Hybrid-Transcriptome Sequencing and Associated Metabolite Analysis Reveal Putative Genes Involved in Flower Color Difference in Rose Mutants


Abstract
1. Introduction
2. Results
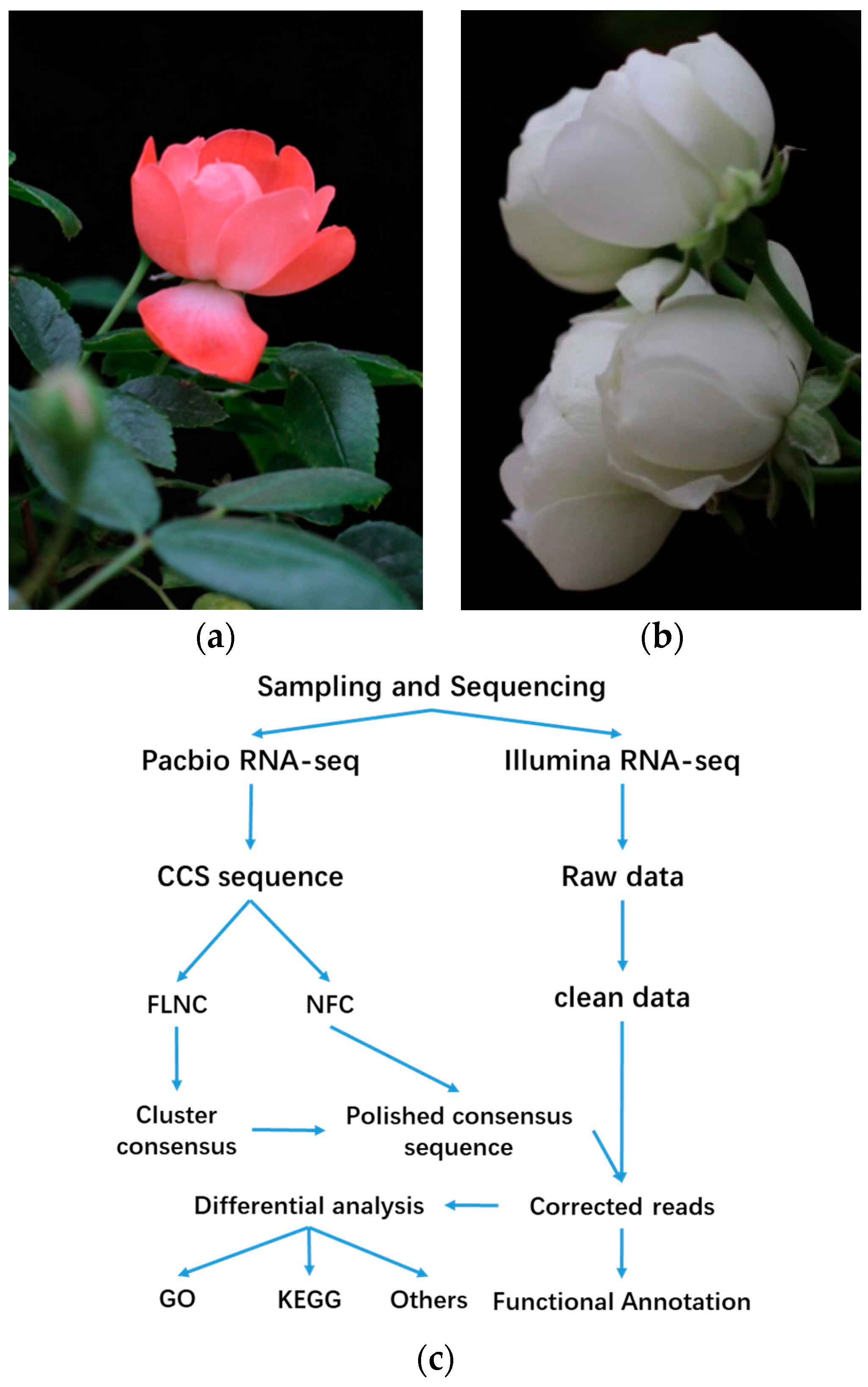
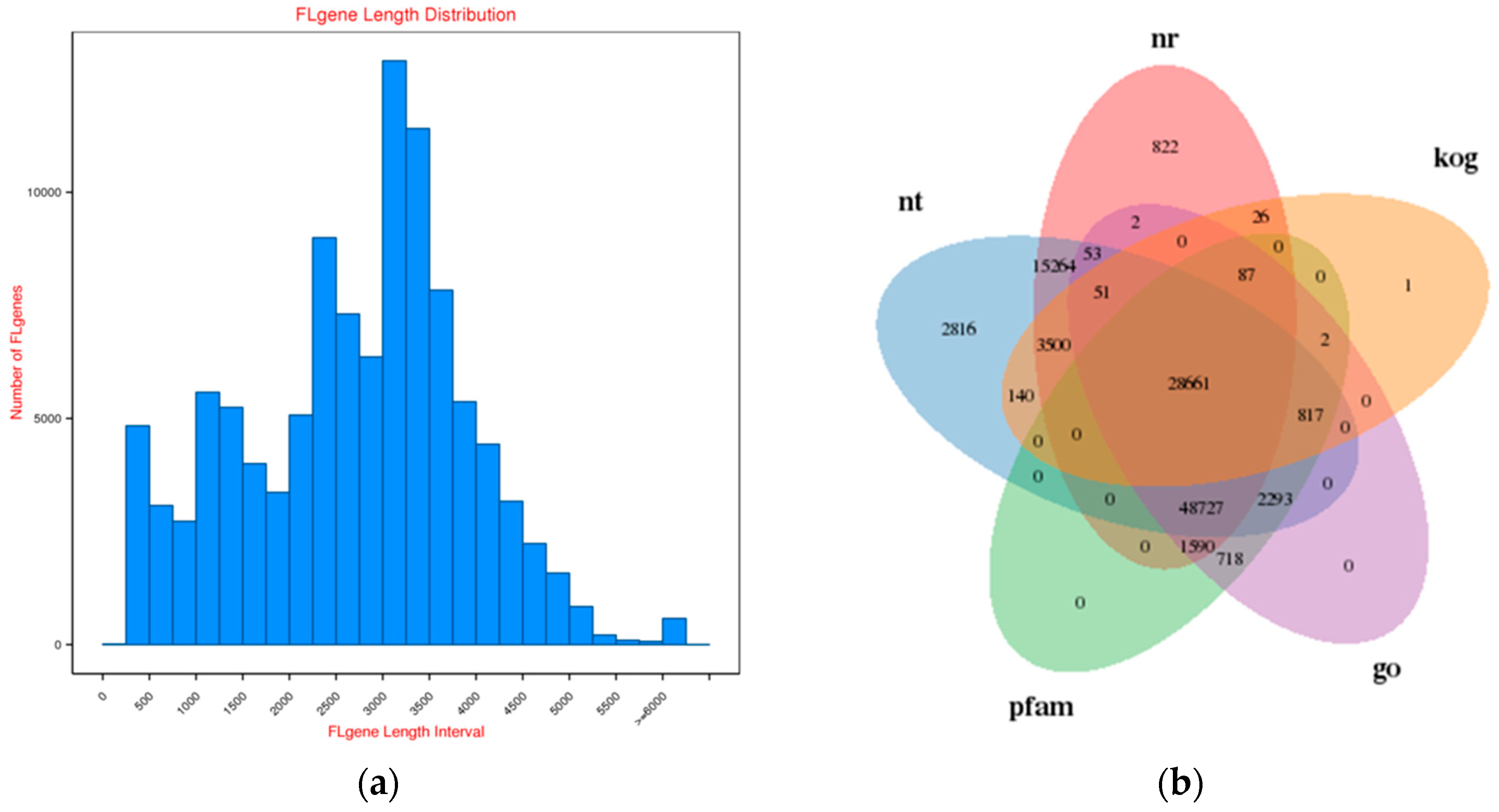
2.1. Survey on Hybrid RNA Sequencing
2.2. Functional Annotation of Rose Variety
2.3. Simple Sequence Repeat (SSR) Detection
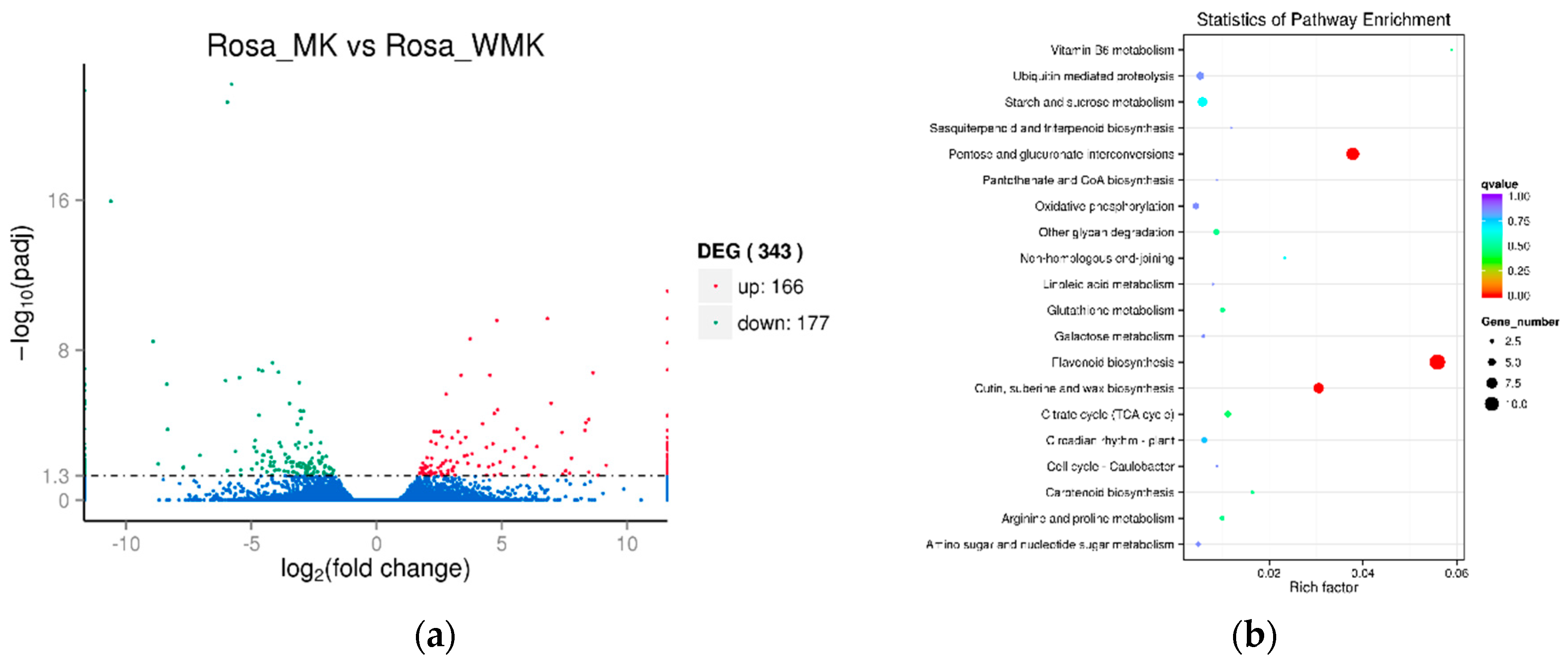
2.4. Analysis of Differentially Expressed Genes (DEGs) Identified in the Two Libraries
2.5. Characterization and Expression Analysis of Genes Involved in Flavonoids and Associated Biosynthesis Pathway
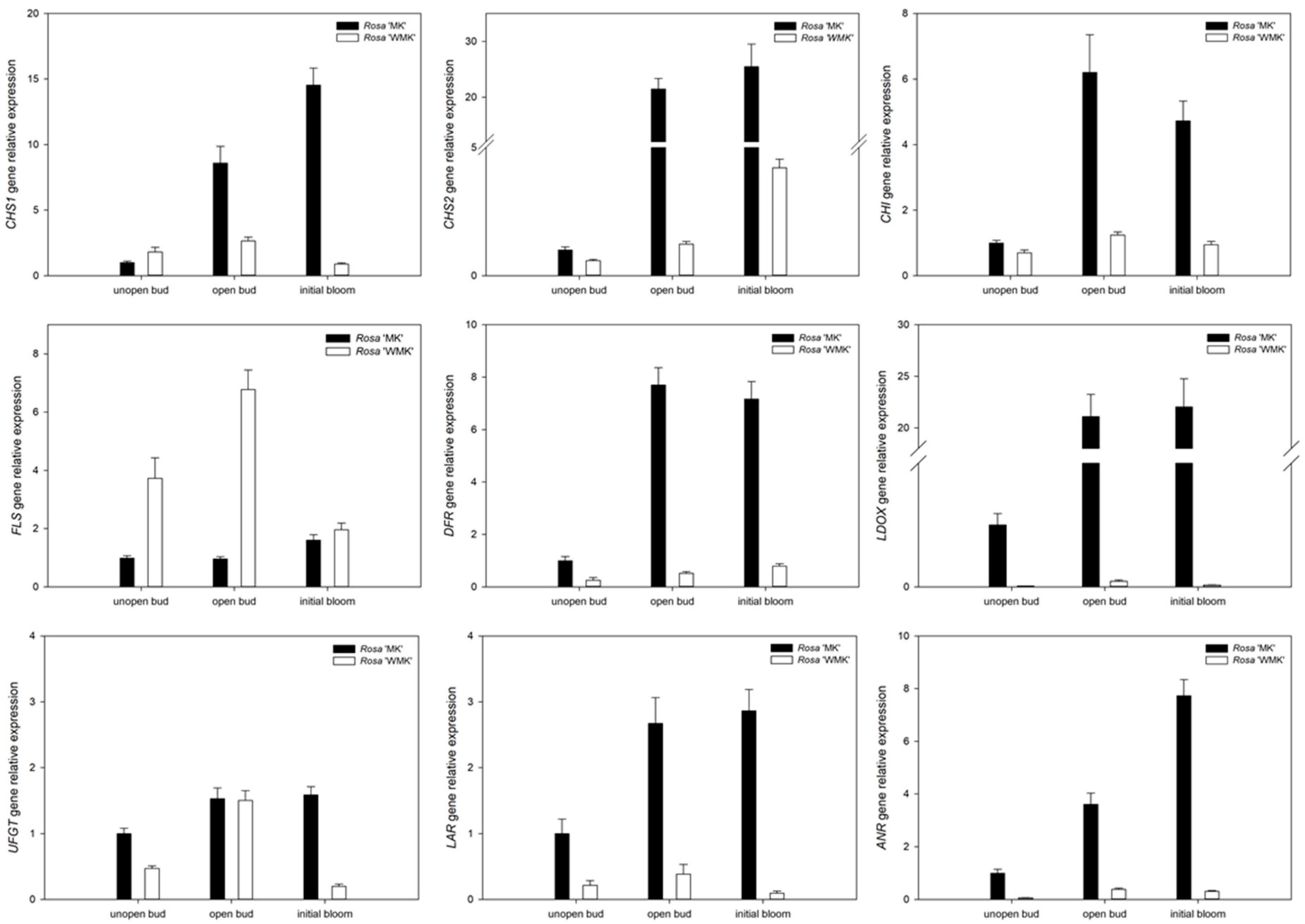
2.6. Expression Profiling of Flavonoid Biosynthesis Related Genes During Flowering
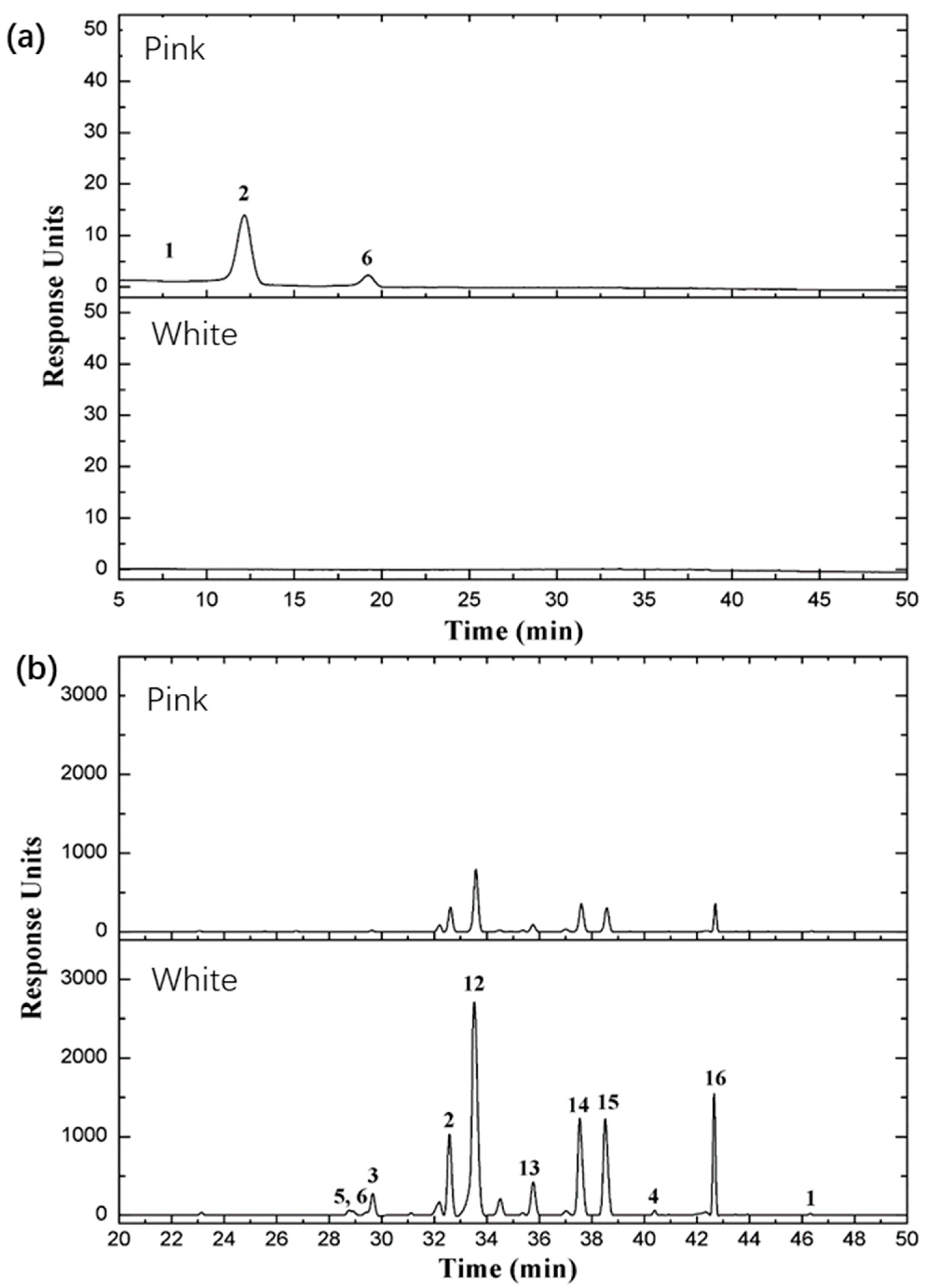
2.7. Major Classes of Flavonoids and Anthocyanins in the Two Rose Varieties
2.8. Alignment of Deduced Amino Acid Sequences of Two R2R3-MYB TFs
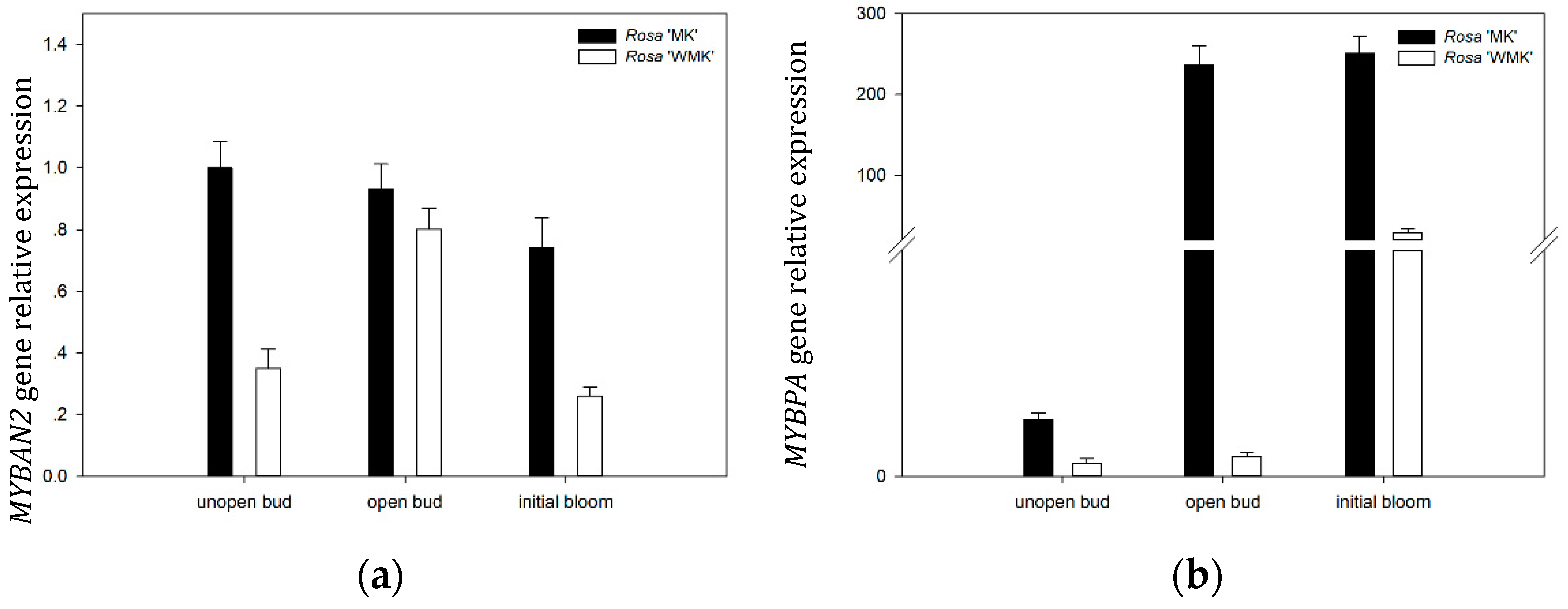
2.9. Expression Patterns of Two R2R3-MYB TFs During Flowering
3. Discussion
3.1. Hybrid Sequencing Strategy Provides More, Better-Quality Information
3.2. Differential Expression of Genes in the Petal of Two Rose Varieties
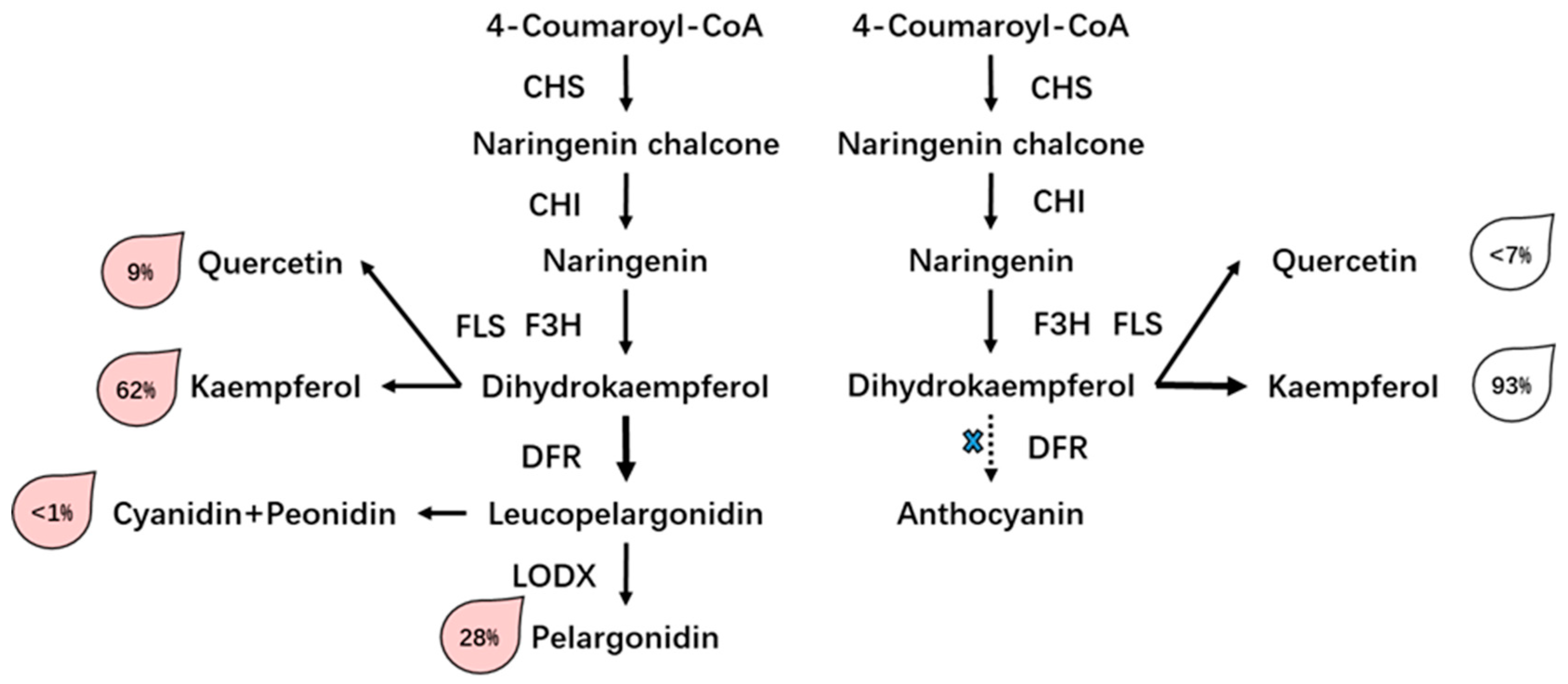
3.3. Differential Accumulation of Flavonoid in Two Rose Varieties
4. Materials and Methods
4.1. Plant Material
4.2. Identification and Quantification of Flavonoids and Anthocyanins in the Rose Petals
4.3. RNA Extraction, NGS Library Construction, and Sequencing
4.4. Library Preparation and PacBio RSII Sequencing
4.5. Data Analysis of the Sequencing Results
4.6. qRT-PCR Validation and Expression Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Availability of Data and Materials
References
- Liu, X.; Cao, X.; Shi, S.; Zhao, N.; Li, D.; Fang, P.; Chen, X.; Qi, W.; Zhang, Z. Comparative RNA-Seq analysis reveals a critical role for brassinosteroids in rose (Rosa hybrida) petal defense against Botrytis cinerea infection. BMC Genet. 2018, 19, 62. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Chen, X.; Fang, P.; Shi, S.; Li, J.; Liu, X.; Cao, X.; Zhao, N.; Hao, H.; Li, Y.; et al. Genomic and transcriptomic sequencing of Rosa hybrida provides microsatellite markers for breeding, flower trait improvement and taxonomy studies. BMC Plant Biol. 2018, 18, 119. [Google Scholar] [CrossRef] [PubMed]
- Debener, T.; Linde, M.; Debener, T. Exploring Complex Ornamental Genomes: The Rose as a Model Plant. Crit. Rev. Plant Sci. 2009, 28, 267–280. [Google Scholar] [CrossRef]
- Kumar, N.; Srivastava, G.C.; Dixit, K. Flower bud opening and senescence in roses (Rosa hybrida L.). Plant Growth Regul. 2008, 55, 81. [Google Scholar] [CrossRef]
- Woźnica, A.; Kalousis, A.; Hilario, M. Genetics and genomics of flower initiation and development in roses. J. Exp. Bot. 2013, 64, 847. [Google Scholar]
- Esselink, G.D.; Smulders, M.J.M.; Vosman, B. Identification of cut rose (Rosa hybrida) and rootstock varieties using robust sequence tagged microsatellite site markers. Theor. Appl. Genet. 2003, 106, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.H.; Byrne, D.H.; Ballard, R.E.; Rajapakse, S. Microsatellite marker development in rose and its application in Tetraploid mapping. J. Am. Soc. Hortic. Sci. 2006, 131, 380–387. [Google Scholar] [CrossRef]
- Koning-Boucoiran, C.F.S.; Esselink, G.D.; Vukosavljev, M.; Westende, W.P.C.V.T.; Gitonga, V.W.; Krens, F.A.; Voorrips, R.E.; Weg, W.E.V.D.; Schulz, D.; Debener, T.; et al. Using RNA-Seq to assemble a rose transcriptome with more than 13,000 full-length expressed genes and to develop the WagRhSNP 68k Axiom SNP array for rose (Rosa L.). Front. Plant Sci. 2015, 6, 249. [Google Scholar] [CrossRef]
- Smulders, M.J.M.; Voorrips, R.E.; Esselink, G.D.; Leonardo, T.M.S.; Westende, W.P.C.V.; Vukosavljev, M.; Koning-Boucoiran, C.F.S.; Weg, W.E.V.D.; Arens, P.; Schulz, D.; et al. Development of the WagRhSNP AXIOM SNP array based on sequences from tetraploid cut roses and garden roses. Acta Hortic. 2015, 1064, 177–184. [Google Scholar] [CrossRef]
- Oyant, H.S.; Crespel, L.; Rajapakse, S.; Zhang, L.; Foucher, F. Genetic linkage maps of rose constructed with new microsatellite markers and locating QTL controlling flowering traits. Tree Genet. Genomes 2008, 4, 11. [Google Scholar] [CrossRef]
- Henz, A.; Debener, T.; Linde, M. Identification of major stable QTLs for flower color in roses. Mol. Breed. 2015, 35, 190. [Google Scholar] [CrossRef]
- Schulz, D.F.; Schott, R.T.; Voorrips, R.E.; Smulders, M.J.M.; Linde, M.; Debener, T. Genome-Wide Association Analysis of the Anthocyanin and Carotenoid Contents of Rose Petals. Front. Plant Sci. 2016, 7, 401. [Google Scholar] [CrossRef]
- Mikanagi, Y.; Saito, N.; Yokoi, M.; Tatsuzawa, F. Anthocyanins in flowers of genus Rosa, sections Cinnamomeae (=Rosa), Chinenses, Gallicanae and some modern garden roses. Biochem. Syst. Ecol. 2000, 28, 887–902. [Google Scholar] [CrossRef]
- Burbulis, I.E.; Winkelshirley, B. Interactions among enzymes of the Arabidopsis flavonoid biosynthetic pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 12929–12934. [Google Scholar] [CrossRef] [PubMed]
- Holton, T.A.; Cornish, E.C. Genetics and Biochemistry of Anthocyanin Biosynthesis. Plant Cell 1995, 7, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Koes, R.; Verweij, W.; Quattrocchio, F. Flavonoids: A colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 2005, 10, 236–242. [Google Scholar] [CrossRef]
- Mol, J.; Grotewold, E.; Koes, R. How gens paint flowers and seeds. Trends Plant Sci. 1998, 6, 212–217. [Google Scholar] [CrossRef]
- Thompson, W.R.; Meinwald, J.; Aneshansley, D.; Eisner, T. Flavonols: Pigments responsible for ultraviolet absorption in nectar guide of flower. Science 1972, 177, 528. [Google Scholar] [CrossRef]
- Maekawa, S.; Inagaki, N.; Terabun, M. Effect of Aluminum Ions on the Bluing of Petal Color in Cut Chinese Bellflower, Platycodon grandiflorum. Plant Cell Physiol. 1983, 24, 759–764. [Google Scholar] [CrossRef]
- Vlaming, P.D.; Schram, A.W.; Wiering, H. Genes affecting flower colour and pH of flower limb homogenates in Petunia hybrida. Theor. Appl. Genet. 1983, 66, 271–278. [Google Scholar] [CrossRef]
- Nakamura, N.; Hirakawa, H.; Sato, S.; Otagaki, S.; Matsumoto, S.; Tabata, S.; Tanaka, Y. Genome structure of Rosa multiflora, a wild ancestor of cultivated roses. DNA Res. 2017, 25, 113–121. [Google Scholar] [CrossRef]
- Raymond, O.; Gouzy, J.; Just, J.; Badouin, H.; Verdenaud, M.; Lemainque, A.; Vergne, P.; Moja, S.; Choisne, N.; Pont, C. The Rosa genome provides new insights into the domestication of modern roses. Nat. Genet. 2018, 50, 772–777. [Google Scholar] [CrossRef]
- Hibrand, S.O.; Ruttink, T.; Hamama, L.; Kirov, I.; Lakhwani, D.; Zhou, N.N.; Bourke, P.M.; Daccord, N.; Leus, L.; Schulz, D. A high-quality genome sequence of Rosa chinensis to elucidate ornamental traits. Nat. Plants 2018, 4, 473–484. [Google Scholar] [CrossRef]
- Guoliang, W. History of roses in cultivation|Ancient Chinese Roses. Encycl. Rose Sci. 2003, 387–395. [Google Scholar] [CrossRef]
- Jing, M.; Fougère-Danezan, M.; Zhang, L.B.; Li, D.Z.; Yi, T.S. Untangling the hybrid origin of the Chinese tea roses: Evidence from DNA sequences of single-copy nuclear and chloroplast genes. Plant Syst. Evol. 2011, 297, 157–170. [Google Scholar]
- Yu, Z.; Liao, Y.; Teixeira da Silva, J.; Yang, Z.; Duan, J. Differential Accumulation of Anthocyanins in Dendrobium officinale Stems with Red and Green Peels. Int. J. Mol. Sci. 2018, 19, 2857. [Google Scholar] [CrossRef]
- Mushtaq, M.A.; Pan, Q.; Chen, D.; Zhang, Q.; Ge, X.; Li, Z. Comparative Leaves Transcriptome Analysis Emphasizing on Accumulation of Anthocyanins in Brassica: Molecular Regulation and Potential Interaction with Photosynthesis. Front. Plant Sci. 2016, 7, 311. [Google Scholar] [CrossRef]
- Xu, Z.; Peters, R.J.; Weirather, J.; Luo, H.; Liao, B.; Zhang, X.; Zhu, Y.; Ji, A.; Zhang, B.; Hu, S. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015, 82, 951–961. [Google Scholar] [CrossRef]
- Massonnet, M.; Fasoli, M.; Tornielli, G.B.; Altieri, M.; Sandri, M.; Zuccolotto, P.; Paci, P.; Gardiman, M.; Zenoni, S.; Pezzotti, M. Ripening Transcriptomic Program in Red and White Grapevine Varieties Correlates with Berry Skin Anthocyanin Accumulation. Plant Physiol. 2017, 174, 2376–2396. [Google Scholar] [CrossRef]
- Xie, L.; Fei, L.; Zhang, S.; Hui, Z.; Wei, Q.; Li, P.; Zhang, S.; Sun, R. Mining for Candidate Genes in an Introgression Line by Using RNA Sequencing: The Anthocyanin Overaccumulation Phenotype in Brassica. Front. Plant Sci. 2016, 7, 1245. [Google Scholar] [CrossRef]
- Dubois, A.; Carrere, S.; Raymond, O.; Pouvreau, B.; Cottret, L.; Roccia, A.; Onesto, J.P.; Sakr, S.; Atanassova, R.; Baudino, S. Transcriptome database resource and gene expression atlas for the rose. BMC Genom. 2012, 13, 638. [Google Scholar] [CrossRef]
- Kim, J.; Park, J.H.; Chan, J.L.; Lim, J.Y.; Ryu, J.Y.; Lee, B.W.; Choi, J.P.; Kim, W.B.; Lee, H.Y.; Choi, Y. Small RNA and transcriptome deep sequencing proffers insight into floral gene regulation in Rosa cultivars. BMC Genom. 2012, 13, 657. [Google Scholar] [CrossRef]
- Pei, H.; Ma, N.; Chen, J.; Zheng, Y.; Tian, J.; Li, J.; Zhang, S.; Fei, Z.; Gao, J. Integrative analysis of miRNA and mRNA profiles in response to ethylene in rose petals during flower opening. PLoS ONE 2013, 8, e64290. [Google Scholar] [CrossRef]
- Guo, X.; Yu, C.; Luo, L.; Wan, H.; Zhen, N.; Xu, T.; Tan, J.; Pan, H.; Zhang, Q. Transcriptome of the floral transition in Rosa chinensis ‘Old Blush’. BMC Genom. 2017, 18, 199. [Google Scholar] [CrossRef]
- Kin Fai, A.; Vittorio, S.; Pegah Tootoonchi, A.; Jens Durruthy, D.; Lawrence, L.; Williams, B.A.; Harm, V.B.; Schadt, E.E.; Reijo-Pera, R.A.; Underwood, J.G. Characterization of the human ESC transcriptome by hybrid sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, e4821–e4830. [Google Scholar]
- Weirather, J.L.; Pegah Tootoonchi, A.; Clark, T.A.; Elizabeth, T.; Powers, L.S.; Underwood, J.G.; Joseph, Z.; Jonas, K.; Wing Hung, W.; Kin Fai, A. Characterization of fusion genes and the significantly expressed fusion isoforms in breast cancer by hybrid sequencing. Nucleic Acids Res. 2015, 43, e116. [Google Scholar] [CrossRef]
- Deonovic, B.; Wang, Y.; Weirather, J.; Wang, X.J.; Au, K.F. IDP-ASE: Haplotyping and quantifying allele-specific expression at the gene and gene isoform level by hybrid sequencing. Nucleic Acids Res. 2017, 45, e32. [Google Scholar] [CrossRef]
- Weirather, J.L.; Cesare, M.D.; Wang, Y.; Piazza, P.; Sebastiano, V.; Wang, X.J.; Buck, D.; Au, K.F. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Research 2017, 6, 100. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sasaki, N.; Ohmiya, A. Biosynthesis of plant pigments: Anthocyanins, betalains and carotenoids. Plant J. 2010, 54, 733–749. [Google Scholar] [CrossRef]
- Tanaka, Y.; Tsuda, S.; Kusumi, T. Metabolic Engineering to Modify Flower Color. Plant Cell Physiol. 1998, 39, 1119–1126. [Google Scholar] [CrossRef]
- Venter, A.; Joubert, E.; de Beer, D. Characterisation of phenolic compounds in South African plum fruits (Prunus salicina Lindl.) using HPLC coupled with diode-array, fluorescence, mass spectrometry and on-line antioxidant detection. Molecules 2013, 18, 5072–5090. [Google Scholar] [CrossRef]
- Jin, H.; Martin, C. Multifunctionality and diversity within the plant MYB-gene family. Plant Mol. Boil. 1999, 41, 577–585. [Google Scholar] [CrossRef]
- Stracke, R.; Werber, M.; Weisshaar, B. The R2R3-MYB gene family in Arabidopsis thaliana. Curr. Opin. Plant Boil. 2001, 4, 447–456. [Google Scholar] [CrossRef]
- Takos, A.M.; Jaffé, F.W.; Jacob, S.R.; Jochen, B.; Robinson, S.P.; Walker, A.R. Light-induced expression of a MYB gene regulates anthocyanin biosynthesis in red apples. Plant Physiol. 2006, 142, 1216–1232. [Google Scholar] [CrossRef]
- Baudry, A.; Heim, M.A.; Dubreucq, B.; Caboche, M.; Weisshaar, B.; Lepiniec, L. TT2, TT8, and TTG1 synergistically specify the expression of BANYULS and proanthocyanidin biosynthesis in Arabidopsis thaliana. Plant J. 2010, 39, 366–380. [Google Scholar] [CrossRef]
- Cone, K.C.; Cocciolone, S.M.; Burr, F.A.; Burr, B. Maize anthocyanin regulatory gene Pl is a duplicate of C1 that functions in the plant. Plant Cell 1993, 5, 1795–1805. [Google Scholar]
- Bogs, J.; Jaffé, F.W.; Takos, A.M.; Walker, A.R.; Robinson, S.P. The Grapevine Transcription Factor VvMYBPA1 Regulates Proanthocyanidin Synthesis during Fruit Development. Plant Physiol. 2007, 143, 1347–1361. [Google Scholar] [CrossRef]
- Xu, Z.; Luo, H.; Ji, A.; Zhang, X.; Song, J.; Chen, S. Global Identification of the Full-Length Transcripts and Alternative Splicing Related to Phenolic Acid Biosynthetic Genes in Salvia miltiorrhiza. Front. Plant Sci. 2016, 7, 100. [Google Scholar] [CrossRef]
- Zhang, J.J.; Su, H.; Zhang, L.; Liao, B.S.; Xiao, S.M.; Dong, L.L.; Hu, Z.G.; Wang, P.; Li, X.W.; Huang, Z.H. Comprehensive Characterization for Ginsenosides Biosynthesis in Ginseng Root by Integration Analysis of Chemical and Transcriptome. Molecules 2017, 22, 889. [Google Scholar] [CrossRef]
- Yukihisa, K.; Masako, F.M.; Yuko, F.; Filippa, B.; Holton, T.A.; Mirko, K.; Noriko, N.; Keiko, Y.S.; Junichi, T.; Alix, P. Engineering of the rose flavonoid biosynthetic pathway successfully generated blue-hued flowers accumulating delphinidin. Plant Cell Physiol. 2007, 48, 1589. [Google Scholar]
- Gonzalez, A. Pigment loss in response to the environment: A new role for the WD/bHLH/MYB anthocyanin regulatory complex. New Phytol. 2009, 182, 1–3. [Google Scholar] [CrossRef]
- Dong, H.S.; Choi, M.G.; Kang, C.S.; Park, C.S.; Choi, S.B.; Park, Y.I. Overexpressing the wheat dihydroflavonol 4-reductase gene TaDFR increases anthocyanin accumulation in an Arabidopsis DFR mutant. Genes Genom. 2016, 38, 333–340. [Google Scholar]
- Watanabe, K.; Kobayashi, A.; Endo, M.; Sageono, K.; Toki, S.; Ono, M. CRISPR/Cas9-mediated mutagenesis of the dihydroflavonol-4-reductase-B (DFR-B) locus in the Japanese morning glory Ipomoea (Pharbitis) nil. Sci. Rep. 2017, 7, 10028. [Google Scholar] [CrossRef]
- Quattrocchio, F.M.; Wing, J.; Van Der Woude, K.; Souer, E.; De Vetten, N.; Mol, J.; Koes, R. Molecular analysis of the anthocyanin2 gene of Petunia and its role in the evolution of flower color. Plant Cell 1999, 11, 1433–1444. [Google Scholar] [CrossRef]
- Davies, K.M.; Albert, N.W.; Schwinn, K.E. From landing lights to mimicry: The molecular regulation of flower colouration and mechanisms for pigmentation patterning. Funct. Plant Boil. 2012, 39, 619–638. [Google Scholar] [CrossRef]
- Chen, K.; Liu, H.; Lou, Q.; Liu, Y. Ectopic Expression of the Grape Hyacinth (Muscari armeniacum) R2R3-MYB Transcription Factor Gene, MaAN2, Induces Anthocyanin Accumulation in Tobacco. Front. Plant Sci. 2017, 8, 965. [Google Scholar] [CrossRef]
- Holton, T.A.; Brugliera, F.; Tanaka, Y. Cloning and expression of flavonol synthase from Petunia hybrida. Plant J. 2010, 4, 1003–1010. [Google Scholar] [CrossRef]
- Ishihara, H.; Tohge, T.; Viehã¶Ver, P.; Fernie, A.R.; Weisshaar, B.; Stracke, R. Natural variation in flavonol accumulation in Arabidopsis is determined by the flavonol glucosyltransferase BGLU6. J. Exp. Bot. 2016, 67, 1505–1517. [Google Scholar] [CrossRef]
- Stracke, R.; Ishihara, H.G.; Barsch, A.; Mehrtens, F.; Niehaus, K.; Weisshaar, B. Differential regulation of closely related R2R3-MYB transcription factors controls flavonol accumulation in different parts of the Arabidopsis thaliana seedling. Plant J. 2010, 50, 660–677. [Google Scholar] [CrossRef]
- Ralf, S.; Oliver, J.; Matthias, K.; Takayuki, T.; Karsten, N.; Fernie, A.R.; Bernd, W. Analysis of PRODUCTION OF FLAVONOL GLYCOSIDES-dependent flavonol glycoside accumulation in Arabidopsis thaliana plants reveals MYB11-, MYB12- and MYB111-independent flavonol glycoside accumulation. New Phytol. 2010, 188, 985–1000. [Google Scholar]
- Frank, M.; Harald, K.; Pawel, B.; Bernd, W. The Arabidopsis transcription factor MYB12 is a flavonol-specific regulator of phenylpropanoid biosynthesis. Plant Physiol. 2005, 138, 1083–1096. [Google Scholar]
- Laurent, D.; Francois, B.; Chloé, M.; Virginie, L.; Alain, D.; Tristan, R.; Jean-Pierre, C.; Jean-Michel, M.; Said, H. Characterization of a grapevine R2R3-MYB transcription factor that regulates the phenylpropanoid pathway. Plant Physiol. 2006, 140, 499–511. [Google Scholar]
- Marles, M.A.S.; Ray, H.; Gruber, M.Y. New perspectives on proanthocyanidin biochemistry and molecular regulation. Phytochemistry 2003, 64, 367–383. [Google Scholar] [CrossRef]
- Debeaujon, I.; Nesi, N.; Perez, P.; Devic, M.; Grandjean, O.; Caboche, M.; Lepiniec, L. Proanthocyanidin-Accumulating Cells in Arabidopsis Testa: Regulation of Differentiation and Role in Seed Development. Plant Cell 2003, 15, 2514–2531. [Google Scholar] [CrossRef]
- Laurent, D.; Jochen, B.; Walker, A.R.; Thilia, F.; Alain, D.; Jean-Michel, M.; Robinson, S.P.; Barrieu, F. The transcription factor VvMYB5b contributes to the regulation of anthocyanin and proanthocyanidin biosynthesis in developing grape berries. Plant Physiol. 2008, 147, 2041–2053. [Google Scholar]
- Koyama, K.; Numata, M.; Nakajima, I.; Gotoyamamoto, N.; Matsumura, H.; Tanaka, N. Functional characterization of a new grapevine MYB transcription factor and regulation of proanthocyanidin biosynthesis in grapes. J. Exp. Bot. 2014, 65, 4433. [Google Scholar] [CrossRef]
- Akagi, T.; Tsujimoto, T.; Ikegami, A.; Yonemori, K. Effects of seasonal temperature changes on DkMyb4 expression involved in proanthocyanidin regulation in two genotypes of persimmon (Diospyros kaki Thunb.) fruit. Planta 2011, 233, 883–894. [Google Scholar] [CrossRef]
- Shamila Weerakoon, A.; Stephen, P.; Ross, C.; Tracie, W.; Simone, R.; Aidyn, M.; German, S. Biosynthesis of proanthocyanidins in white clover flowers: Cross talk within the flavonoid pathway. Plant Physiol. 2012, 158, 666–678. [Google Scholar]
- Su, F.; Zhang, Q.; Luo, Z. Isolation and characterisation of a Myb transcription factor DkPA1 related to proanthocyanidin biosynthesis in C-PCNA and non-PCNA persimmon (Diospyros kaki Thunb.) fruit. Acta Physiol. Plant. 2014, 36, 1831–1839. [Google Scholar] [CrossRef]
- Iwashina, T. Contribution to flower colors of flavonoids including anthocyanins: A review. Nat. Prod. Commun. 2015, 10, 529. [Google Scholar] [CrossRef]
- Jun, O.; Yoshiaki, K.; Yoshio, I.; Hidehito, T.; Masahiko, S. Plant biochemistry: Anthocyanin biosynthesis in roses. Nature 2005, 435, 757–758. [Google Scholar]
- Pollastri, S.; Tattini, M. Flavonols: Old compounds for old roles. Ann. Bot. 2011, 108, 1225. [Google Scholar] [CrossRef]
- Luo, P.; Ning, G.; Wang, Z.; Shen, Y.; Jin, H.; Li, P.; Huang, S.; Zhao, J.; Bao, M. Disequilibrium of Flavonol Synthase and Dihydroflavonol-4-Reductase Expression Associated Tightly to White vs. Red Color Flower Formation in Plants. Front. Plant Sci. 2015, 6, 1257. [Google Scholar] [CrossRef]
- Hanny, B.W. Gossypol, flavonoid, and condensed tannin content of cream and yellow anthers of five cotton (Gossypium hirsutum L.) cultivars. J. Agric. Food Chem. 1980, 28, 504–506. [Google Scholar] [CrossRef]
- Veberic, R.; Slatnar, A.; Bizjak, J.; Stampar, F.; Mikulic-Petkovsek, M. Anthocyanin composition of different wild and cultivated berry species. LWT Food Sci. Technol. 2015, 60, 509–517. [Google Scholar] [CrossRef]
- Hackl, T.; Hedrich, R.; Schultz, J.; Förster, F. proovread: Large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 2014, 30, 3004. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Yi, Z.; Chen, J.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J. iTAK: A Program for Genome-wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol. Plants 2016, 9, 1667–1670. [Google Scholar]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Anders, S. Analysing RNA-Seq data with the DESeq package. Mol. Biol. 2010, 43, 1–7. [Google Scholar]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Ramakers, C.; Ruijter, J.M.; Deprez, R.H.; Moorman, A.F. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett. 2003, 339, 62–66. [Google Scholar] [CrossRef]
- Wang, W.; Cai, J.; Wang, P.; Tian, S.; Qin, G. Post-transcriptional regulation of fruit ripening and disease resistance in tomato by the vacuolar protease SlVPE3. Genome Boil. 2017, 18, 47. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RNA-Seq | Sample | Sub-Reads Base (Gb) | Sub-Reads Number | Circular Consensus Sequence | Full Length | Flnc | Average Flnc Length (bp) |
|---|---|---|---|---|---|---|---|
| Pacbio RSII | Mixture petal of two rose varieties at blooming stage | 10.55 | 5,383,989 | 463,753 | 370,571 | 365,407 | 2591 |
| RNA-seq | Sample | Repetition | Read Number | Read Bases (G) | GC (%) | Q30 (%) | Total Mapping (%) |
|---|---|---|---|---|---|---|---|
| IlluminaHiSeq 2500 | Petal of Rosa ‘Margo Koster’ at blooming stage | Rep1 | 52,204,870 | 6.52 6.55 6.54 6.54 6.53 6.55 | 46.3 | 96.8 | 44,427,346 (85.10%) |
| Rep2 | 52,393,148 | 6.55 6.54 6.54 6.53 6.55 | 46.42 | 97.08 | 45,811,502 (87.44%) | ||
| Rep3 | 52,361,786 | 6.54 | 46.46 | 97.05 | 43,569,408 (83.21%) | ||
| Petal of Rosa ‘White Margo Koster’ at blooming stage | Rep1 | 52,317,316 | 6.54 | 46.29 | 97.06 | 45,439,584 (86.85%) | |
| Rep2 | 52,252,330 | 6.53 | 46.41 | 97.04 | 45,425,380 (86.93%) | ||
| Rep3 | 52,424,294 | 6.55 | 46.52 | 97.1 | 43,782,470 (83.52%) |
| Parameters | Counts | Motifs | No. of SSR |
|---|---|---|---|
| Total number of sequences examined | 107,280 | Mononucleotide | 56,828 |
| Total size of examined sequences (bp) | 290,704,013 | Dinucleotide | 25,261 |
| Total number of identified SSRs | 96,902 | Trinucleotide | 13,799 |
| Number of SSR containing sequences | 45,782 | Tetranucleotide | 550 |
| Number of sequences containing more than one SSR | 20,252 | Pentanucleotide | 114 |
| Number of SSRs present in compound formation | 28,391 | Hexanucleotide | 350 |
| Gene Name | #ID | Expression Profile (FPKM) | Log2 (Change Fold) | |
|---|---|---|---|---|
| Rosa ‘Margo Koster’ | Rosa ‘White Margo Koster’ | |||
| Rm4CL | c75759/f1p0/2016 * | 35.59 | 72.72 | 0.97 |
| RmCHS | c73992/f1p0/1241 | 79.55 | 37.44 | −0.92 |
| RmF3H | c18601/f3p3/1313 * | 28.11 | 12.28 | −0.84 |
| RmDFR | c10949/f1p0/1197 | 44.07 | 11.44 | −0.51 |
| RmFLS | c70511/f47p0/1468 | 4.42 | 249.11 | 0.17 |
| RmLDOX | c68764/f1p8/1370 | 1240.90 | 5.67 | −0.13 |
| RmANR | c64060/f1p0/1292 | 169.81 | 42.56 | −0.50 |
| RmLAR | c21707/f3p7/1533 * | 86.88 | 29.34 | −0.64 |
| RmGT | c41272/f1p0/1762 | 8.53 | 35.75 | 0.48 |
| RmCCD | c22761/f1p25/2265 * | 11.20 | 5.35 | −0.94 |
| No. | Retention Time (min) | λmax (nm) | [M]+ | Fragment Ions | Tentative Identification | Rosa ‘MK’ (μg/g) | Rosa ‘WMK’ (μg/g) | Reference | Catalogue |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 9.681 | 280, 520 | 611.161 | 287.055 | Cyanidin 3,5 diglucoside | 138.679 | 0.000 | STD | AC |
| 2 | 12.396 | 280, 520 | 595.167 | 271.059 | Pelargonidin 3,5 diglucoside | 5004.735 | 0.000 | STD | AC |
| 3 | 13.569 | 280, 520 | 625.177 | 301.070 | Peonidin, 3,5-diglucoside | 5.794 | 0.000 | STD | AC |
| 4 | 18.955 | 280, 520 | 433.112 | 271.059 | Pelargonidin 3-glucoside | 618.789 | 0.000 | [41] | AC |
| 5 | 26.356 | 280, 520 | 519.114 | 271.059 | Pelargonidin 3-glucoside-carboxyacetyl | 0.000 | 0.000 | [41] | AC |
| 6 | 11.678 | 280 | 579.149 | 291.086 | Procyanindin B1 | 63.072 | 168.931 | STD | PC |
| 7 | 32.757 | 280 | 595.166 | 566.426 | Proanthocyanidins | 22,932.771 | 27,408.189 | STD | PC |
| 8 | 46.510 | 264, 365 | 287.057 | 153.017 | Kaempferol | 28.553 | 74.894 | STD | FL |
| 9 | 42.813 | 265, 350 | 637.177 | 287.057 | Multiflorin A | 12,585.927 | 22,779.216 | [41] | FL |
| 10 | 42.590 | 254, 370 | 303.049 | 153.017 | Quercetin | 1.432 | 11.934 | STD | FL |
| 11 | 33.688 | 265, 350 | 449.108 | 287.057 | Kaempferol-3-glucoside | 3275.087 | 6992.866 | [41] | FL |
| 12 | 38.683 | 265, 350 | 433.113 | 287.057 | Kaempferol-3-rhamnopyranoside (Afzelin) | 4189.292 | 7339.989 | [41] | FL |
| 13 | 37.703 | 265, 350 | 419.097 | 287.057 | Kaempferol-3-O-β-glucopyranosyl-7-O-α-rhamnopyranoside | 1905.600 | 1880.046 | [41] | FL |
| 14 | 35.872 | 265, 350 | 418.090 | 287.057 | Kaempferol-3-O-α-d-Arabinoside | 1409.055 | 2876.430 | [41] | FL |
| 15 | 32.689 | 264, 348 | 595.166 | 287.057 | Kaempfeol-3-rutinoside | 2701.640 | 3043.795 | STD | FL |
| 16 | 29.689 | 255, 353 | 465.102 | 303.049 | Quercetin 3-glucoside | 260.776 | 1623.913 | STD | FL |
| 17 | 28.741 | 254, 350 | 611.160 | 303.049 | Rutin | 34.425 | 320.854 | STD | FL |
| 18 | 28.883 | 255, 352 | 465.102 | 303.049 | Hyperoside | 0.000 | 181.090 | STD | FL |
| 19 | 19.050 | 280 | 291.084 | 139.038 | (−)-Epicatechin | 0.000 | 6.821 | STD | FL |
| 20 | 13.429 | 280 | 291.085 | 139.038 | (+/−)-Catechin hydrate | 282.540 | 1534.824 | STD | FL |
| 21 | 14.455 | 221, 260, 292 | 169.049 | 125.060 | Vanillic acid | 141.047 | 72.536 | STD | FL |
| 22 | 14.184 | 240, 325 | 355.102 | 163.039 | Chlorogenic acid | 1.465 | 0.765 | STD | FL |
| 23 | 14.184 | 243, 323 | 355.102 | 163.038 | Neochlorogenic acid | 14.655 | 7.647 | STD | FL |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, P.; Lin, F.; Li, B.; Zheng, Y. Hybrid-Transcriptome Sequencing and Associated Metabolite Analysis Reveal Putative Genes Involved in Flower Color Difference in Rose Mutants. Plants 2019, 8, 267. https://doi.org/10.3390/plants8080267
Huang P, Lin F, Li B, Zheng Y. Hybrid-Transcriptome Sequencing and Associated Metabolite Analysis Reveal Putative Genes Involved in Flower Color Difference in Rose Mutants. Plants. 2019; 8(8):267. https://doi.org/10.3390/plants8080267
Chicago/Turabian StyleHuang, Ping, Furong Lin, Bin Li, and Yongqi Zheng. 2019. "Hybrid-Transcriptome Sequencing and Associated Metabolite Analysis Reveal Putative Genes Involved in Flower Color Difference in Rose Mutants" Plants 8, no. 8: 267. https://doi.org/10.3390/plants8080267
APA StyleHuang, P., Lin, F., Li, B., & Zheng, Y. (2019). Hybrid-Transcriptome Sequencing and Associated Metabolite Analysis Reveal Putative Genes Involved in Flower Color Difference in Rose Mutants. Plants, 8(8), 267. https://doi.org/10.3390/plants8080267