
Genomic Position and Markers Associated with the Hull-Less Seed Trait in Pumpkin


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material, Population Development and Phenotyping
2.2. DNA Extraction and Whole Genome Re-Sequencing
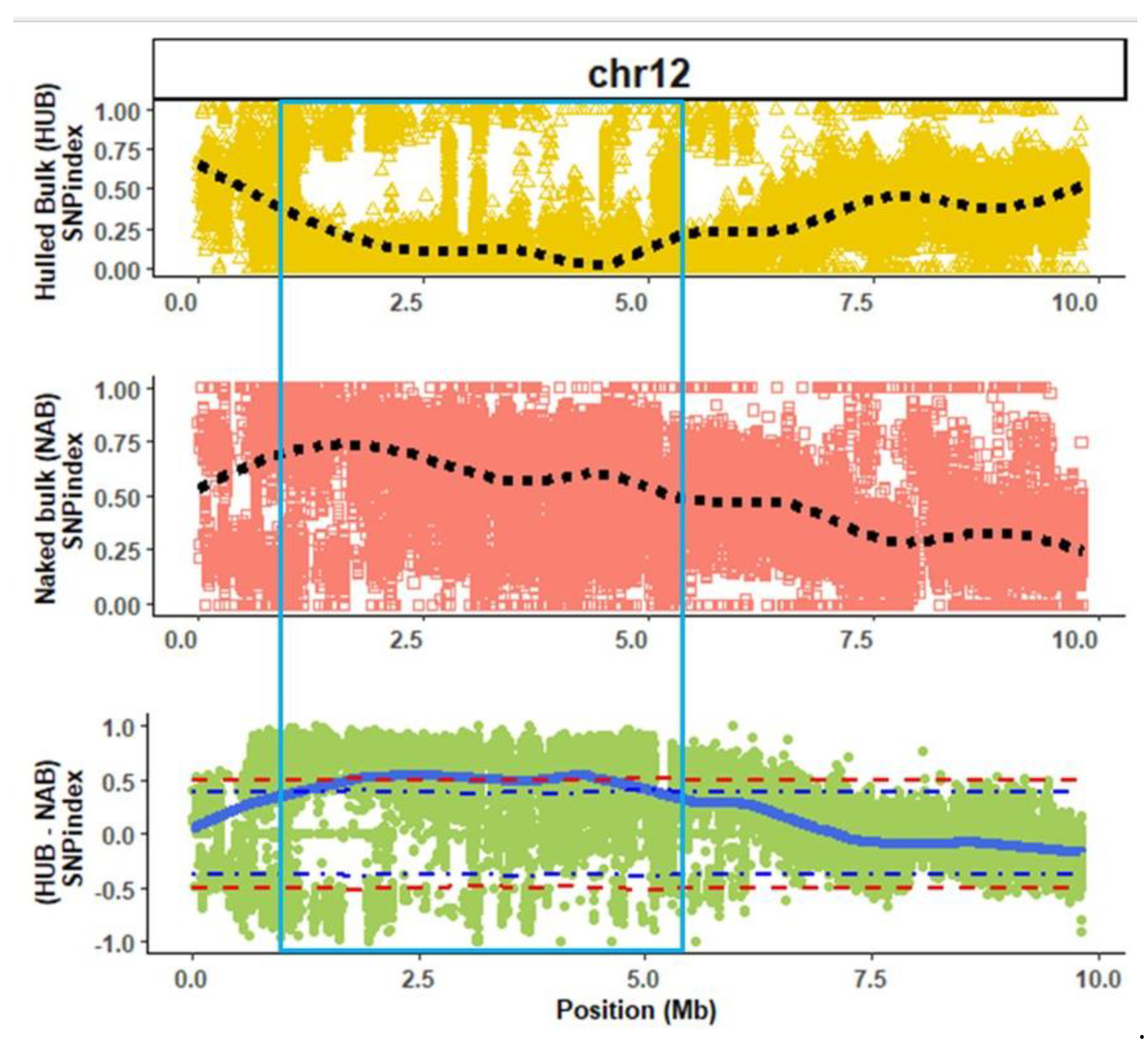
2.3. QTL-Seq Analysis
2.4. Marker Test and Candidate Genes
3. Results
3.1. Phenotypic Data
3.2. QTL Analysis
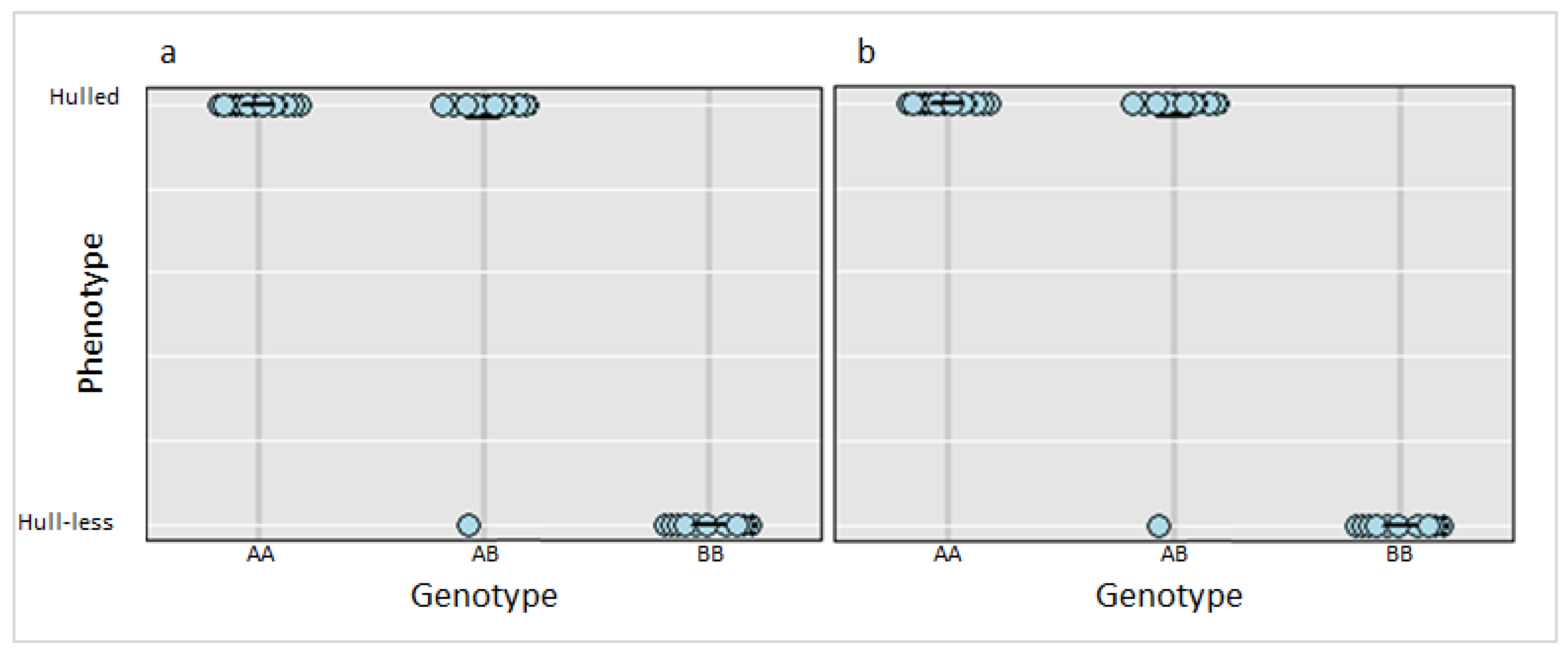
3.3. Marker Validation and Candidate Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Loy, J.B. Morpho-physiological aspects of productivity and quality in squash and pumpkins (Cucurbita spp.). Crit. Rev. Plant Sci. 2004, 23, 337–363. [Google Scholar] [CrossRef]
- Baxter, G.G.; Murphy, K.; Paech, A. The potential to produce pumpkin seed for processing in North East Victoria. Rural. Ind. Res. Dev. Corp. 2012, 11, 5–36. [Google Scholar]
- Stevenson, D.G.; Eller, F.J.; Wang, L.; Jane, J.L.; Wang, T.; Inglett, G.E. Oil and tocopherol content and composition of pumpkin seed oil in 12 cultivars. J. Agric. Food Chem. 2007, 55, 4005–4013. [Google Scholar] [CrossRef] [PubMed]
- Fruhwirth, G.O.; Hermetter, A. Seeds and oil of the Styrian oil pumpkin: Components and biological activities. Eur. J. Lipid Sci. Technol. 2007, 109, 1128–1140. [Google Scholar] [CrossRef]
- Lelley, T.; Loy, B.L.; Murkovic, M. Hull-Less oil seed pumpkin. In Oil Crops, Handbook of Plant Breeding; Vollmann, J., Rajcan, I., Eds.; Springer: New York, NY, USA, 2009; pp. 469–492. [Google Scholar]
- Meru, G.; Fu, Y.; Leyva, D.; Sarnoski, P.; Yagiz, Y. Phenotypic relationships among oil, protein, fatty acid composition and seed size traits in Cucurbita pepo. Sci. Hortic. 2018, 233, 47–53. [Google Scholar] [CrossRef]
- Nesaretnam, K.; Gomez, P.A.; Selvaduray, K.R.; Razak, G.A. Tocotrienol levels in adipose tissue of benign and malignant breast lumps in patients in Malaysia. Asia Pac. J. Clin. Nutr. 2007, 16, 498–504. [Google Scholar]
- Wassom, J.J.; Mikkelineni, V.; Bohn, M.O.; Rocheford, T.R. QTL for fatty acid composition of maize kernel oil in Illinois high oil B73 backcross-derived lines. Crop Sci. 2008, 48, 69–78. [Google Scholar] [CrossRef]
- Teppner, H. Notes on Lagenaria and Cucurbita (Cucurbitaceae) review and new contributions. Phyton 2008, 44, 245–308. [Google Scholar]
- McGregor, C.; Meru, G. Translational genomics of cucurbit oil seeds. In Oil Crop Genomics; Tombuloglu, H., Unver, T., Tombuloglu, G., Hakeem, K.R., Eds.; Springer Nature: Cham, Switzerland, 2021. [Google Scholar]
- Teppner, H. Cucurbita pepo (Cucurbitaceae)-history, seed coat types, thin coated seeds and their genetics. Phyton Horn 2000, 40, 1–42. [Google Scholar]
- Zraidi, A.; Obermayer, R.; Pachner, M.; Lelley, T. On the genetics and histology of the hull–less character of Styrian Oil–Pumpkin (Cucurbita pepo L.). Cucurbit Genet. Coop. Rep. 2003, 26, 57–61. [Google Scholar]
- Collard, B.C.; Jahufer, M.Z.Z.; Brouwer, J.B.; Pang, E.C.K. An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: The basic concepts. Euphytica 2005, 142, 169–196. [Google Scholar] [CrossRef]
- Martinez, I.; Michael, V.N.; Fu, Y.; Shrestha, S.; Meru, G. DNA Extraction from a Single Seed for Marker-Assisted Selection in Squash. Am. J. Plant Sci. 2021, 12, 1912–1925. [Google Scholar] [CrossRef]
- Gong, L.; Stift, G.; Kofler, R.; Pachner, M.; Lelley, T. Microsatellites for the Genus Cucurbita and an SSR-Based Genetic Linkage Map of Cucurbita pepo L. Theor. Appl. Genet. 2008, 117, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Blacket, M.J.; Robin, C.; Good, R.T.; Lee, S.F.; Miller, A. Universal primers for fluorescent labelling of PCR fragments-an efficient and cost-effective approach to genotyping by fluorescence. Mol. Ecol. Resour. 2012, 12, 456–463. [Google Scholar] [CrossRef]
- Wyatt, L.E.; Strickler, S.R.; Mueller, L.A.; Mazourek, M. Comparative analysis of Cucurbita pepo metabolism throughout fruit development in acorn squash and oilseed pumpkin. Hortic. Res. 2016, 3, 16045. [Google Scholar] [CrossRef]
- Michelmore, R.; Paran, I.; Kesseli, R.V. Identification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. USA 1991, 88, 9828–9832. [Google Scholar] [CrossRef]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef]
- Ramos, A.; Fu, Y.; Michael, V.; Meru, G. QTL-seq for identification of loci associated with resistance to Phytophthora crown rot in squash. Sci. Rep. 2020, 10, 5326. [Google Scholar] [CrossRef]
- Shrestha, S.; Michael, V.N.; Fu, Y.; Meru, G. Genetic Loci Associated with Resistance to Zucchini Yellow Mosaic Virus in Squash. Plants 2021, 10, 1935. [Google Scholar] [CrossRef]
- Vogel, G.; LaPlant, K.E.; Mazourek, M.; Gore, M.A.; Smart, C.D. A combined BSA-Seq and linkage mapping approach identifies genomic regions associated with Phytophthora root and crown rot resistance in squash. Theor. Appl. Genet. 2021, 134, 1015–1031. [Google Scholar] [CrossRef]
- Semagn, K.; Babu, R.; Hearne, S.; Olsen, M. Single nucleotide polymorphism genotyping using Kompetitive Allele Specific PCR (KASP): Overview of the technology and its application in crop improvement. Mol. Breed. 2014, 33, 1–14. [Google Scholar] [CrossRef]
- Freeman, J.H.; McAvoy, E.J.; Boyd, N.S.; Ozores-Hampton, M.; Paret, M.L.; Wang, Q.; Miller, C.F.; Desaeger, J.; Noling, J.W.; Xavier, M. Cucurbit Production. In Vegetable Production Handbook of Florida 2018–2019 Edition; Univerity of Florida Institute of Food and Agricultural Sciences: Gainesville, FL, USA, 2021; p. 55. [Google Scholar]
- Montero-Pau, J.; Blanca, J.; Bombarely, A.; Ziarsolo, P.; Esteras, C.; Martí-Gómez, C.; Ferriol, M.; Gómez, P.; Jamilena, M.; Mueller, L.; et al. De novo assembly of the zucchini genome reveals a whole-genome duplication associated with the origin of the Cucurbita genus. Plant Biotechnol. J. 2018, 16, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Branham, S.E.; Wechter, W.P.; Lambel, S.; Massey, L.; Ma, M.; Fauve, J.; Farnham, M.W.; Levi, A. QTL-seq and marker development for resistance to Fusarium oxysporum f. sp. niveum race 1 in cultivated watermelon. Mol. Breed. 2018, 38, 139. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Mansfeld, B.N.; Grumet, R. QTLseqr: An R Package for bulk segregant analysis with next-generation sequencing. Plant Genome 2018, 11, 180006. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 7 December 2021).
- Broman, K.W.; Sen, S. A Guide to QTL Mapping with R/qtl; Springer: New York, NY, USA, 2009; pp. 21–177. [Google Scholar]
- Kumpatla, S.; Buyyarapu, R.; Abdurakhmonov, I.; Mammadov, J. Genomics-assisted plant breeding in the 21st century: Technological advances and progress. In Plant Breeding; Abdurakhmonov, I.Y., Ed.; IntechOpen: London, UK, 2012. [Google Scholar]
- Paris, H.S.; Yonash, N.; Portnoy, V.; Mozes-Daube, N.; Tzuri, G.; Katzir, N. Assessment of genetic relationships in Cucurbita pepo (Cucurbitaceae) using DNA markers. Theor. Appl. Genet. 2003, 106, 971–978. [Google Scholar] [CrossRef]
- Yang, Y.; Yoo, C.G.; Rottmann, W.; Winkeler, K.A.; Collins, C.M.; Gunter, L.E.; Jawdy, S.S.; Yang, X.; Pu, Y.; Ragauskas, A.J.; et al. PdWND3A, a wood-associated NAC domain-containing protein, affects lignin biosynthesis and composition in Populus. BMC Plant Biol. 2019, 19, 486. [Google Scholar] [CrossRef]
- Yu, A.; Wang, Z.; Zhang, Y.; Li, F.; Liu, A. Global gene expression of seed coat tissues reveals a potential mechanism of regulating seed size formation in castor bean. Int. J. Mol. Sci. 2019, 20, 1282. [Google Scholar] [CrossRef]
- Li, X.; Wu, H.X.; Southerton, S.G. Identification of putative candidate genes for juvenile wood density in Pinus radiata. Tree Physiol. 2012, 32, 1046–1057. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Genotype | Hulled | Hull-Less | Expected Ratio (Hulled: Hull-Less) | χ2 | p Value |
|---|---|---|---|---|---|
| Kakai | 0 | 12 | - | - | |
| Table Gold Acorn | 12 | 0 | - | - | |
| F1: Kakai × TGA | 12 | 0 | - | - | |
| F2: Kakai × TGA | 112 | 31 | 3:1 | 0.841 | 0.3590 (NS) |
| Genotype | Mapping Rate (%) | Sequencing Depth |
|---|---|---|
| Kakai | 98.91 | 57.78 |
| Table Gold Acorn | 97.24 | 68.79 |
| Hulled bulk | 95.31 | 67.71 |
| Hull-less bulk | 97.81 | 70.26 |
| Marker | Genomic Position (bp) 1 | Mutation | p-Value * | LOD * |
|---|---|---|---|---|
| Ch12_2976476 | 2976476 | T/C | 0.0006 | 4.72 |
| Ch12_3412046 | 3412046 | C/T | 0.0006 | 4.72 |
| Ch12_3412133 | 3412133 | C/T | 0.0006 | 4.72 |
| Ch12_3412203 | 3412203 | C/T | 0.002 | 4.70 |
| Ch12_3412341 | 3412341 | T/C | 0.0006 | 4.72 |
| Ch12_3417142 | 3417142 | A/C | 0.0006 | 4.72 |
| Ch12_4323103 | 4323103 | A/G | 0.0001 | 6.01 |
| Ch12_4326161 | 4326161 | C/T | 0.0001 | 6.01 |
| Ch12_4326524 | 4326524 | A/G | 0.0001 | 6.01 |
| Ch12_4327827 | 4327827 | C/T | 0.04 | 3.20 |
| Ch12_4328336 | 4328336 | T/C | 0.03 | 3.04 |
| Ch12 _4382130 | 4382130 | C/T | 0.0001 | 5.99 |
| Marker | Genomic Position (bp) 1 | p-Value * |
|---|---|---|
| Ch12_2976476 | 2976476 | 0.31 |
| Ch12_3412046 | 3412046 | 0.008 * |
| Ch12_3412344 | 3412344 | 0.31 |
| Ch12_3412203 | 3412203 | 0.31 |
| Ch12_3412341 | 3412341 | 0.80 |
| Ch12_3417142 | 3417142 | 0.002 * |
| Ch12_4323103 | 4323103 | 0.22 |
| Ch12_4326161 | 4326161 | 0.08 |
| Ch12_4326524 | 4326524 | 0.056 |
| Ch12_4327827 | 4327827 | 0.06 |
| Ch12_4328336 | 4328336 | 0.09 |
| Ch12 _4382130 | 4382130 | 0.09 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meru, G.; Fu, Y.; Shrestha, S.; Michael, V.N.; Dorval, M.; Mainviel, R. Genomic Position and Markers Associated with the Hull-Less Seed Trait in Pumpkin. Plants 2022, 11, 1238. https://doi.org/10.3390/plants11091238
Meru G, Fu Y, Shrestha S, Michael VN, Dorval M, Mainviel R. Genomic Position and Markers Associated with the Hull-Less Seed Trait in Pumpkin. Plants. 2022; 11(9):1238. https://doi.org/10.3390/plants11091238
Chicago/Turabian StyleMeru, Geoffrey, Yuqing Fu, Swati Shrestha, Vincent Njung’e Michael, Marie Dorval, and Riphine Mainviel. 2022. "Genomic Position and Markers Associated with the Hull-Less Seed Trait in Pumpkin" Plants 11, no. 9: 1238. https://doi.org/10.3390/plants11091238
APA StyleMeru, G., Fu, Y., Shrestha, S., Michael, V. N., Dorval, M., & Mainviel, R. (2022). Genomic Position and Markers Associated with the Hull-Less Seed Trait in Pumpkin. Plants, 11(9), 1238. https://doi.org/10.3390/plants11091238