Abstract
Chromosomal microarray (CMA) is considered a first-tier test for genetic analysis as it can be used to examine gene copy number variations (CNVs) throughout the entire genome, with enhanced sensitivity for detecting submicroscopic deletions and duplications. However, its cost can represent a heavy burden. Moreover, the diagnostic yield of CMA in infants with developmental delay (DD) was reported to be less than 10%. Therefore, we aimed to investigate the relationship between CMA results and clinical features and risk factors of DD. The study included 59 infants with DD who were recruited between August 2019 and February 2020 during a visit to the outpatient clinic of a rehabilitation department. We reviewed the clinical records of the infants regarding gender, age, body weight at birth, delivery method, brain imaging data, perinatal history, and parent-related clinical parameters, such as mother and father age at birth. The infants were categorized according to CMA results, and differences in clinical parameters were evaluated. Except for brain anomalies, there was no statistically significant differences between infants who had pathogenic and variants of unknown significance (VOUS)-likely pathogenic CNVs groups compared with those within the VOUS-likely no sub-classification, VOUS-likely benign, benign, and normal CNVs groups. The incidence of brain anomalies was significantly higher within infants with pathogenic and VOUS-likely pathogenic CNVs groups (p < 0.05). Our study suggests that infants with DD who present dysmorphism or brain anomaly may benefit from early CMA analysis, for adequate diagnosis and timely treatment. Further studies are warranted to confirm the relationship between DD clinical parameters and CMA results.
1. Introduction
A developmental delay (DD) is characterized by noticeable shortage of developmental milestones achieved by a child throughout development [1]. Although there is no exact definition, the DD concept is generally used to describe delays in major criteria of the developmental process, including cognitive, physical, communication, emotional, and social impairments [1]. DD incidence rate in the general population is close to 3% [2,3]. Early screening of DD may represent an important strategy to support early therapeutic intervention and enhance the prognosis of those affected [4,5]. However, only 30% of DD affected children are diagnosed at pre-school age [6]. In many cases, late recognition can be explained by the absence of definite clinical symptoms of DD and its comorbidities, making it difficult for caregivers and parents to perceive this condition and seek clinical help [7]. Recent findings on genetic defects in infants with unexplained DD have provided important clues on the diagnostic value of screening tests for early diagnosis of DD [8].
Several cytogenetic and molecular biology techniques are used to identify the underlying genetic cause of DD, from conventional approaches (such as molecular karyotyping) to state-of-the-art chromosomal microarray (CMA) and next-generation sequencing (NGS) [9]. Recently, CMA has been widely used as a first-tier of genetic analysis in place of G-banded karyotyping as it can examine gene copy number variations (CNVs) throughout the genome with improved sensitivity for submicroscopic deletions and duplications [9,10]. However, this diagnostic improvement is associated with a significant economic burden to the caregivers [9]. Nevertheless, Farooqi et al. showed that the diagnostic yield of CMAs, with identification of pathogenic CNVs or variants of unknown significance (VOUS)-likely pathogenic, was less than 10% in infants with DD [11]. This finding highlights the clear need of a better understanding of DD clinical features and its association with CMA data to reduce unnecessary CMA testing. In this study, we investigated the correlation between various clinical parameters, including risk factors of DD, and CMA results collected from infants with DD.
2. Methods
2.1. Infants with DD
The study included 59 infants who visited the outpatient department of rehabilitation medicine at our hospital from August 2019 to February 2020 for suspected DD. Most of the infants showed delayed gross motor function or impaired language performance compared to age-matched children with no developmental problems. According to the after Korean Developmental Screening Test (K-DST) manual, the children can be categorized into four groups based on their individual K-DST result: further evaluation (<−2 standard deviation [−2SD]), follow-up test (−2SD to −1SD), peer-level (−1SD to 1SD), and high level (>1SD) [12,13]. We recommended CMA testing for children classified within the ‘further evaluation’ or ‘follow-up test’ groups. The K-DST includes 6 sections that assess developmental areas of gross motor, fine motor, cognition, communication, social interaction, and self-control. Additional questions are designed to take into account clinically important diseases, such as cerebral palsy, language delay, and autism [12,13]. Therefore, all infants with DD who included in this study were classified within the ‘further evaluation’ or ‘follow-up test’ groups. We also reviewed the clinical records of the infants regarding their sex, age, body weight at birth, gestational age, delivery method, perinatal history, as well as parent-related clinical parameters, including mother and father age at the time of the child birth. This study was approved by the Institutional Review Board of our hospital (DFE200RIO074.). A detailed description of the CMA test and an agreement to perform the test was obtained from the parents of all infants. The parents also agreed to release the results of the CMA test for research purposes.
2.2. CMA Protocol
Peripheral venous blood was collected into an ethylenediaminetetraacetic acid (EDTA) collection tube, and DNA was extracted from peripheral blood leukocytes. CytoScanDx assay (Affymetrix, Santa Clara, CA, USA) was used for CNVs analysis and it was performed according to the instructions of the manufacturer [10]. The assay comprises more than 750,436 CNV markers, including 200,436 genotype-able single nucleotide polymorphisms probes and more than 550,000 non-polymorphism probes. The overall average marker space was 4127 base pairs [10]. All data were visualized and analyzed through the GeneChip System 3000Dx (Affymetrix) platform using the human genome assembly GRCh37 (hg19) as reference [10]. All deletions and duplications with more than 400 kb were reported, and those with 400 kb or less, well-known microdeletion, duplication syndrome sites, or affecting clinically relevant gene sites were also reported [14]. The CMA results were reported according to the following classifications: pathogenic CNVs, variant of unknown significance (VOUS)-likely pathogenic CNVs, VOUS with no sub-classification, VOUS-likely benign, benign, or normal CNVs [10].
2.3. Statistical Analysis
The Kolmogorov–Smirnov test was used to determine whether the data conformed to a normal distribution. Pearson Chi-square test, independent t-test, and Mann–Whitney U-test were used to evaluate the differences between pathogenic, VOUS-likely pathogenic CNVs, and other groups, and between two groups (pathogenic, VOUS with no sub-classification, VOUS-likely benign versus benign and normal CNVs group). Categorical variables included gender, brain anomalies, cardiac anomalies, dysmorphism, autism spectrum disorder, cerebral palsy, delivery method, and mode of conception. Continuous variables included age, father and mother age, body weight at birth, and gestational age. Statistical analyses were performed using SPSS 25.0 for Windows (IBM, Armonk, NY, USA) and R software for Windows (version 2.15.2; R Foundation for Statistical Computing, Vienna, Austria). A p-value < 0.05 was considered to be statistically significant in all tests.
3. Results
3.1. Demographic and Genetic Characterization of the Patients
CMA data were collected from 59 infants, of whom 41 were males and 18 were females. All 59 infants with DD who included in this study were classified within the ‘further evaluation’ or ‘follow-up test’ groups according to K-DST (‘further evaluation’: 23 infants; ‘follow-up test’: 36 infants). The average age of the infants was 38.7 ± 18.50 months, and the average age of their fathers and mothers at the time of birth was 35.7 ± 5.23 and 32.2 ± 4.04 years, respectively. The average birthweight of the infants was 2921.0 ± 716.48 g, their gestational age was 37.4 ± 2.72 weeks, and 61% of children were born by cesarean section (Table 1).

Table 1.
Characteristics of 59 infants with developmental delay who included in this study.
We found 41 CNVs in 35 infants, including 4 (9.8%) pathogenic CNVs, 2 (4.9%) VOUS-likely pathogenic CNVs, 16 (39.0%) VOUS with no sub-classification, 16 (39.0%) VOUS-likely benign CNVs, and 3 (7.3%) benign CNVs (Figure 1). CMA results and affected genes, clinical features, and other risk factors are summarized in Table 2. Typical genetic problems that were identified among pathogenic CNVs, such as Klinefelter’s syndrome, mosaicism of Turner’s syndrome, or genetic problems associated with incest history, are also reported. The remaining 24 infants had normal CNV findings.
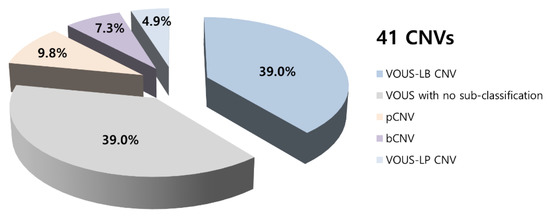
Figure 1.
Summary of chromosomal microarray results. CNV—copy number variations, bCNV—benign copy number variations, pCNV—pathogenic copy number variations, LB—likely-benign, LP—likely-pathogenic, VOUS—Variant of unknown significance.
Table 2.
Clinical features and parameters with chromosomal microarray (CMA) in 35 infants with developmental delay and abnormalities in copy number variations.
3.2. CMA Results Comparisons
No significant difference was observed between pathogenic and VOUS-likely pathogenic CNV groups compared with VOUS with no sub-classification, VOUS-likely benign, benign, and normal CNVs groups, except for brain anomalies (Figure 2). Significantly more infants classified within the pathogenic and VOUS-likely pathogenic CNV groups had brain anomalies (p < 0.05). Moreover, the rate of infants with cardiac and/or brain anomalies, and dysmorphism was higher in the pathogenic and VOUS-likely pathogenic CNV groups than in the VOUS like no sub-classification, VOUS-likely benign, benign, and normal CNVs groups. However, this difference did not reach statistical significance (p = 0.053) (Figure 2). Additional analyses showed that gender, age, body weight at birth, gestational age, delivery method, mode of conception, result of brain magnetic resonance imaging, and perinatal history of the infants, as well as parent-related clinical parameters, were that there were no significant difference observed between pathogenic and VOUS-likely pathogenic CNV groups compared with VOUS with no sub-classification, VOUS-likely benign, benign, and normal CNVs groups, except for brain anomalies (Figure 2).
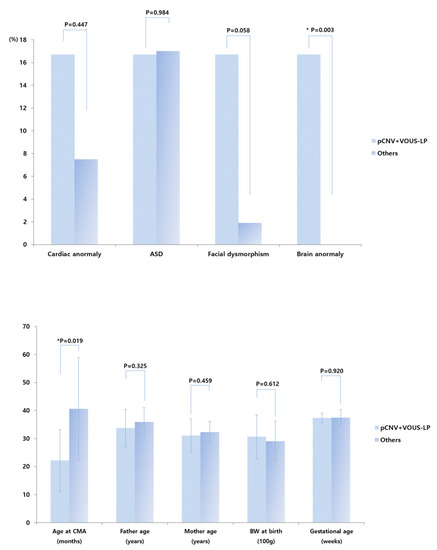
Figure 2.
Prevalence of cardiac anomaly, autism spectrum disorder, facial dysmorphism, and brain anomaly in pathogenic and VOUS-likely pathogenic CNV groups versus VOUS with no sub-classification, VOUS-likely benign, benign, and normal CNVs groups. CMA—chromosomal microarray analysis, BW—body weight, ASD—autism spectrum disorder, pCNV—pathogenic copy number variations, LP—likely-pathogenic, VOUS—Variant of unknown significance. * p < 0.05.
Between pathogenic, VOUS-likely pathogenic, and VOUS with no sub-classification versus VOUS-likely benign, benign, and normal CNVs groups, additional analyses showed that gender, age, body weight at birth, gestational age, delivery method, mode of conception, result of brain magnetic resonance imaging, and perinatal history of the infants, as well as parent-related clinical parameters, showed no significant differences (Figure 3).
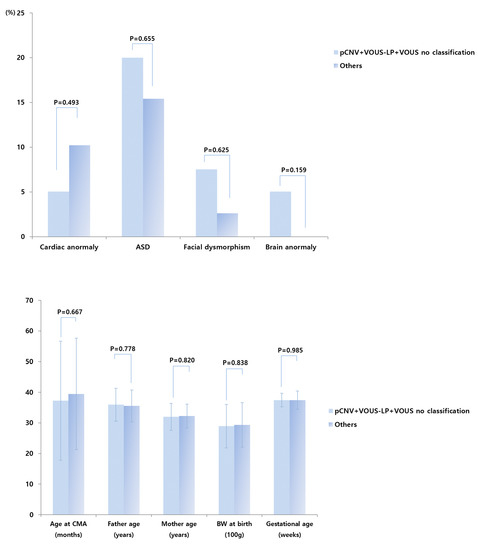
Figure 3.
Prevalence of cardiac anomaly, autism spectrum disorder, facial dysmorphism, and brain anomaly in pathogenic, VOUS-likely pathogenic CNV groups, and VOUS with no sub-classification versus VOUS-likely benign, benign, and normal CNVs groups. CMA—chromosomal microarray analysis, BW—body weight, ASD—autism spectrum disorder, pCNV—pathogenic copy number variations, LP—likely-pathogenic, VOUS—Variant of unknown significance, VOUS no classification—VOUS with no-subclassification.
4. Discussion
We found four pathogenic CNVs and two VOUS-likely pathogenic CNVs in six infants, which indicated that 10.2% of the infants had genetic alterations associated with DD and comorbid conditions. This finding was in concordance with the findings of a previous CMA study conducted in infants with DD [11].
We identified a variety of CMA results; however, most of these genetic alterations were of unknown phenotype (39% of VOUS with no sub-classification). Nevertheless, we expected to find more abnormal CNVs within our study population. Therefore, we also evaluated potential correlations between demographic and clinical parameters of the infants and their parents, including risk factors known to cause DD, and the genetic findings. This analysis failed to find significant differences between pathogenic and VOUS-likely pathogenic CNV groups versus VOUS-likely no sub-classification, VOUS-likely benign, benign, and normal CNVs group, with exception of brain anomalies. Indeed, our data showed a tendency of slightly higher rate of infants with brain and cardiac anomalies and dysmorphism within the pathogenic and VOUS-likely pathogenic CNVs groups. Nevertheless, only one and two infants included in the study had brain anomalies and dysmorphism, respectively, which may contribute to the low statistical power of the analysis. Kim et al. reported that dysmorphism was significantly higher in infants with pathogenic CNVs as compared with those with normal CMA results [10]. Moreover, Gilissen et al. also reported that infants with intellectual disability and multiple congenital anomalies have higher burden of CNVs than those with intellectual disability alone [15]. Altogether, these findings support the important role of CMA for assessing DD infants with anomalies and dysmorphism.
Interestingly, the average age of infants with pathogenic or VOUS likely-pathogenic CNVs was 22.1 months, which was much lower than the overall average age of the study population (Figure 2). This result suggests that children with pathogenic or VOUS likely-pathogenic CNVs may have a more prominent developmental delay, leading them to visit the hospital earlier and to receive an earlier diagnosis, which is definitely advantageous for planning their rehabilitation program. In such cases, as well as in cases of identified VOUS no sub-classification, VOUS likely-benign, and even benign CNVs, an early diagnosis allows clinicians to discuss with the parents about existing genetic risks and provide them genetic counseling before having another child.
Despite of these findings, our study has some limitations. First, we investigated CMA results within a small number of infants with DD. However, we expanded our analysis to include additional demographic and clinical parameters, such as body weight at birth, delivery method, and parent-related clinical parameters, which was not performed in the previous study [10]. Contrary to our expectation, the age of the parents seemed to not have a significant impact on the incidence of clinically significant CNVs. Nevertheless, more CMA studies are warranted with larger cohorts of infants with DD to further confirm this finding. Secondly, although CMA offers the sensitivity of high-resolution genome-wide detection of clinically significant CNVs, there is an additional challenge for interpreting VOUS, which is the preferred terminology based on a recent study of variant terminology [9]. Moreover, there are definite limitations of CMA for genetic analysis. For example, CMA cannot detect balanced chromosome rearrangements, such as inversions or translocations, which do not result in deletion or duplication of genetic material, or cases of low-level tissue mosaicism, although balanced rearrangements rarely are associated with disease unless there is disruption of a critical gene. Additionally, CMA may not identify low levels of tissue mosaicism in the fetus [9,16]. Additionally, uniparental disomy may also not be detected when the region for which heterozygosity was lost is small or in cases of heterodisomy. Furthermore, 13p, 14p, 15p, 21p, 22p, Yq11.23, Yq12, and pericentric heterochromatin region of all chromosomes are regions that are undetectable by CMA [9,16]. Despite these technical limitations, CMA is still used as a primary method for detecting gene deletions and duplications throughout the entire genome. Therefore, our study may provide helpful information for selecting infants with DD whose families may benefit of CMA testing. More studies addressing data collected from multiple genetic analysis platforms, such as CMA and next-generation sequencing, in infants with DD maybe helpful to further confirm the relationship between genetic and clinical features of DD.
5. Conclusions
In conclusion, our research suggests that infants with DD who present dysmorphism or brain anomaly may benefit from early CMA analysis for adequate diagnosis and timely treatment. However, further studies are needed with focus on CMA data in infants with DD.
Author Contributions
Data curation, Z.L., B.J.L.; writing—original draft preparation, S.P., D.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Petersen, M.C.; Kube, D.A.; Palmer, F.B. Classification of developmental delays. Semin. Pediatr. Neurol. 1998, 5, 2–14. [Google Scholar] [CrossRef]
- Shevell, M.; Ashwal, S.; Donley, D.; Flint, J.; Gingold, M.; Hirtz, D.; Majnemer, A.; Noetzel, M.; Sheth, R.D.; Quality Standards Subcommittee of the American Academy of N; et al. Practice parameter: Evaluation of the child with global developmental delay: Report of the Quality Standards Subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society. Neurology 2003, 60, 367–380. [Google Scholar] [PubMed]
- Whelen, M.A. Practice parameter: Evaluation of the child with global developmental delay. Neurology 2003, 61, 1315. [Google Scholar] [CrossRef] [PubMed]
- Guralnick, M.J. Early Intervention Approaches to Enhance the Peer-Related Social Competence of Young Children With Developmental Delays: A Historical Perspective. Infants Young Child 2010, 23, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Guralnick, M.J. Why Early Intervention Works: A Systems Perspective. Infants Young Child 2011, 24, 6–28. [Google Scholar] [CrossRef] [PubMed]
- Palfrey, J.S.; Singer, J.D.; Walker, D.K.; Butler, J.A. Early identification of children’s special needs: A study in five metropolitan communities. J. Pediatr. 1987, 111, 651–659. [Google Scholar] [CrossRef]
- Chung, C.Y.; Liu, W.Y.; Chang, C.J.; Chen, C.L.; Tang, S.F.; Wong, A.M. The relationship between parental concerns and final diagnosis in children with developmental delay. J. Child Neurol. 2011, 26, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Moeschler, J.B.; Shevell, M. American Academy of Pediatrics Committee on G: Clinical genetic evaluation of the child with mental retardation or developmental delays. Pediatrics 2006, 117, 2304–2316. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Park, C.I.; Lim, J.W.; Lee, G.M.; Cho, E.; Kim, H.J. Phenotypic Analysis of Korean Patients with Abnormal Chromosomal Microarray in Patients with Unexplained Developmental Delay/Intellectual Disability. Yonsei Med. J. 2018, 59, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, M.S.; Figueroa, S.; Gotway, G.; Wang, J.; Luu, H.S.; Park, J.Y. Reinterpretation of Chromosomal Microarrays with Detailed Medical History. J. Pediatr. 2020, 222, 180–185.e1. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.H.; Kim, S.W.; Jeon, H.R.; Jung, D.W.; Cho, H.E.; Kim, J.; Lee, J.W. Clinical Usefulness of the Korean Developmental Screening Test (K-DST) for Developmental Delays. Ann. Rehabil. Med. 2019, 43, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Yim, C.H.; Kim, G.H.; Eun, B.L. Usefulness of the Korean Developmental Screening Test for infants and children for the evaluation of developmental delay in Korean infants and children: A single-center study. Korean J. Pediatr. 2017, 60, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.T.; Marques-Bonet, T.; Sharp, A.J.; Mefford, H.C. The genetics of microdeletion and microduplication syndromes: An update. Annu. Rev. Genom. Hum. Genet. 2014, 15, 215–244. [Google Scholar] [CrossRef] [PubMed]
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; van de Vorst, M.; van Bon, B.W.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A.; et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Gajecka, M. Unrevealed mosaicism in the next-generation sequencing era. Mol. Genet. Genom. 2016, 291, 513–530. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).