
Piperine and Its Metabolite’s Pharmacology in Neurodegenerative and Neurological Diseases




Abstract
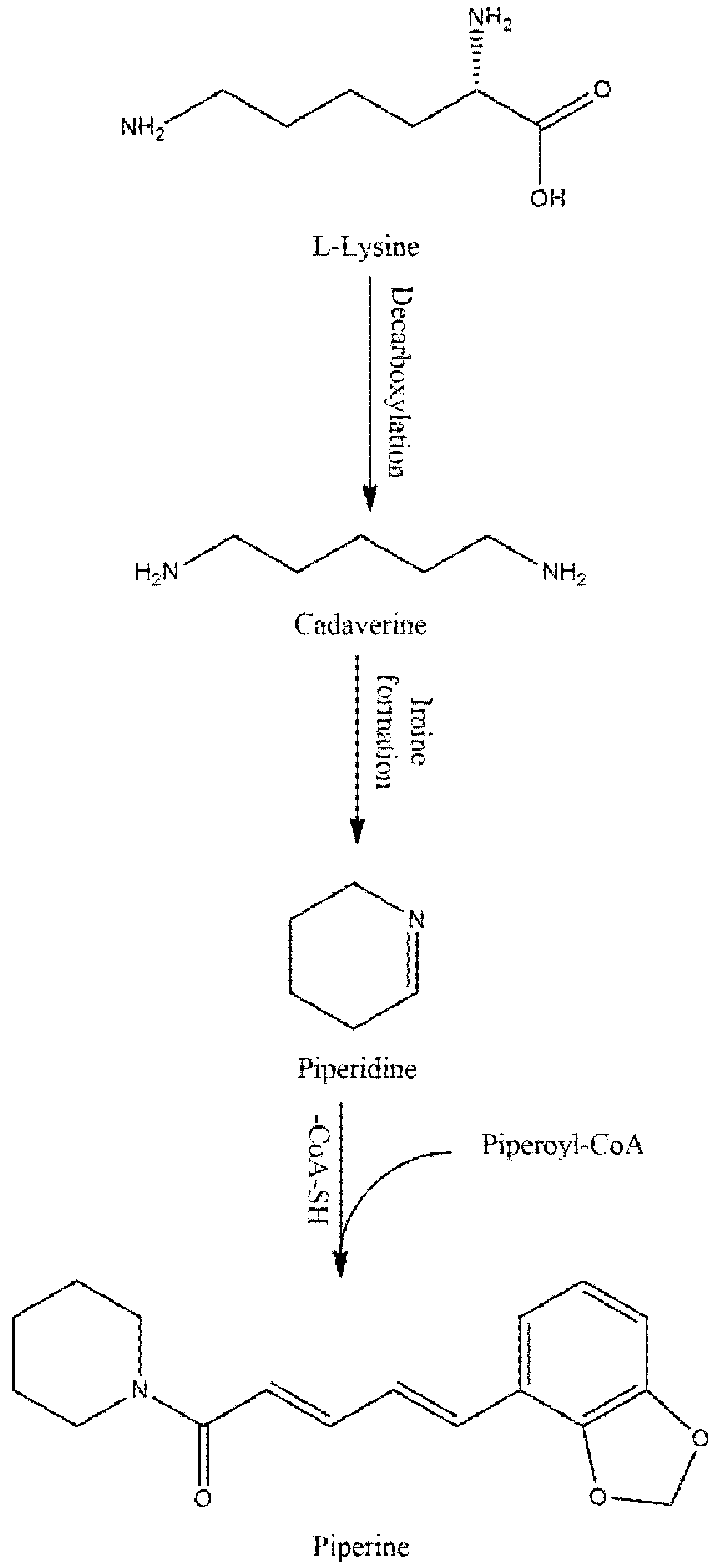
:1. Introduction
2. Pharmacokinetic and Pharmacodynamic Properties of PIP
2.1. PIP Interaction with Cytochrome P450 Superfamily Enzymes
2.2. PIP-Mediated Regulation of Drug Pharmacology
2.3. PIP Effect on Other Drug Metabolizing Enzyme
3. PIP Metabolism and Effect on Neurodegeneration
3.1. Catabolism of PIP
3.2. Excretion of Metabolites
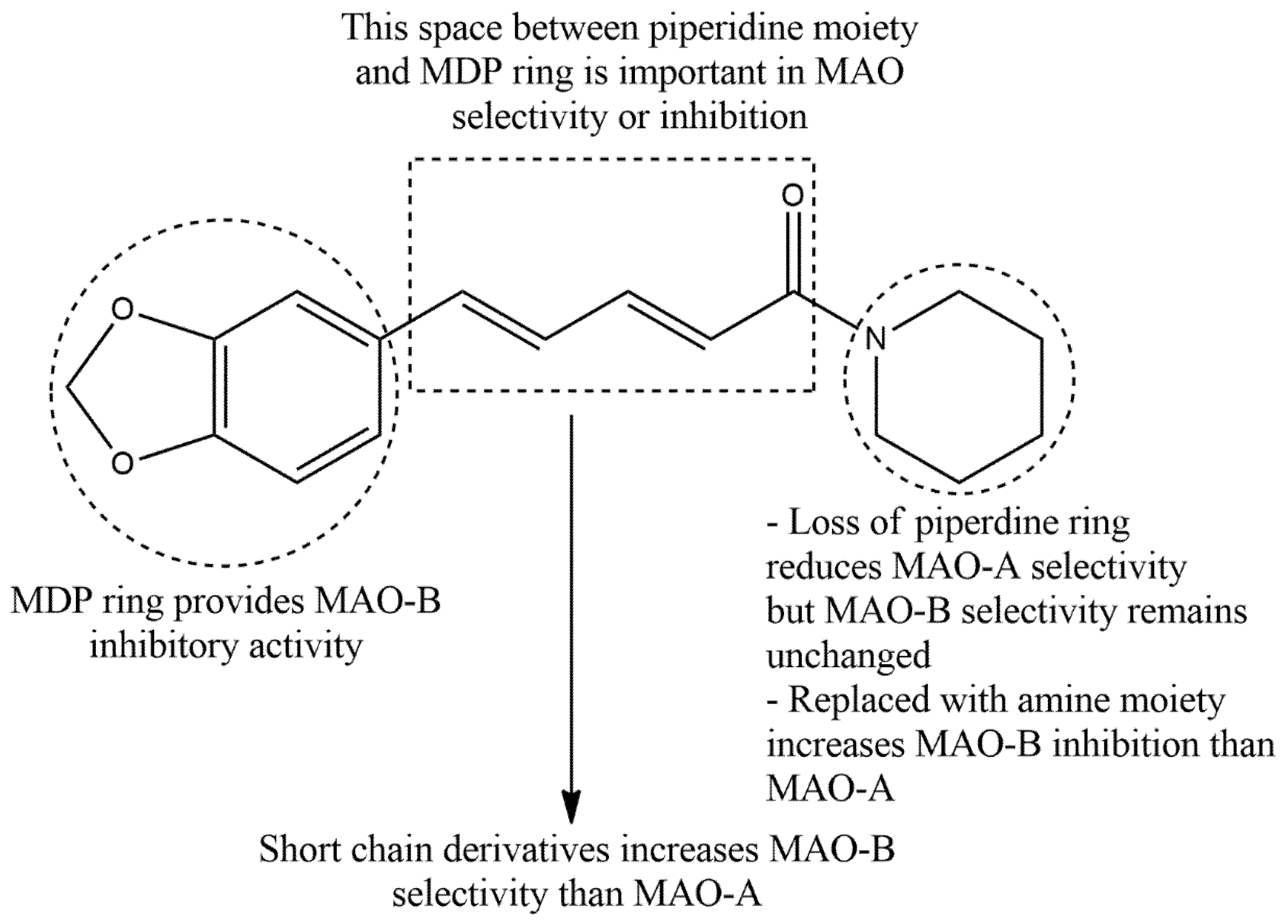
3.3. SAR of PIP in Neuropharmacology
4. PIP in Neurological and Psychiatric Diseases
4.1. AD
4.2. PD
4.3. Huntington’s Disease
4.4. Epilepsy
4.5. Other Neurological Diseases
5. Therapeutic Index and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Khan, A.; Jahan, S.; Imtiyaz, Z.; Alshahrani, S.; Antar Makeen, H.; Mohammed Alshehri, B.; Kumar, A.; Arafah, A.; Rehman, M.U. Neuroprotection: Targeting Multiple Pathways by Naturally Occurring Phytochemicals. Biomedicines 2020, 8, 284. [Google Scholar] [CrossRef]
- Mohapatra, M.; Basak, U. Evaluation of Piperine Content from Roots of Piper Longum Linn., Originated from Different Sources with Comparison of Zonal Variation in Odisha, India. Int. J. Pharma Res. Rev. 2015, 4, 1–8. [Google Scholar]
- Rameshkumar, K.B.; Aravind, A.P.A.; Mathew, P.J. Comparative Phytochemical Evaluation and Antioxidant Assay of Piper longum L. and Piper chaba Hunter Used in Indian Traditional Systems of Medicine. J. Herbs Spices Med. Plants 2011, 17, 351–360. [Google Scholar] [CrossRef]
- Khan, M. Comparative Physicochemical Evaluation of Fruits and Anti depressant Potential of volatile oils of fruits of Local Piper Species. Orient. J. Chem. 2015, 31, 541–545. [Google Scholar] [CrossRef] [Green Version]
- Juliani, H.R.; Koroch, A.R.; Giordano, L.; Amekuse, L.; Koffa, S.; Asante-Dartey, J.; Simon, J.E. Piper guineense (Piperaceae): Chemistry, Traditional Uses, and Functional Properties of West African Black Pepper. In African Natural Plant Products Volume II: Discoveries and Challenges in Chemistry, Health, and Nutrition; American Chemical Society: Washington, DC, USA, 2013; Volume 1127, pp. 33–48. [Google Scholar]
- Hussain, K.; Ismail, Z.; Sadikun, A.; Ibrahim, P. Antioxidant, anti-TB activities, phenolic and amide contents of standardised extracts of Piper sarmentosum Roxb. Nat. Prod. Res. 2009, 23, 238–249. [Google Scholar] [CrossRef]
- Bhat, B.G.; Chandrasekhara, N. Studies on the metabolism of piperine: Absorption, tissue distribution and excretion of urinary conjugates in rats. Toxicology 1986, 40, 83–92. [Google Scholar] [CrossRef]
- Suresh, D.; Srinivasan, K. Tissue distribution & elimination of capsaicin, piperine & curcumin following oral intake in rats. Indian J. Med. Res. 2010, 131, 682–691. [Google Scholar]
- Liu, H.; Luo, R.; Chen, X.; Liu, J.; Bi, Y.; Zheng, L.; Wu, X. Tissue distribution profiles of three antiparkinsonian alkaloids from Piper longum L. in rats determined by liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 928, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Haq, I.-U.; Imran, M.; Nadeem, M.; Tufail, T.; Gondal, T.A.; Mubarak, M.S. Piperine: A review of its biological effects. Phytother. Res. 2021, 35, 680–700. [Google Scholar] [CrossRef] [PubMed]
- Bajad, S.; Coumar, M.; Khajuria, R.; Suri, O.P.; Bedi, K.L. Characterization of a new rat urinary metabolite of piperine by LC/NMR/MS studies. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2003, 19, 413–421. [Google Scholar] [CrossRef]
- Ren, T.; Hu, M.; Cheng, Y.; Shek, T.L.; Xiao, M.; Ho, N.J.; Zhang, C.; Leung, S.S.Y.; Zuo, Z. Piperine-loaded nanoparticles with enhanced dissolution and oral bioavailability for epilepsy control. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2019, 137, 104988. [Google Scholar] [CrossRef] [PubMed]
- Zafar, F.; Jahan, N.; Bhatti, H.N. Increased Oral Bioavailability of Piperine from an Optimized Piper nigrum Nanosuspension. Planta Med. 2019, 85, 249–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, X.; Yuan, Z.; Qu, B.; Zhou, H.; Liu, Z.; Xie, Y. Piperine enhances the bioavailability of silybin via inhibition of efflux transporters BCRP and MRP2. Phytomed. Int. J. Phytother. Phytopharm. 2019, 54, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Yang, M.; Xiao, M.; Zhu, J.; Xie, W.; Zuo, Z. Time-dependent inhibition of carbamazepine metabolism by piperine in anti-epileptic treatment. Life Sci. 2019, 218, 314–323. [Google Scholar] [CrossRef]
- Pannu, N.; Bhatnagar, A. Combinatorial therapeutic effect of resveratrol and piperine on murine model of systemic lupus erythematosus. Inflammopharmacology 2020, 28, 401–424. [Google Scholar] [CrossRef]
- Dudhatra, G.B.; Mody, S.K.; Awale, M.M.; Patel, H.B.; Modi, C.M.; Kumar, A.; Kamani, D.R.; Chauhan, B.N. A comprehensive review on pharmacotherapeutics of herbal bioenhancers. Sci. World J. 2012, 2012, 637953. [Google Scholar] [CrossRef]
- Stojanović-Radić, Z.; Pejčić, M.; Dimitrijević, M.; Aleksić, A.; Anil Kumar, N.V.; Salehi, B.; Cho, W.C.; Sharifi-Rad, J. Piperine-A Major Principle of Black Pepper: A Review of Its Bioactivity and Studies. Appl. Sci. 2019, 9, 4270. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Lee, S.Y.; Oh, S.J.; Lee, K.H.; Jung, Y.S.; Kim, S.K. Assessment of drug-drug interactions caused by metabolism-dependent cytochrome P450 inhibition. Chem.-Biol. Interact. 2012, 198, 49–56. [Google Scholar] [CrossRef]
- Cui, T.; Wang, Q.; Tian, X.; Zhang, K.; Peng, Y.; Zheng, J. Piperine Is a Mechanism-Based Inactivator of CYP3A. Drug Metab. Dispos. Biol. Fate Chem. 2020, 48, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Buening, M.K.; Franklin, M.R. SKF 525-A inhibition, induction, and 452-nm complex formation. Drug Metab. Dispos. Biol. Fate Chem. 1976, 4, 244–255. [Google Scholar]
- Syed, S.B.; Arya, H.; Fu, I.H.; Yeh, T.K.; Periyasamy, L.; Hsieh, H.P.; Coumar, M.S. Targeting P-glycoprotein: Investigation of piperine analogs for overcoming drug resistance in cancer. Sci. Rep. 2017, 7, 7972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.B.; Kuo, C.L.; Lien, L.L.; Lien, E.J. Structure-activity relationship: Analyses of p-glycoprotein substrates and inhibitors. J. Clin. Pharm. Ther. 2003, 28, 203–228. [Google Scholar] [CrossRef] [PubMed]
- Pellicani, R.Z.; Stefanachi, A.; Niso, M.; Carotti, A.; Leonetti, F.; Nicolotti, O.; Perrone, R.; Berardi, F.; Cellamare, S.; Colabufo, N.A. Potent galloyl-based selective modulators targeting multidrug resistance associated protein 1 and P-glycoprotein. J. Med. Chem. 2012, 55, 424–436. [Google Scholar] [CrossRef]
- Bedada, S.K.; Appani, R.; Boga, P.K. Effect of Piperine on the Metabolism and Pharmacokinetics of Carbamazepine in Healthy Volunteers. Drug Res. 2017, 67, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Reen, R.K.; Jamwal, D.S.; Taneja, S.C.; Koul, J.L.; Dubey, R.K.; Wiebel, F.J.; Singh, J. Impairment of UDP-glucose dehydrogenase and glucuronidation activities in liver and small intestine of rat and guinea pig in vitro by piperine. Biochem. Pharmacol. 1993, 46, 229–238. [Google Scholar] [CrossRef]
- Srinivasan, K. Black pepper and its pungent principle-piperine: A review of diverse physiological effects. Crit. Rev. Food Sci. Nutr. 2007, 47, 735–748. [Google Scholar] [CrossRef]
- Li, Y.; Li, M.; Wang, Z.; Wen, M.; Tang, J. Identification of the metabolites of piperine via hepatocyte incubation and liquid chromatography combined with diode-array detection and high-resolution mass spectrometry. Rapid Commun. Mass Spectrom. RCM 2020, 34, e8947. [Google Scholar] [CrossRef]
- Gao, T.; Xue, H.; Lu, L.; Zhang, T.; Han, H. Characterization of piperine metabolites in rats by ultra-high-performance liquid chromatography with electrospray ionization quadruple time-of-flight tandem mass spectrometry. Rapid Commun. Mass Spectrom. RCM 2017, 31, 901–910. [Google Scholar] [CrossRef]
- Praneetha, P.; Balhara, A.; Ladumor, M.K.; Singh, D.K.; Patil, A.; Preethi, J.; Pokharkar, S.; Deshpande, A.Y.; Giri, S.; Singh, S. Characterization of stable and reactive metabolites of piperine formed on incubation with human liver microsomes. J. Mass Spectrom. 2019, 54, 738–749. [Google Scholar] [CrossRef]
- Chavarria, D.; Silva, T.; Magalhães e Silva, D.; Remião, F.; Borges, F. Lessons from black pepper: Piperine and derivatives thereof. Expert Opin. Ther. Pat. 2016, 26, 245–264. [Google Scholar] [CrossRef]
- Mu, L.H.; Wang, B.; Ren, H.Y.; Liu, P.; Guo, D.H.; Wang, F.M.; Bai, L.; Guo, Y.S. Synthesis and inhibitory effect of piperine derivates on monoamine oxidase. Bioorg. Med. Chem. Lett. 2012, 22, 3343–3348. [Google Scholar] [CrossRef]
- Borroto-Escuela, D.O.; Hinz, S.; Navarro, G.; Franco, R.; Müller, C.E.; Fuxe, K. Understanding the Role of Adenosine A2AR Heteroreceptor Complexes in Neurodegeneration and Neuroinflammation. Front. Neurosci. 2018, 12, 43. [Google Scholar] [CrossRef] [Green Version]
- Pretorius, J.; Malan, S.F.; Castagnoli, N., Jr.; Bergh, J.J.; Petzer, J.P. Dual inhibition of monoamine oxidase B and antagonism of the adenosine A(2A) receptor by (E,E)-8-(4-phenylbutadien-1-yl)caffeine analogues. Bioorg. Med. Chem. 2008, 16, 8676–8684. [Google Scholar] [CrossRef]
- Dhiman, P.; Malik, N.; Khatkar, A. Natural based piperine derivatives as potent monoamine oxidase inhibitors: An in silico ADMET analysis and molecular docking studies. BMC Chem. 2020, 14, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binda, C.; Hubálek, F.; Li, M.; Edmondson, D.E.; Mattevi, A. Crystal structure of human monoamine oxidase B, a drug target enzyme monotopically inserted into the mitochondrial outer membrane. FEBS Lett. 2004, 564, 225–228. [Google Scholar] [CrossRef] [Green Version]
- Nazifi, M.; Oryan, S.; Esfahani, D.E.; Ashrafpoor, M. The functional effects of piperine and piperine plus donepezil on hippocampal synaptic plasticity impairment in rat model of Alzheimer’s disease. Life Sci. 2021, 265, 118802. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Cai, Z.; Wang, W.; Wei, M.; Kou, D.; Li, T.; Yang, Z.; Guo, H.; Le, W.; Li, S. Piperine attenuates cognitive impairment in an experimental mouse model of sporadic Alzheimer’s disease. J. Nutr. Biochem. 2019, 70, 147–155. [Google Scholar] [CrossRef]
- Khalili-Fomeshi, M.; Azizi, M.G.; Esmaeili, M.R.; Gol, M.; Kazemi, S.; Ashrafpour, M.; Moghadamnia, A.A.; Hosseinzadeh, S. Piperine restores streptozotocin-induced cognitive impairments: Insights into oxidative balance in cerebrospinal fluid and hippocampus. Behav. Brain Res. 2018, 337, 131–138. [Google Scholar] [CrossRef]
- Hsieh, T.Y.; Chang, Y.; Wang, S.J. Piperine-mediated suppression of voltage-dependent Ca(2+) influx and glutamate release in rat hippocampal nerve terminals involves 5HT(1A) receptors and G protein βγ activation. Food Funct. 2019, 10, 2720–2728. [Google Scholar] [CrossRef]
- Andreza, S.F.; Matheus, G.d.O.; Giuliana, M.V.V.S.; Carlos, H.T.d.P.d.S.; Vinícius, B.d.S.; Carlton, A.T.; Gilberto, L.B.d.A. The Natural Alkaloid Piperine and its Acid and Ester Synthetic Derivatives are Acetylcholinesterase Inhibitors. Curr. Phys. Chem. 2015, 5, 294–300. [Google Scholar] [CrossRef]
- Ravelli, K.G.; Rosário, B.D.; Camarini, R.; Hernandes, M.S.; Britto, L.R. Intracerebroventricular Streptozotocin as a Model of Alzheimer’s Disease: Neurochemical and Behavioral Characterization in Mice. Neurotox. Res. 2017, 31, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Matsumura, S.; Yoshioka, Y.; Ueno, Y.; Matsuda, H. Screening of β-secretase and acetylcholinesterase inhibitors from plant resources. J. Nat. Med. 2015, 69, 123–129. [Google Scholar] [CrossRef]
- Kumar, S.; Chowdhury, S.; Razdan, A.; Kumari, D.; Purty, R.S.; Ram, H.; Kumar, P.; Nayak, P.; Shukla, S.D. Downregulation of Candidate Gene Expression and Neuroprotection by Piperine in Streptozotocin-Induced Hyperglycemia and Memory Impairment in Rats. Front. Pharmacol. 2020, 11, 595471. [Google Scholar] [CrossRef]
- Head, E.; Murphey, H.L.; Dowling, A.L.; McCarty, K.L.; Bethel, S.R.; Nitz, J.A.; Pleiss, M.; Vanrooyen, J.; Grossheim, M.; Smiley, J.R.; et al. A combination cocktail improves spatial attention in a canine model of human aging and Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2012, 32, 1029–1042. [Google Scholar] [CrossRef] [Green Version]
- Elnaggar, Y.S.; Etman, S.M.; Abdelmonsif, D.A.; Abdallah, O.Y. Intranasal Piperine-Loaded Chitosan Nanoparticles as Brain-Targeted Therapy in Alzheimer’s Disease: Optimization, Biological Efficacy, and Potential Toxicity. J. Pharm. Sci. 2015, 104, 3544–3556. [Google Scholar] [CrossRef]
- Yusuf, M.; Khan, M.; Khan, R.A.; Ahmed, B. Preparation, characterization, in vivo and biochemical evaluation of brain targeted Piperine solid lipid nanoparticles in an experimentally induced Alzheimer’s disease model. J. Drug Target. 2013, 21, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Etman, S.M.; Elnaggar, Y.S.R.; Abdelmonsif, D.A.; Abdallah, O.Y. Oral Brain-Targeted Microemulsion for Enhanced Piperine Delivery in Alzheimer’s Disease Therapy: In Vitro Appraisal, In Vivo Activity, and Nanotoxicity. AAPS PharmSciTech 2018, 19, 3698–3711. [Google Scholar] [CrossRef]
- Gad, S.C.; Cassidy, C.D.; Aubert, N.; Spainhour, B.; Robbe, H. Nonclinical vehicle use in studies by multiple routes in multiple species. Int. J. Toxicol. 2006, 25, 499–521. [Google Scholar] [CrossRef] [Green Version]
- Hong, R.; Li, X. Discovery of monoamine oxidase inhibitors by medicinal chemistry approaches. MedChemComm 2019, 10, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Al-Baghdadi, O.B.; Prater, N.I.; Van der Schyf, C.J.; Geldenhuys, W.J. Inhibition of monoamine oxidase by derivatives of piperine, an alkaloid from the pepper plant Piper nigrum, for possible use in Parkinson’s disease. Bioorg. Med. Chem. Lett. 2012, 22, 7183–7188. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, P.; Vaibhav, K.; Tabassum, R.; Khan, A.; Ishrat, T.; Khan, M.M.; Ahmad, A.; Islam, F.; Safhi, M.M.; Islam, F. Anti-apoptotic and anti-inflammatory effect of Piperine on 6-OHDA induced Parkinson’s rat model. J. Nutr. Biochem. 2013, 24, 680–687. [Google Scholar] [CrossRef]
- Liu, J.; Chen, M.; Wang, X.; Wang, Y.; Duan, C.; Gao, G.; Lu, L.; Wu, X.; Wang, X.; Yang, H. Piperine induces autophagy by enhancing protein phosphotase 2A activity in a rotenone-induced Parkinson’s disease model. Oncotarget 2016, 7, 60823–60843. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liu, J.; Gao, G.; Wu, X.; Wang, X.; Yang, H. Protection effect of piperine and piperlonguminine from Piper longum L. alkaloids against rotenone-induced neuronal injury. Brain Res. 2016, 1639, 214–227. [Google Scholar] [CrossRef]
- Wang, L.; Cai, X.; Shi, M.; Xue, L.; Kuang, S.; Xu, R.; Qi, W.; Li, Y.; Ma, X.; Zhang, R.; et al. Identification and optimization of piperine analogues as neuroprotective agents for the treatment of Parkinson’s disease via the activation of Nrf2/keap1 pathway. Eur. J. Med. Chem. 2020, 199, 112385. [Google Scholar] [CrossRef]
- Li, R.; Lu, Y.; Zhang, Q.; Liu, W.; Yang, R.; Jiao, J.; Liu, J.; Gao, G.; Yang, H. Piperine promotes autophagy flux by P2RX4 activation in SNCA/α-synuclein-induced Parkinson disease model. Autophagy 2021, 1–17. [Google Scholar] [CrossRef]
- Huang, L.; Zhong, X.; Luo, Q.; Zhang, Q.; Deng, M. Autophagic activity of piperine on small intestine in dementia model mice with Parkinson’s disease. Zhongguo Zhong Yao Za Zhi Zhongguo Zhongyao Zazhi China J. Chin. Mater. Med. 2020, 45, 5238–5247. [Google Scholar] [CrossRef]
- Salman, M.; Tabassum, H.; Parvez, S. Piperine mitigates behavioral impairments and provides neuroprotection against 3-nitropropinoic acid-induced Huntington disease-like symptoms. Nutr. Neurosci. 2020, 1–10. [Google Scholar] [CrossRef]
- Pal, A.; Nayak, S.; Sahu, P.K.; Swain, T. Piperine protects epilepsy associated depression: A study on role of monoamines. Eur. Rev. Med. Pharmacol. Sci. 2011, 15, 1288–1295. [Google Scholar]
- da Cruz, G.M.; Felipe, C.F.; Scorza, F.A.; da Costa, M.A.; Tavares, A.F.; Menezes, M.L.; de Andrade, G.M.; Leal, L.K.; Brito, G.A.; da Graça Naffah-Mazzacoratti, M.; et al. Piperine decreases pilocarpine-induced convulsions by GABAergic mechanisms. Pharmacol. Biochem. Behav. 2013, 104, 144–153. [Google Scholar] [CrossRef] [Green Version]
- Mishra, A.; Punia, J.K.; Bladen, C.; Zamponi, G.W.; Goel, R.K. Anticonvulsant mechanisms of piperine, a piperidine alkaloid. Channels 2015, 9, 317–323. [Google Scholar] [CrossRef]
- Chen, C.Y.; Li, W.; Qu, K.P.; Chen, C.R. Piperine exerts anti-seizure effects via the TRPV1 receptor in mice. Eur. J. Pharmacol. 2013, 714, 288–294. [Google Scholar] [CrossRef]
- Dong, Y.; Yin, Y.; Vu, S.; Yang, F.; Yarov-Yarovoy, V.; Tian, Y.; Zheng, J. A distinct structural mechanism underlies TRPV1 activation by piperine. Biochem. Biophys. Res. Commun. 2019, 516, 365–372. [Google Scholar] [CrossRef]
- Pattanaik, S.; Hota, D.; Prabhakar, S.; Kharbanda, P.; Pandhi, P. Pharmacokinetic interaction of single dose of piperine with steady-state carbamazepine in epilepsy patients. Phytother. Res. PTR 2009, 23, 1281–1286. [Google Scholar] [CrossRef]
- Verma, A.K.; Khan, E.; Mishra, S.K.; Jain, N.; Kumar, A. Piperine Modulates Protein Mediated Toxicity in Fragile X-Associated Tremor/Ataxia Syndrome through Interacting Expanded CGG Repeat (r(CGG)(exp)) RNA. ACS Chem. Neurosci. 2019, 10, 3778–3788. [Google Scholar] [CrossRef]
- Mao, Q.Q.; Huang, Z.; Ip, S.P.; Xian, Y.F.; Che, C.T. Role of 5-HT(1A) and 5-HT(1B) receptors in the antidepressant-like effect of piperine in the forced swim test. Neurosci. Lett. 2011, 504, 181–184. [Google Scholar] [CrossRef]
- Mao, Q.Q.; Huang, Z.; Zhong, X.M.; Xian, Y.F.; Ip, S.P. Brain-derived neurotrophic factor signalling mediates the antidepressant-like effect of piperine in chronically stressed mice. Behav. Brain Res. 2014, 261, 140–145. [Google Scholar] [CrossRef]
- Khom, S.; Strommer, B.; Schöffmann, A.; Hintersteiner, J.; Baburin, I.; Erker, T.; Schwarz, T.; Schwarzer, C.; Zaugg, J.; Hamburger, M.; et al. GABAA receptor modulation by piperine and a non-TRPV1 activating derivative. Biochem. Pharmacol. 2013, 85, 1827–1836. [Google Scholar] [CrossRef] [Green Version]
- Wang-Sheng, C.; Jie, A.; Jian-Jun, L.; Lan, H.; Zeng-Bao, X.; Chang-Qing, L. Piperine attenuates lipopolysaccharide (LPS)-induced inflammatory responses in BV2 microglia. Int. Immunopharmacol. 2017, 42, 44–48. [Google Scholar] [CrossRef]
- Vaibhav, K.; Shrivastava, P.; Javed, H.; Khan, A.; Ahmed, M.E.; Tabassum, R.; Khan, M.M.; Khuwaja, G.; Islam, F.; Siddiqui, M.S.; et al. Piperine suppresses cerebral ischemia-reperfusion-induced inflammation through the repression of COX-2, NOS-2, and NF-κB in middle cerebral artery occlusion rat model. Mol. Cell. Biochem. 2012, 367, 73–84. [Google Scholar] [CrossRef]
- Wattanathorn, J.; Chonpathompikunlert, P.; Muchimapura, S.; Priprem, A.; Tankamnerdthai, O. Piperine, the potential functional food for mood and cognitive disorders. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2008, 46, 3106–3110. [Google Scholar] [CrossRef]
- Li, S.; Wang, C.; Li, W.; Koike, K.; Nikaido, T.; Wang, M.W. Antidepressant-like effects of piperine and its derivative, antiepilepsirine. J. Asian Nat. Prod. Res. 2007, 9, 421–430. [Google Scholar] [CrossRef]
- Gilhotra, N.; Dhingra, D. Possible involvement of GABAergic and nitriergic systems for antianxiety-like activity of piperine in unstressed and stressed mice. Pharmacol. Rep. 2014, 66, 885–891. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [Green Version]
- Shwaireb, M.H.; Wrba, H.; el-Mofty, M.M.; Dutter, A. Carcinogenesis induced by black pepper (Piper nigrum) and modulated by vitamin A. Exp. Pathol. 1990, 40, 233–238. [Google Scholar] [CrossRef]
- Unchern, S.; Saito, H.; Nishiyama, N. Death of cerebellar granule neurons induced by piperine is distinct from that induced by low potassium medium. Neurochem. Res. 1998, 23, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.Y.; Chang, J.P.; Wang, C.J. Modulatory effect of piperine on benzo[a]pyrene cytotoxicity and DNA adduct formation in V-79 lung fibroblast cells. Food Chem. Toxicol. 1994, 32, 373–377. [Google Scholar] [CrossRef]
- Dogra, R.K.; Khanna, S.; Shanker, R. Immunotoxicological effects of piperine in mice. Toxicology 2004, 196, 229–236. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Food Contact Materials, Enzymes, Flavourings and Processing Aids (CEF). Scientific Opinion on Flavouring Group Evaluation 86, Revision 2 (FGE.86Rev2): Consideration of aliphatic and arylalkyl amines and amides evaluated by JECFA (65th meeting). EFSA J. 2015, 13, 3998. [Google Scholar] [CrossRef] [Green Version]
- Elnaggar, Y.S.; Etman, S.M.; Abdelmonsif, D.A.; Abdallah, O.Y. Novel piperine-loaded Tween-integrated monoolein cubosomes as brain-targeted oral nanomedicine in Alzheimer’s disease: Pharmaceutical, biological, and toxicological studies. Int. J. Nanomed. 2015, 10, 5459–5473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moradi, S.Z.; Momtaz, S.; Bayrami, Z.; Farzaei, M.H.; Abdollahi, M. Nanoformulations of Herbal Extracts in Treatment of Neurodegenerative Disorders. Front. Bioeng. Biotechnol. 2020, 8, 238. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sources | Plant Parts | Estimated Amount (%) |
|---|---|---|
| Piper nigrum [2] | Fruit | 1.7–7.4 |
| Piper longum [3] | Spike and root | 5.9 |
| Fruit | 0.03 | |
| Piper chaba [3,4] | Fruit | 0.95–1.32 |
| Piper guineense [5] | Fruit | 0.23–1.1 |
| Piper sarmentosum [6] | Root | 0.20 |
| Stem | 1.59 | |
| Leaf | 0.104 | |
| Fruit | 2.75 |
| Metabolites | Mouse | Rat | Dog | Human |
|---|---|---|---|---|
| C1 | ||||
| C2 | ||||
| C3 | ||||
| C4 | ||||
| C5 | ||||
| C6 | ||||
| C7 | ||||
| C8 | ||||
| C9 | ||||
| C10 | ||||
| C11 | ||||
| C12 | ||||
| C13 | ||||
| C14 | ||||
| C15 | ||||
| C16 | ||||
| C17 | ||||
| C18 | ||||
| C19 | ||||
| C20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azam, S.; Park, J.-Y.; Kim, I.-S.; Choi, D.-K. Piperine and Its Metabolite’s Pharmacology in Neurodegenerative and Neurological Diseases. Biomedicines 2022, 10, 154. https://doi.org/10.3390/biomedicines10010154
Azam S, Park J-Y, Kim I-S, Choi D-K. Piperine and Its Metabolite’s Pharmacology in Neurodegenerative and Neurological Diseases. Biomedicines. 2022; 10(1):154. https://doi.org/10.3390/biomedicines10010154
Chicago/Turabian StyleAzam, Shofiul, Ju-Young Park, In-Su Kim, and Dong-Kug Choi. 2022. "Piperine and Its Metabolite’s Pharmacology in Neurodegenerative and Neurological Diseases" Biomedicines 10, no. 1: 154. https://doi.org/10.3390/biomedicines10010154
APA StyleAzam, S., Park, J.-Y., Kim, I.-S., & Choi, D.-K. (2022). Piperine and Its Metabolite’s Pharmacology in Neurodegenerative and Neurological Diseases. Biomedicines, 10(1), 154. https://doi.org/10.3390/biomedicines10010154