano, G.M.C.; Biering-Sørensen, T. The significance of left ventricular ejection time in heart failure with reduced ejection fraction. Eur. J. Heart Fail. 2021, 23, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Boudoulas, H. Systolic time intervals. Eur. Heart J. 1990, 11 Suppl I, 93–104. [Google Scholar] [CrossRef]
- Vistisen, S.T.; Juhl-Olsen, P.; Frederiksen, C.A.; Kirkegaard, H. Variations in the pre-ejection period induced by deep breathing do not predict the hemodynamic response to early haemorrhage in healthy volunteers. J. Clin. Monit. Comput. 2014, 28, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Politi, M.T.; Ghigo, A.; Fernández, J.M.; Khelifa, I.; Gaudric, J.; Fullana, J.M.; Lagrée, P.-Y. The dicrotic notch analyzed by a numerical model. Comput. Biol. Med. 2016, 72, 54–64. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 439501, O.R.J. 2022. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Ouabain (accessed on 18 January 2022).
- Wittenberg, S.M. Chronotropic Effects of Ouabain and Heart Rate on Canine Atrium in Vivo. Circ. Res. 1974, 34, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Furukawa, Y.; Kobayashi, M. Direct positive chronotropic and inotropic effects of ouabain in the isolated and blood-perfused canine atrium. Jpn. Heart J. 1978, 19, 877–885. [Google Scholar] [CrossRef]
- Salahudeen, M.S.; Nishtala, P.S. An overview of pharmacodynamic modelling, ligand-binding approach and its application in clinical practice. Saudi. Pharm. J. 2017, 25, 165–175. [Google Scholar] [CrossRef]
- Matchkov, V.V.; Krivoi, I.I. Specialized Functional Diversity and Interactions of the Na,K-ATPase. Front. Physiol. 2016, 7, 179. [Google Scholar] [CrossRef]
- Ferrandi, M.; Manunta, P.; Balzan, S.; Hamlyn, J.M.; Bianchi, G.; Ferrari, P. Ouabain-like Factor Quantification in Mammalian Tissues and Plasma. Hypertension 1997, 30, 886–896. [Google Scholar] [CrossRef]
- Goto, A.; Yamada, K.; Nagoshi, H.; Terano, Y.; Omata, M. Stress-Induced Elevation of Ouabainlike Compound in Rat Plasma and Adrenal. Hypertension 1995, 26, 1173–1176. [Google Scholar] [CrossRef]
- Bai, Y.-L.; Chu, Q.-J.; Li, J.; Chen, Y.-Y.; Li, W.-J.; Zhang, Q. A comparison of the effects of digoxin, ouabain and milrinone on naloxone-precipitated withdrawal syndrome in mice. Eur. J. Pharmacol. 2012, 694, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Akiyama, T.; Kitagawa, H.; Komaki, F.; Mori, H.; Kawada, T.; Sunagawa, K.; Sugimachi, M. Characterization of ouabain-induced noradrenaline and acetylcholine release from in situ cardiac autonomic nerve endings. Acta Physiol. 2007, 191, 275–284. [Google Scholar] [CrossRef]
- Manunta, P.; Rogowski, A.C.; Hamilton, B.P.; Hamlyn, J.M. Ouabain-induced hypertension in the rat: Relationships among plasma and tissue ouabain and blood pressure. J. Hypertens. 1994, 12, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Cooke, J.P.; Shepherd, J.T.; Vanhoutte, P.M. Vasoconstriction induced by ouabain in the canine coronary artery: Contribution of adrenergic and nonadrenergic responses. Cardiovasc. Drugs Ther. 1988, 2, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Woolfson, R.G.; Hilton, P.J.; Poston, L. Effects of ouabain and low sodium on contractility of human resistance arteries. Hypertension 1990, 15, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wang, Y.; Summer, W.R.; Greenberg, S.S. Ouabain Enhances Basal Release of Nitric Oxide from Carotid Artery. Am. J. Med. Sci. 1993, 305, 157–163. [Google Scholar] [CrossRef]
- Zhang, L.; Aalkjaer, C.; Matchkov, V.V. The Na,K-ATPase-Dependent Src Kinase Signaling Changes with Mesenteric Artery Diameter. Int. J. Mol. Sci. 2018, 19, 2489. [Google Scholar] [CrossRef]
- Zulian, A.; Linde, C.I.; Pulina, M.V.; Baryshnikov, S.G.; Papparella, I.; Hamlyn, J.M.; Golovina, V.A. Activation of c-SRC underlies the differential effects of ouabain and digoxin on Ca(2+) signaling in arterial smooth muscle cells. Am. J. Physiol. Cell Physiol. 2013, 304, C324–C333. [Google Scholar] [CrossRef]
- Zhang, J.; Lee, M.Y.; Cavalli, M.; Chen, L.; Berra-Romani, R.; Balke, C.W.; Bianchi, G.; Ferrari, P.; Hamlyn, J.M.; Iwamoto, T.; et al. Sodium pump α2 subunits control myogenic tone and blood pressure in mice. Physiol. J. 2005, 569, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Matchkov, V.V.; Moeller-Nielsen, N.; Dam, V.S.; Nourian, Z.; Briggs Boedtkjer, D.M.; Aalkjaer, C. The α2 isoform of the Na,K-pump is important for intercellular communication, agonist-induced contraction, and EDHF-like response in rat mesenteric arteries. Am. J. Physiol.-Heart Circ. Physiol. 2012, 303, H36–H46. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ceretti, E.; Samson, J.P.; Reisin, I.; Schanne, O.F. Ionic and electrical effects of quabain on isolated rabbit hearts. J. Mol. Cell. Cardiol. 1977, 9, 51–61. [Google Scholar] [CrossRef]
- Psotka, M.A.; Gottlieb, S.S.; Francis, G.S.; Allen, L.A.; Teerlink, J.R.; Adams, K.F., Jr.; Rosano, G.M.C.; Lancellotti, P. Cardiac Calcitropes, Myotropes, and Mitotropes: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 2345–2353. [Google Scholar] [CrossRef] [PubMed]
- Hamlin, R.L.; del Rio, C. dP/dtmax — A measure of ‘baroinometry’. J. Pharmacol. Toxicol. Methods 2012, 66, 63–65. [Google Scholar] [CrossRef]
- Warrington, S.J.; Weerasuriya, K.; Burgess, C.D. Correction of systolic time intervals for heart rate: A comparison of individual with population derived regression equations. Br. J. Clin. Pharmacol. 1988, 26, 155–165. [Google Scholar] [CrossRef]
- Wolf, G.K.; Belz, G.G.; Stauch, M. Systolic time intervals—Correction for heart rate. Basic Res. Cardiol. 1978, 73, 85–96. [Google Scholar] [CrossRef]
- Mertens, H.M.; Mannebach, H.; Trieb, G.; Gleichmann, U. Influence of heart rate on systolic time intervals: Effects of atrial pacing versus dynamic exercise. Clin. Cardiol. 1981, 4, 22–27. [Google Scholar] [CrossRef]
- Ritterhoff, J.; Tian, R. Metabolism in cardiomyopathy: Every substrate matters. Cardiovasc. Res. 2017, 113, 411–421. [Google Scholar] [CrossRef]
- Sabbah, H.N. Silent disease progression in clinically stable heart failure. Eur. J. Heart Fail. 2017, 19, 469–478. [Google Scholar] [CrossRef]
- Hippisley-Cox, J.; Coupland, C.; Brindle, P. Development and validation of QRISK3 risk prediction algorithms to estimate future risk of cardiovascular disease: Prospective cohort study. BMJ 2017, 357, j2099. [Google Scholar] [CrossRef] [PubMed]
- Adelborg, K.; Szépligeti, S.K.; Holland-Bill, L.; Ehrenstein, V.; Horváth-Puhó, E.; Henderson, V.W.; Sørensen, H.T. Migraine and risk of cardiovascular diseases: Danish population based matched cohort study. BMJ 2018, 360, k96. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, H.G.; Griffiths, L.R. Genetics of Migraine: Insights into the Molecular Basis of Migraine Disorders. Headache J. Head Face Pain 2017, 57, 537–569. [Google Scholar] [CrossRef]
- Pellacani, S.; Sicca, F.; Di Lorenzo, C.; Grieco, G.S.; Valvo, G.; Cereda, C.; Rubegni, A.; Santorelli, F.M. The Revolution in Migraine Genetics: From Aching Channels Disorders to a Next-Generation Medicine. Front. Cell. Neurosci. 2016, 10, 156. [Google Scholar] [CrossRef] [PubMed]
- Tietjen, G. Migraine as a Systemic Vasculopathy. Cephalalgia 2009, 29, 989–996. [Google Scholar] [CrossRef]
- Venugopal, N.; Sriram Gopal, M.R. Migraine is a marker for systemic disease. Indian J. Ophthalmol. 2013, 61, 613. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wild Type (n = 12) | α2+/G301R (n = 12) | p-Value | |
|---|---|---|---|
| Age (weeks) | 41 ± 2 | 45 ± 3 | 0.23 |
| Body mass (g) | 35.3 ± 1.6 | 35.3 ± 0.8 | >0.99 |
| Cardiovascular variables and respiratory parameters | |||
| Systolic pressure (mmHg) | 76.09 ± 2.97 | 72.09 ± 4.68 | 0.48 |
| Diastolic pressure (mmHg) | 49.71 ± 3.53 | 46.16 ± 5.00 | 0.57 |
| Total peripheral resistance (mmHg•min/mL) | 6.89 ± 0.71 | 7.21 ± 0.73 | 0.76 |
| Cardiac output (mL/min) | 9.13 ± 0.75 | 8.28 ± 0.93 | 0.48 |
| Stroke volume (µL) | 17.20 ± 1.50 | 15.66 ± 1.83 | 0.52 |
| Heart rate (BPM) | 534 ± 8 | 530 ± 10 | 0.81 |
| End-tidal CO2 (%) | 3.69 ± 0.13 | 3.76 ± 0.11 | 0.69 |
| Ventilation rate (min−1) | 116 ± 6 | 114 ± 7 | 0.87 |
| Systolic time interval ratio (AU.) | 0.59 ± 0.04 | 0.54 ± 0.03 | 0.27 |
| Pre-ejection period (ms) | 19.2 ± 0.8 | 18.2 ± 0.5 | 0.28 |
| Left ventricular ejection time (ms) | 33.2 ± 1.5 | 34.4 ± 1.6 | 0.58 |
| Systolic duration (ms) | 52.4 ± 1.6 | 52.6 ± 1.8 | 0.82 |
| Arterial dP/dtmax (mmHg/s) | 3705 ± 298.9 | 3142 ± 197.9 | 0.13 |
| Arterial dP/dtmin (mmHg/s) | −2157 ± 92.78 | −2106 ± 134.4 | 0.76 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajanathan, R.; Pedersen, T.M.; Guldbrandsen, H.O.; Olesen, L.F.; Thomsen, M.B.; Bøtker, H.E.; Matchkov, V.V. Augmented Ouabain-Induced Vascular Response Reduces Cardiac Efficiency in Mice with Migraine-Associated Mutation in the Na+, K+-ATPase α2-Isoform. Biomedicines 2023, 11, 344. https://doi.org/10.3390/biomedicines11020344
Rajanathan R, Pedersen TM, Guldbrandsen HO, Olesen LF, Thomsen MB, Bøtker HE, Matchkov VV. Augmented Ouabain-Induced Vascular Response Reduces Cardiac Efficiency in Mice with Migraine-Associated Mutation in the Na+, K+-ATPase α2-Isoform. Biomedicines. 2023; 11(2):344. https://doi.org/10.3390/biomedicines11020344
Chicago/Turabian StyleRajanathan, Rajkumar, Tina Myhre Pedersen, Halvor Osterby Guldbrandsen, Lenette Foldager Olesen, Morten B. Thomsen, Hans Erik Bøtker, and Vladimir V. Matchkov. 2023. "Augmented Ouabain-Induced Vascular Response Reduces Cardiac Efficiency in Mice with Migraine-Associated Mutation in the Na+, K+-ATPase α2-Isoform" Biomedicines 11, no. 2: 344. https://doi.org/10.3390/biomedicines11020344
APA StyleRajanathan, R., Pedersen, T. M., Guldbrandsen, H. O., Olesen, L. F., Thomsen, M. B., Bøtker, H. E., & Matchkov, V. V. (2023). Augmented Ouabain-Induced Vascular Response Reduces Cardiac Efficiency in Mice with Migraine-Associated Mutation in the Na+, K+-ATPase α2-Isoform. Biomedicines, 11(2), 344. https://doi.org/10.3390/biomedicines11020344