Chaperoning Proteins for Destruction: Diverse Roles of Hsp70 Chaperones and their Co-Chaperones in Targeting Misfolded Proteins to the Proteasome


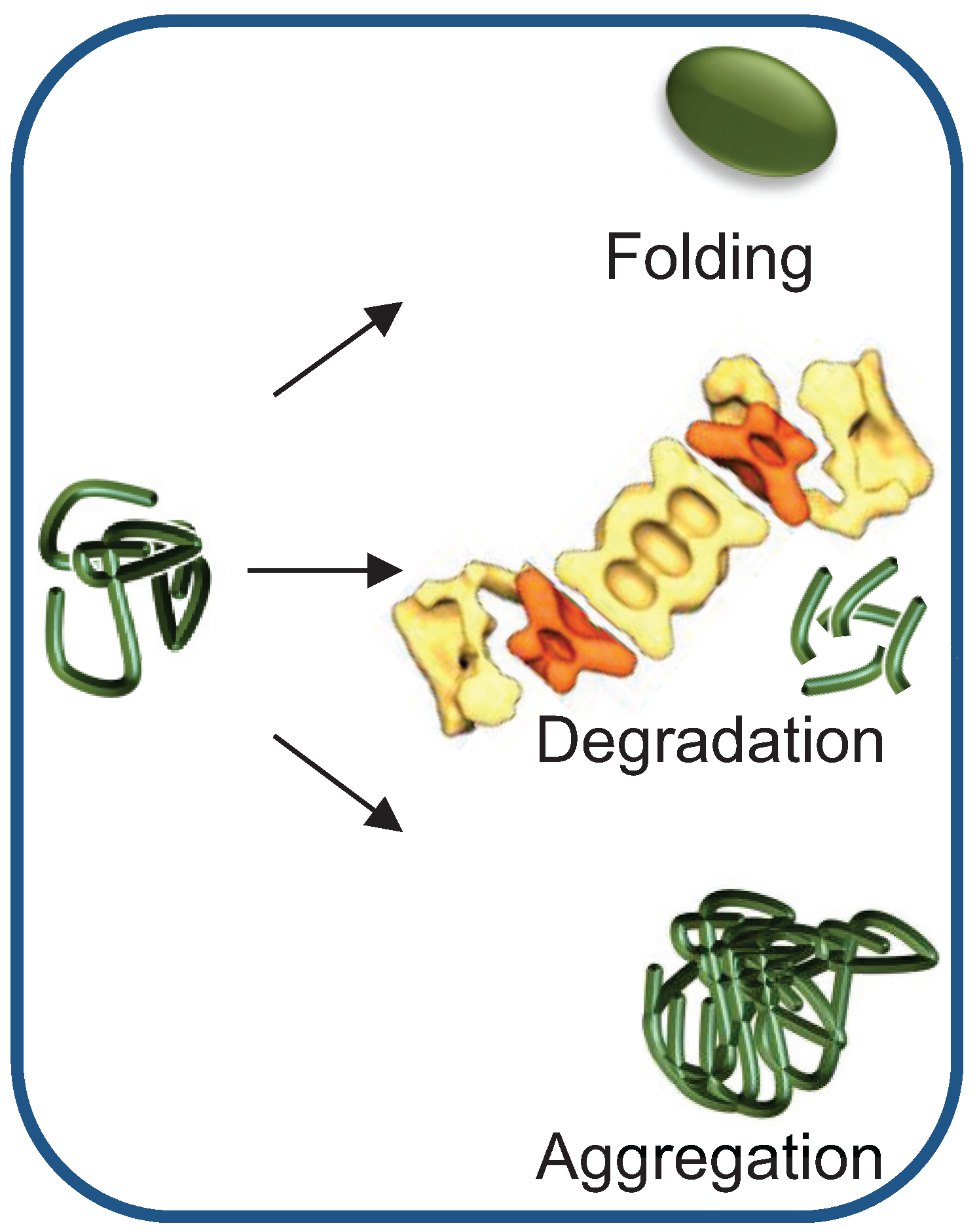{kind=link}
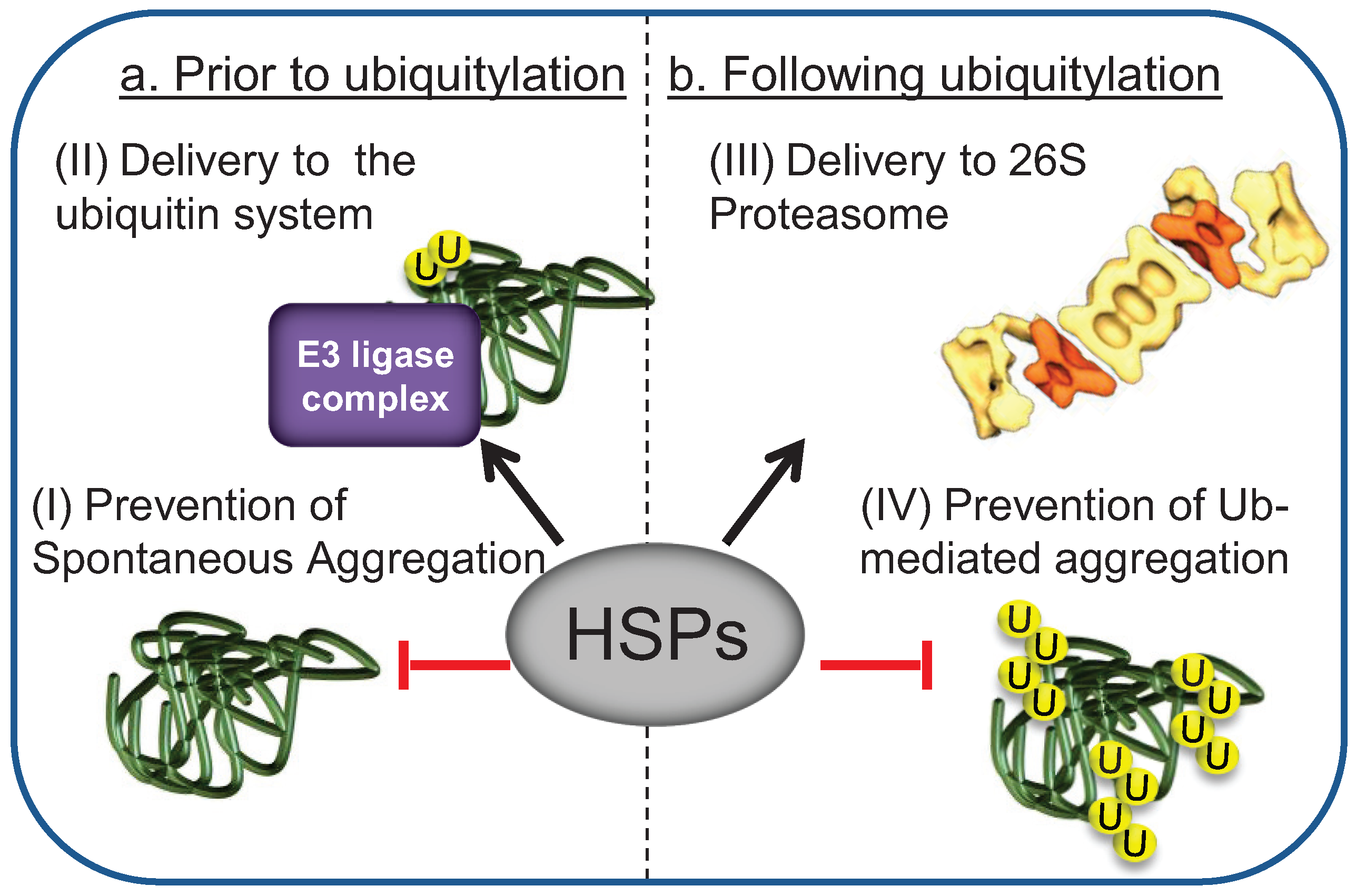{kind=link}
{kind=link}
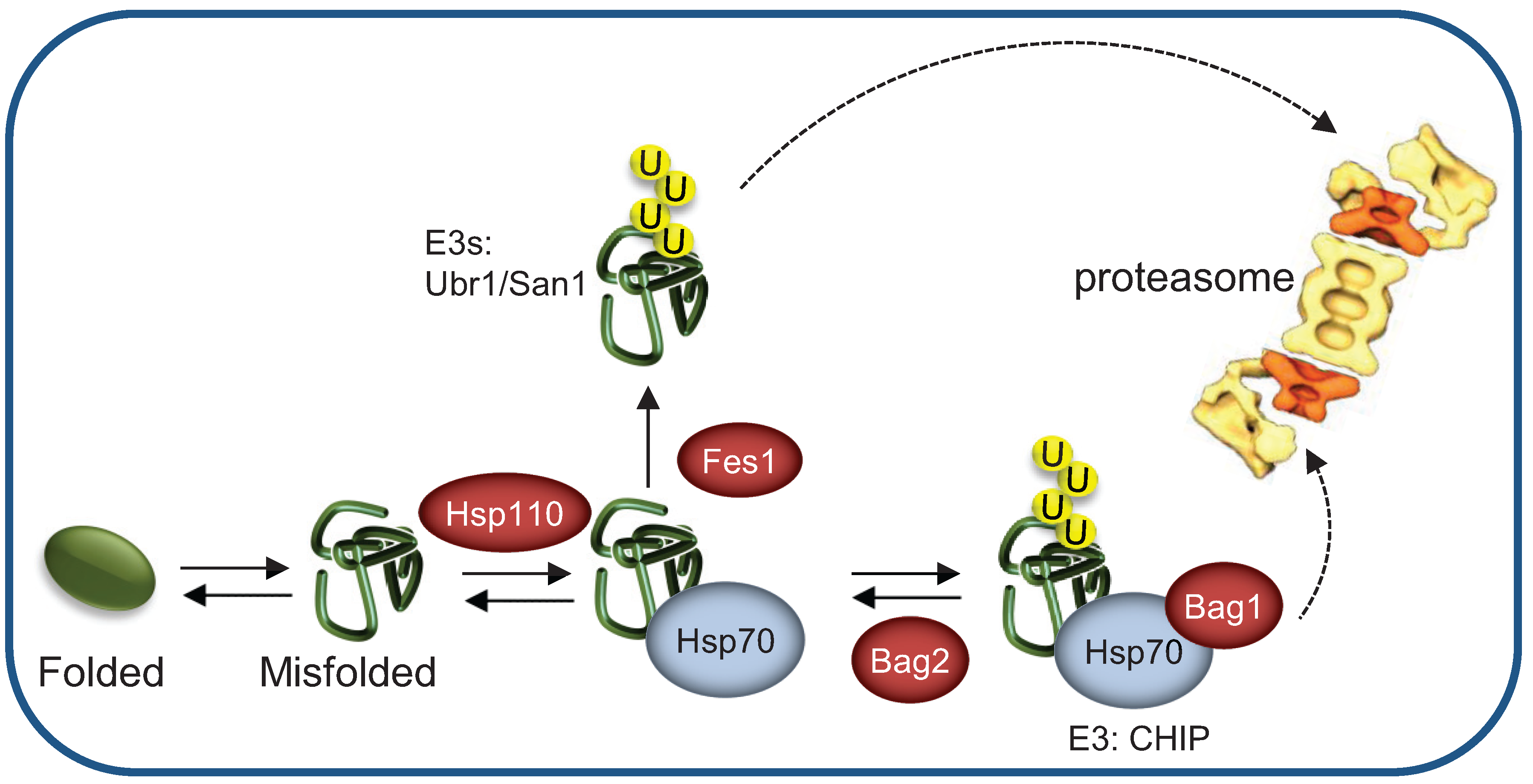{kind=link}
{kind=link}
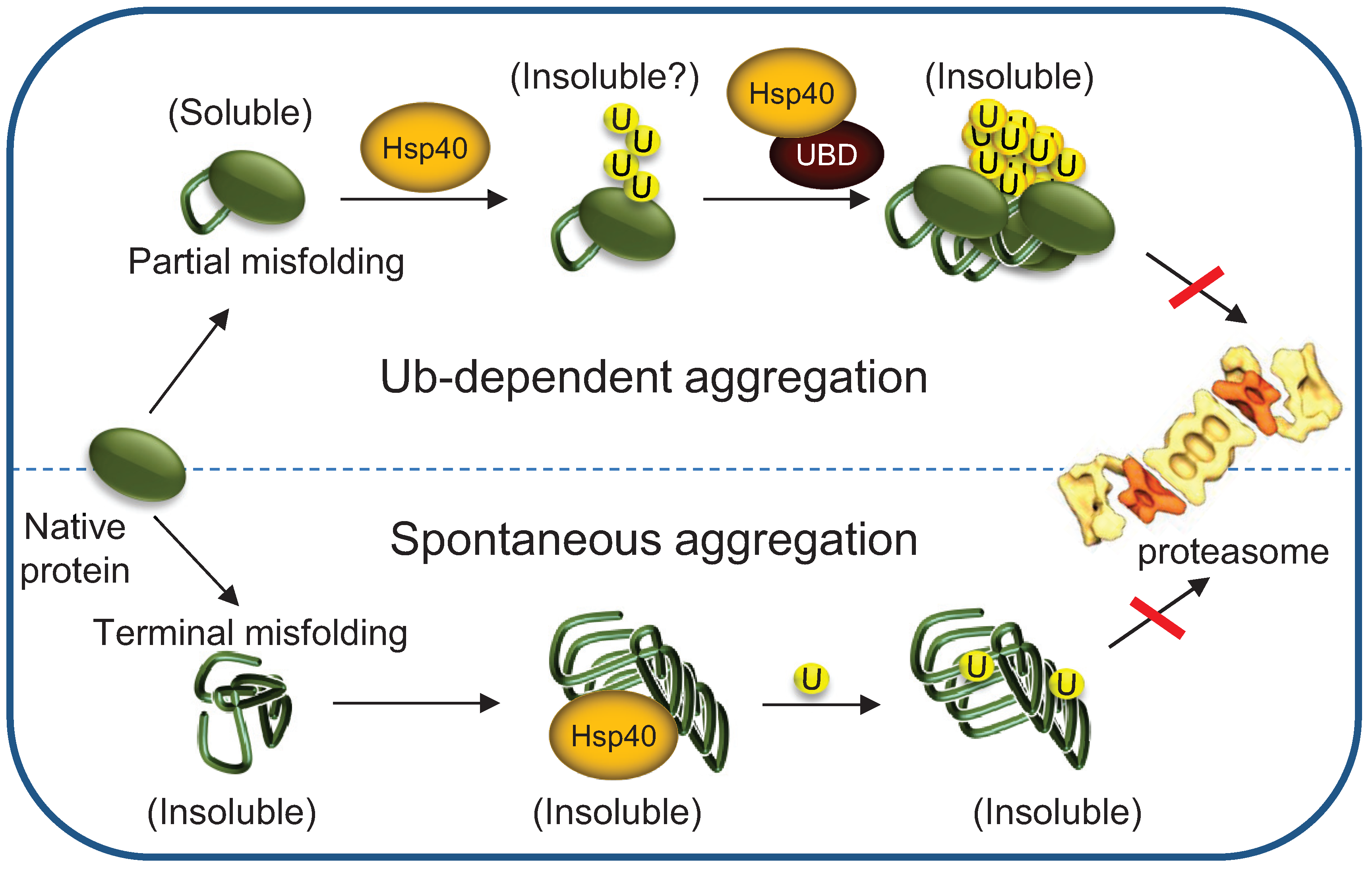{kind=link}
Abstract
:1. Introduction
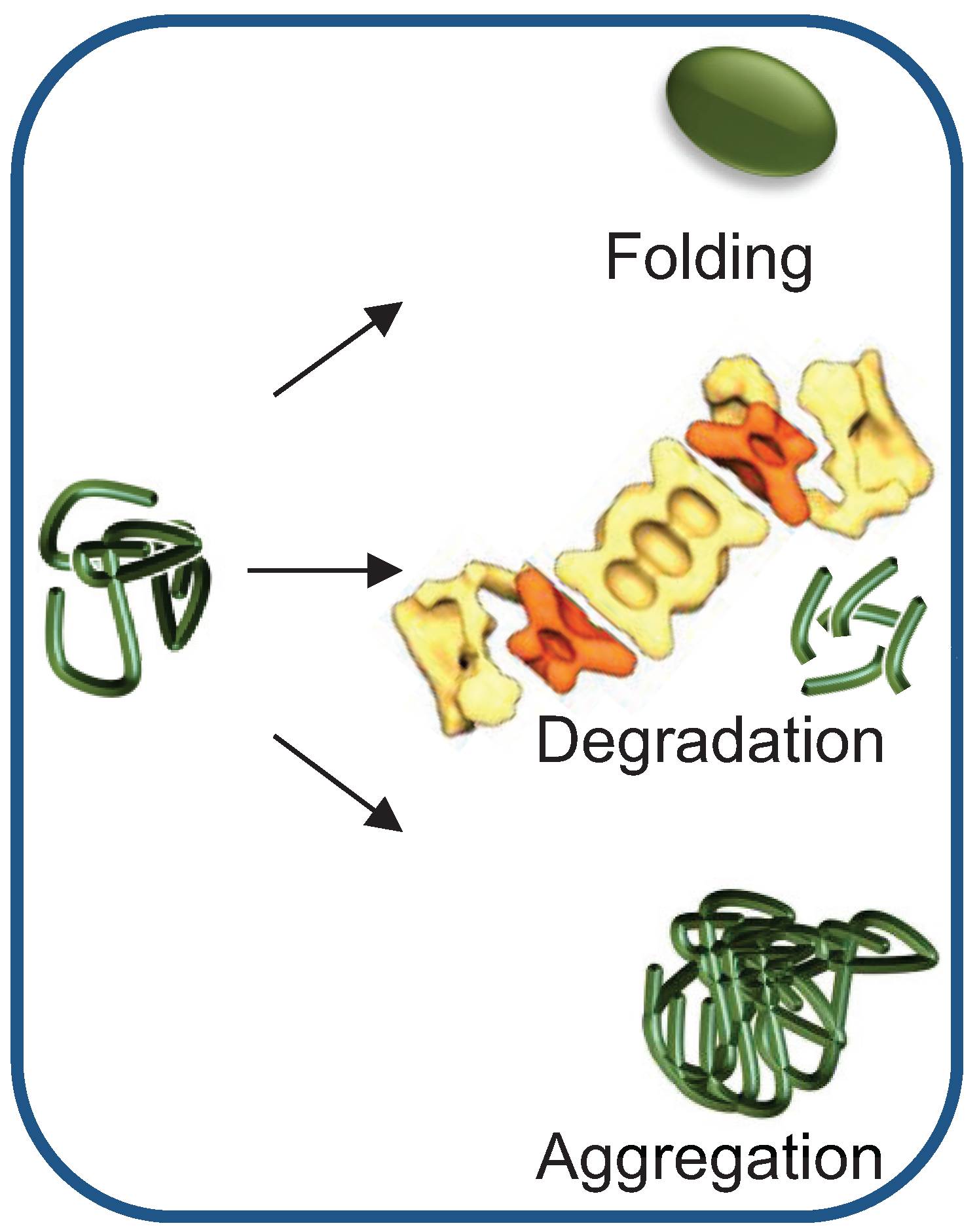
2. Chaperone-Assisted Proteasomal Degradation: Hsp40/70s’ Role in Presenting Misfolded Substrates to the UPS
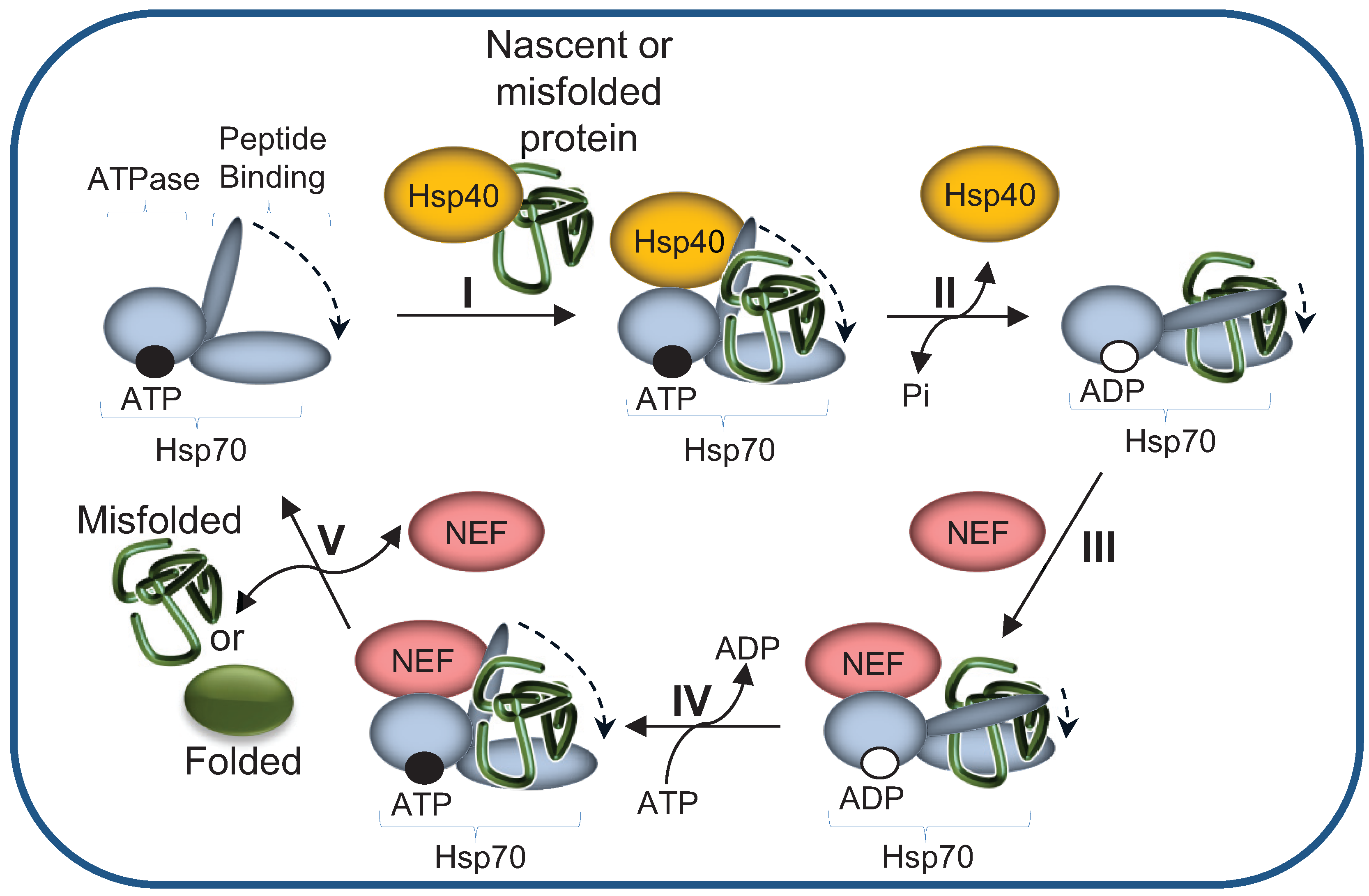
2.1. The Hsp70 Machinery Promotes UPS-Mediated Degradation by Maintaining Misfolded Protein Solubility
2.2. Hsp40 Co-Chaperones Are Essential Co-Factors of Hsp70s
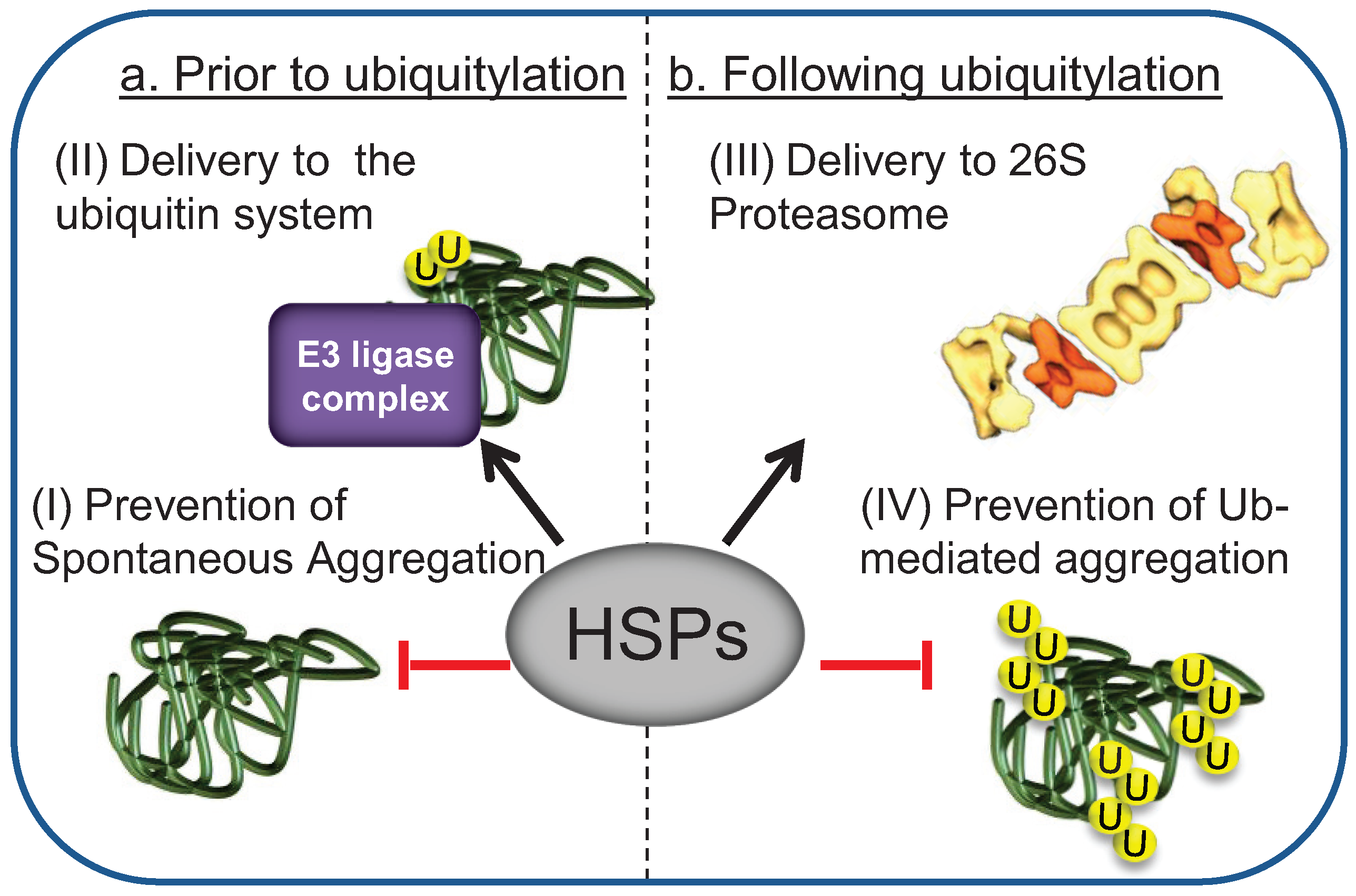
3. HSPs Mediate the Interaction of Misfolded Substrates with E3 Ligase Ubiquitylation Complexes
3.1. The E3 Ub Ligase CHIP Directly Binds Hsp70-Substrate Complexes
3.2. Dedicated Co-Chaperones Direct a Slow-Folding Protein to the E3 Ligase Complex
3.3. Nucleotide Exchange Factors Regulate Hsp70-Mediated Substrate Association with E3 Ligases
3.4. Spatial Regulation of Misfolded Protein Degradation by HSPs
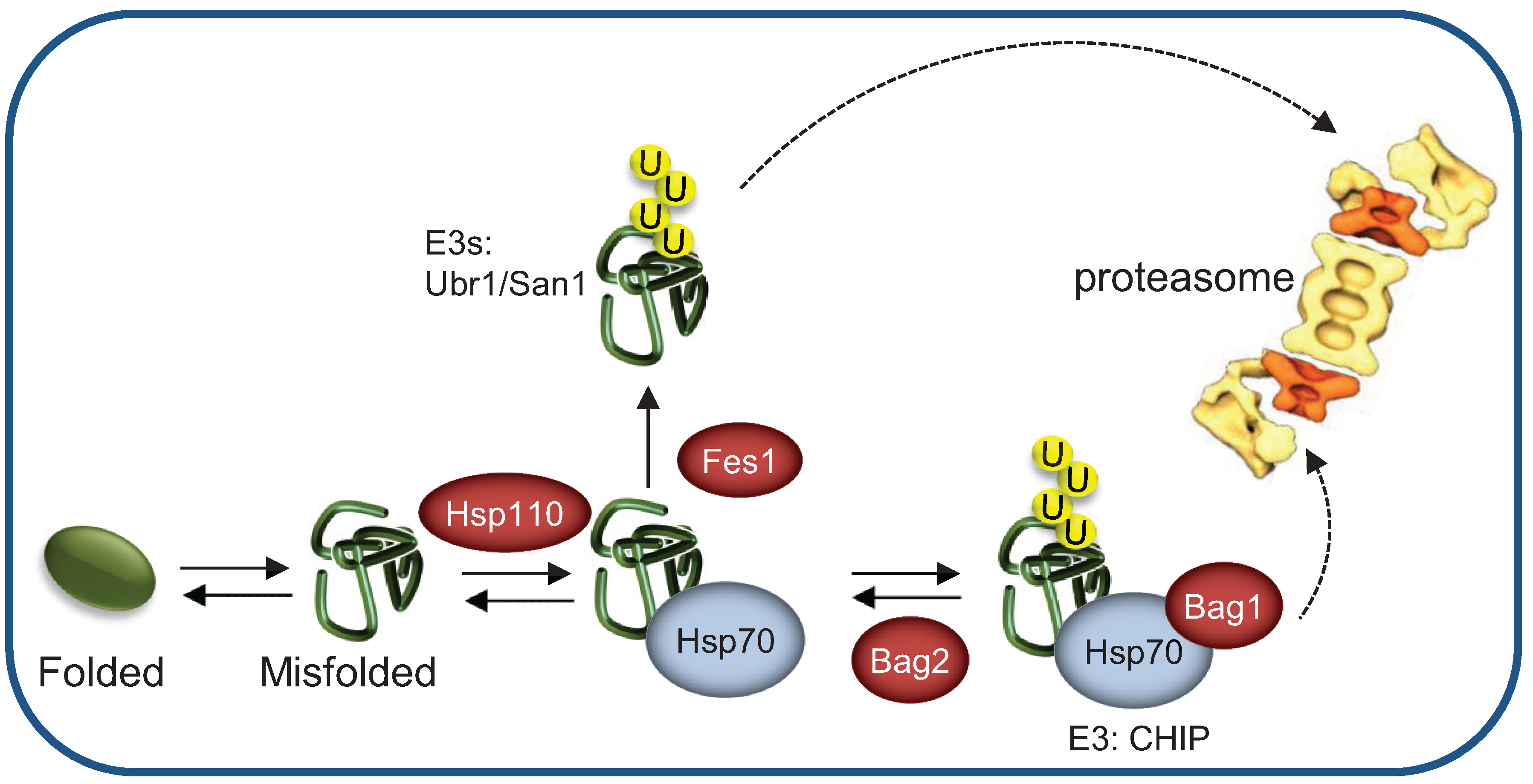
3.5. Terminally- and Partially-Misfolded Proteins are Targeted for Proteasomal Degradation by Distinct HSP-Dependent Pathways
3.6. Hsp40s May Act as Hsp70-Independent Ubiquitylation Factors—the Case of Sis1
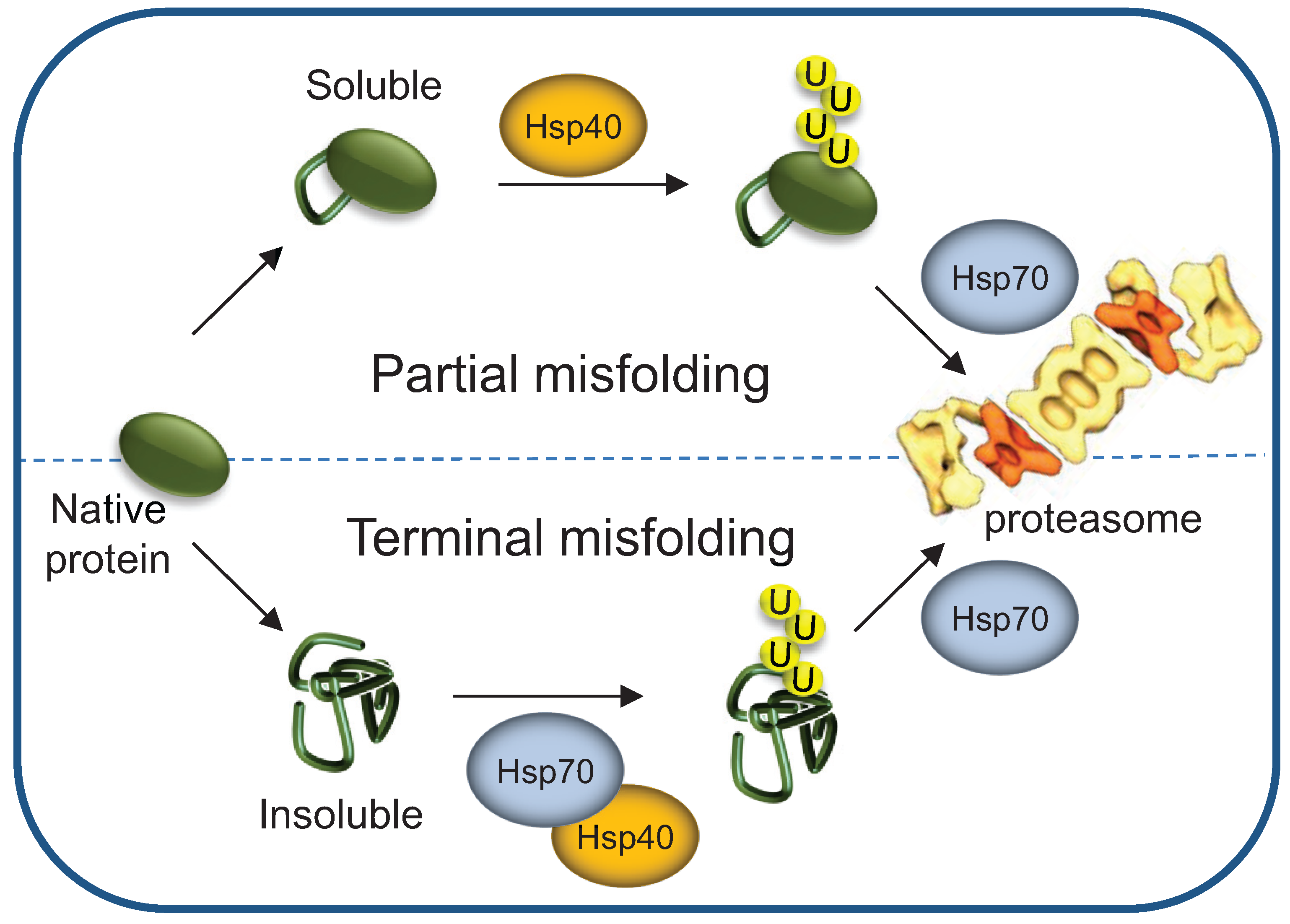
4. HSPs’ Roles as Proteasome Shuttling and Docking Factors
4.1. The Co-Chaperone BAG1 Mediates Binding of Hsp70-Substrate Complexes to the Proteasome
4.2. HSJ1 Is a Tissue-Specific Hsp40 Required for Shuttling Substrates to the Proteasome
4.3. Hsp70 Ubiquitylation Mediates Hsp70-Substrate Complex Docking at the Proteasome
5. Chaperones Prevent the Aggregation of Misfolded Degradation Substrates
5.1. Hsp70s Cellular Levels Determine the Fate of Polyubiquitylated Proteins
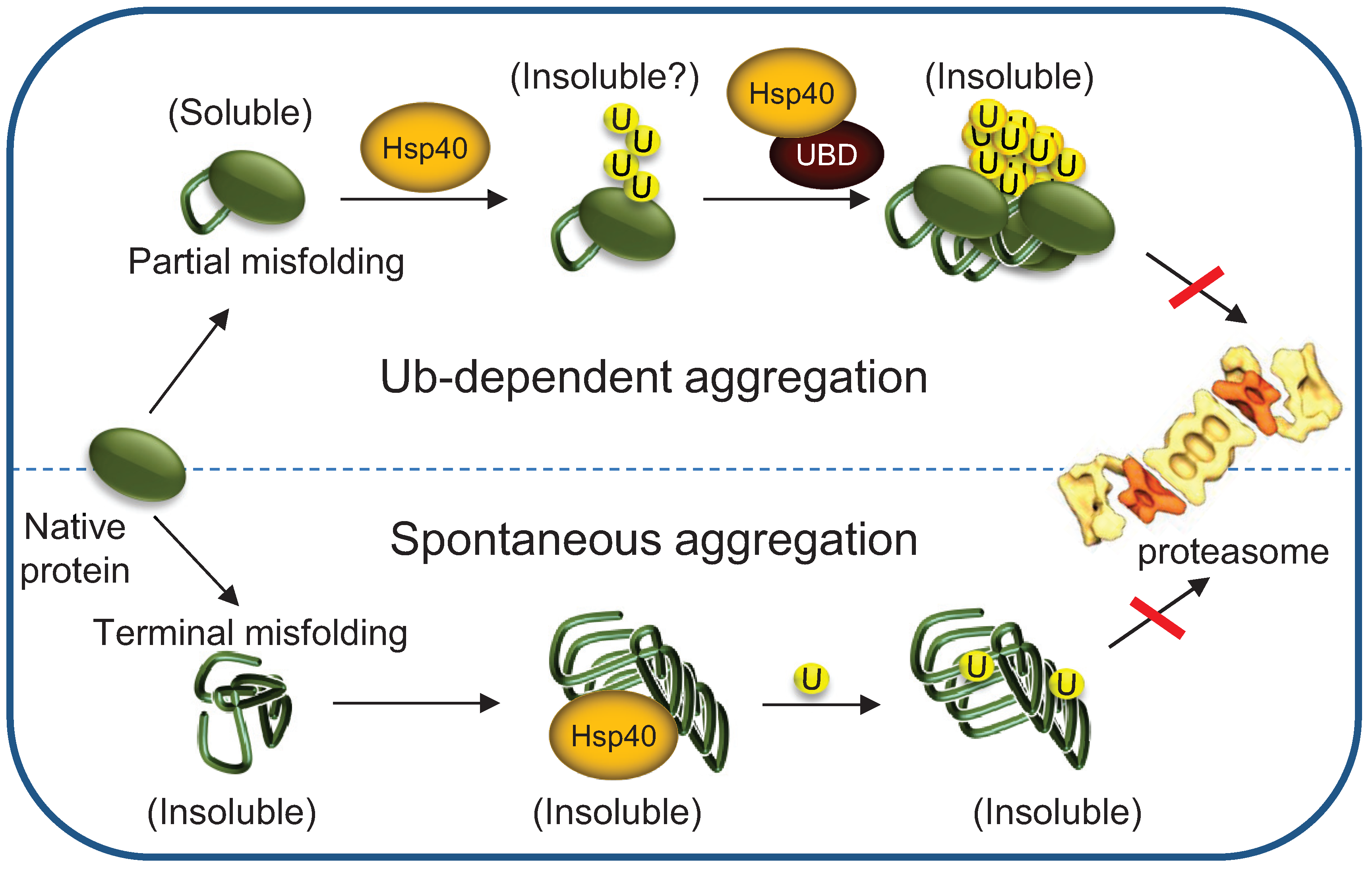
5.2. Implications to Protein Misfolding and Degenerative Diseases
6. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hartl, F.U.; Hayer-Hartl, M. Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 2009, 16, 574–581. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Nickell, S.; Beck, F.; Scheres, S.H.; Korinek, A.; Forster, F.; Lasker, K.; Mihalache, O.; Sun, N.; Nagy, I.; Sali, A.; et al. Insights into the molecular architecture of the 26S proteasome. Proc. Natl. Acad. Sci. USA 2009, 106, 11943–11947. [Google Scholar] [CrossRef]
- Ritossa, F. A new puffing pattern induced by temperature shock and DNP in Drosophila. Experientia 1962, 18, 571–573. [Google Scholar] [CrossRef]
- Spradling, A.; Penman, S.; Pardue, M.L. Analysis of Drosophila mRNA by in situ hybridization: Sequences transcribed in normal and heat shocked cultured cells. Cell 1975, 4, 395–404. [Google Scholar] [CrossRef]
- Schedl, P.; Artavanis-Tsakonas, S.; Steward, R.; Gehring, W.J.; Mirault, M.E.; Goldschmidt-Clermont, M.; Moran, L.; Tissieres, A. Two hybrid plasmids with D. Melanogaster DNA sequences complementary to mRNA coding for the major heat shock protein. Cell 1978, 14, 921–929. [Google Scholar] [CrossRef]
- Ritossa, F. Discovery of the heat shock response. Cell Stress Chaperones 1996, 1, 97–98. [Google Scholar] [CrossRef]
- Picard, D. Heat-shock protein 90, a chaperone for folding and regulation. Cell. Mol. Life Sci. 2002, 59, 1640–1648. [Google Scholar] [CrossRef]
- Dworniczak, B.; Mirault, M.E. Structure and expression of a human gene coding for a 71 kd heat shock “cognate” protein. Nucleic Acids Res. 1987, 15, 5181–5197. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Buchner, J. Supervising the fold: Functional principles of molecular chaperones. FASEB J. 1996, 10, 10–19. [Google Scholar]
- Muchowski, P.J.; Wacker, J.L. Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 2005, 6, 11–22. [Google Scholar] [CrossRef]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef]
- Hochstrasser, M. Ubiquitin-dependent protein degradation. Annu. Rev. Genet. 1996, 30, 405–439. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Bercovich, B.; Stancovski, I.; Mayer, A.; Blumenfeld, N.; Laszlo, A.; Schwartz, A.L.; Ciechanover, A. Ubiquitin-dependent degradation of certain protein substrates in vitro requires the molecular chaperone Hsc70. J. Biol. Chem. 1997, 272, 9002–9010. [Google Scholar] [CrossRef]
- Kriegenburg, F.; Ellgaard, L.; Hartmann-Petersen, R. Molecular chaperones in targeting misfolded proteins for ubiquitin-dependent degradation. FEBS J. 2012, 279, 532–542. [Google Scholar] [CrossRef]
- Shiber, A.; Breuer, W.; Brandeis, M.; Ravid, T. Ubiquitin conjugation triggers misfolded protein sequestration into quality control foci when Hsp70 chaperone levels are limiting. Mol. Biol. Cell 2013, 24, 2076–2087. [Google Scholar] [CrossRef]
- Park, S.-H.; Bolender, N.; Eisele, F.; Kostova, Z.; Takeuchi, J.; Coffino, P.; Wolf, D.H. The cytoplasmic Hsp70 chaperone machinery subjects misfolded and endoplasmic reticulum import-incompetent proteins to degradation via the ubiquitin-proteasome system. Mol. Biol. Cell 2007, 18, 153–165. [Google Scholar]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Alpha-synuclein and protein degradation systems: A reciprocal relationship. Mol. Neurobiol. 2013, 47, 537–551. [Google Scholar] [CrossRef]
- Ellis, R.J.; van der Vies, S.M.; Hemmingsen, S.M. The molecular chaperone concept. Biochem. Soc. Symp. 1989, 55, 145–153. [Google Scholar]
- Hartl, F.U.; Hayer-Hartl, M. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science 2002, 295, 1852–1858. [Google Scholar] [CrossRef]
- Morimoto, R.I.; Kline, M.P.; Bimston, D.N.; Cotto, J.J. The heat-shock response: Regulation and function of heat-shock proteins and molecular chaperones. Essays Biochem. 1997, 32, 17–29. [Google Scholar]
- Esser, C.; Alberti, S.; Hohfeld, J. Cooperation of molecular chaperones with the ubiquitin/proteasome system. Biochim. Biophys. Acta 2004, 1695, 171–188. [Google Scholar]
- Stolz, A.; Wolf, D.H. Endoplasmic reticulum associated protein degradation: A chaperone assisted journey to hell. Biochim. Biophys. Acta 2010, 1803, 694–705. [Google Scholar] [CrossRef]
- Ravid, T.; Hochstrasser, M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell Biol. 2008, 9, 679–689. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Craig, E.A. The Hsp70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef]
- Qiu, X.B.; Shao, Y.M.; Miao, S.; Wang, L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell. Mol. Life Sci. 2006, 63, 2560–2570. [Google Scholar] [CrossRef]
- Kettern, N.; Dreiseidler, M.; Tawo, R.; Hohfeld, J. Chaperone-assisted degradation: Multiple paths to destruction. Biol. Chem. 2010, 391, 481–489. [Google Scholar]
- Arndt, V.; Rogon, C.; Hohfeld, J. To be, or not to be—Molecular chaperones in protein degradation. Cell. Mol. Life Sci. 2007, 64, 2525–2541. [Google Scholar] [CrossRef]
- Simons, J.F.; Ferro-Novick, S.; Rose, M.D.; Helenius, A. BiP/Kar2p serves as a molecular chaperone during carboxypeptidase Y folding in yeast. J. Cell Biol. 1995, 130, 41–49. [Google Scholar] [CrossRef]
- Plemper, R.K.; Bohmler, S.; Bordallo, J.; Sommer, T.; Wolf, D.H. Mutant analysis links the translocon and bip to retrograde protein transport for ER degradation. Nature 1997, 388, 891–895. [Google Scholar] [CrossRef]
- Nishikawa, S.; Brodsky, J.L.; Nakatsukasa, K. Roles of molecular chaperones in endoplasmic reticulum (ER) quality control and ER-associated degradation (ERAD). J. Biochem. 2005, 137, 551–555. [Google Scholar] [CrossRef]
- Venkatraman, P.; Wetzel, R.; Tanaka, M.; Nukina, N.; Goldberg, A.L. Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol. Cell 2004, 14, 95–104. [Google Scholar] [CrossRef]
- Nakatsukasa, K.; Huyer, G.; Michaelis, S.; Brodsky, J.L. Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell 2008, 132, 101–112. [Google Scholar] [CrossRef]
- McClellan, A.J.; Scott, M.D.; Frydman, J. Folding and quality control of the VHL tumor suppressor proceed through distinct chaperone pathways. Cell 2005, 121, 739–748. [Google Scholar] [CrossRef]
- Huyer, G.; Piluek, W.F.; Fansler, Z.; Kreft, S.G.; Hochstrasser, M.; Brodsky, J.L.; Michaelis, S. Distinct machinery is required in saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J. Biol. Chem. 2004, 279, 38369–38378. [Google Scholar]
- Metzger, M.B.; Maurer, M.J.; Dancy, B.M.; Michaelis, S. Degradation of a cytosolic protein requires endoplasmic reticulum-associated degradation machinery. J. Biol. Chem. 2008, 283, 32302–32316. [Google Scholar] [CrossRef]
- Liberek, K.; Marszalek, J.; Ang, D.; Georgopoulos, C.; Zylicz, M. Escherichia coli DnaJ and GrpE heat shock proteins jointly stimulate ATPase activity of DnaK. Proc. Natl. Acad. Sci. USA 1991, 88, 2874–2878. [Google Scholar] [CrossRef]
- Buck, T.M.; Kolb, A.R.; Boyd, C.R.; Kleyman, T.R.; Brodsky, J.L. The endoplasmic reticulum-associated degradation of the epithelial sodium channel requires a unique complement of molecular chaperones. Mol. Biol. Cell 2010, 21, 1047–1058. [Google Scholar] [CrossRef]
- Cyr, D.M. Cooperation of the molecular chaperone Ydj1 with specific Hsp70 homologs to suppress protein aggregation. FEBS Lett. 1995, 359, 129–132. [Google Scholar] [CrossRef]
- Summers, D.W.; Wolfe, K.J.; Ren, H.Y.; Cyr, D.M. The type II Hsp40 Sis1 cooperates with Hsp70 and the E3 ligase Ubr1 to promote degradation of terminally misfolded cytosolic protein. PLoS One 2013, 8, e52099. [Google Scholar] [CrossRef]
- Lee, D.H.; Sherman, M.Y.; Goldberg, A.L. Involvement of the molecular chaperone Ydj1 in the ubiquitin-dependent degradation of short-lived and abnormal proteins in saccharomyces cerevisiae. Mol. Cell. Biol. 1996, 16, 4773–4781. [Google Scholar]
- Connell, P.; Ballinger, C.A.; Jiang, J.; Wu, Y.; Thompson, L.J.; Hohfeld, J.; Patterson, C. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 2001, 3, 93–96. [Google Scholar] [CrossRef]
- Murata, S.; Minami, Y.; Minami, M.; Chiba, T.; Tanaka, K. CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep. 2001, 2, 1133–1138. [Google Scholar] [CrossRef]
- Ballinger, C.A.; Connell, P.; Wu, Y.; Hu, Z.; Thompson, L.J.; Yin, L.Y.; Patterson, C. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol. 1999, 19, 4535–4545. [Google Scholar]
- Jiang, J.; Ballinger, C.A.; Wu, Y.; Dai, Q.; Cyr, D.M.; Hohfeld, J.; Patterson, C. CHIP is a U-box-dependent E3 ubiquitin ligase: Identification of Hsc70 as a target for ubiquitylation. J. Biol. Chem. 2001, 276, 42938–42944. [Google Scholar]
- Zhou, P.; Fernandes, N.; Dodge, I.L.; Reddi, A.L.; Rao, N.; Safran, H.; DiPetrillo, T.A.; Wazer, D.E.; Band, V.; Band, H. ErbB2 degradation mediated by the co-chaperone protein CHIP. J. Biol. Chem. 2003, 278, 13829–13837. [Google Scholar] [CrossRef]
- Shimura, H.; Schwartz, D.; Gygi, S.P.; Kosik, K.S. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J. Biol. Chem. 2004, 279, 4869–4876. [Google Scholar]
- Meacham, G.C.; Patterson, C.; Zhang, W.; Younger, J.M.; Cyr, D.M. The Hsc70 co-chaperone chip targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 2001, 3, 100–105. [Google Scholar] [CrossRef]
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar]
- Lukacs, G.L.; Mohamed, A.; Kartner, N.; Chang, X.B.; Riordan, J.R.; Grinstein, S. Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 1994, 13, 6076–6086. [Google Scholar]
- Du, K.; Sharma, M.; Lukacs, G.L. The deltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat. Struct. Mol. Biol. 2005, 12, 17–25. [Google Scholar] [CrossRef]
- Younger, J.M.; Chen, L.; Ren, H.Y.; Rosser, M.F.; Turnbull, E.L.; Fan, C.Y.; Patterson, C.; Cyr, D.M. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell 2006, 126, 571–582. [Google Scholar] [CrossRef]
- Szabo, A.; Langer, T.; Schröder, H.; Flanagan, J.; Bukau, B.; Hartl, F.U. The ATP hydrolysis-dependent reaction cycle of the Escherichia coli Hsp70 system DnaK, DnaJ, and GrpE. Proc. Natl. Acad. Sci. USA 1994, 91, 10345–10349. [Google Scholar]
- Alberti, S.; Bohse, K.; Arndt, V.; Schmitz, A.; Hohfeld, J. The cochaperone HspBP1 inhibits the CHIP ubiquitin ligase and stimulates the maturation of the cystic fibrosis transmembrane conductance regulator. Mol. Biol. Cell 2004, 15, 4003–4010. [Google Scholar] [CrossRef]
- Dai, Q.; Qian, S.B.; Li, H.H.; McDonough, H.; Borchers, C.; Huang, D.; Takayama, S.; Younger, J.M.; Ren, H.Y.; Cyr, D.M.; et al. Regulation of the cytoplasmic quality control protein degradation pathway by BAG2. J. Biol. Chem. 2005, 280, 38673–38681. [Google Scholar] [CrossRef]
- Arndt, V.; Daniel, C.; Nastainczyk, W.; Alberti, S.; Hohfeld, J. BAG-2 acts as an inhibitor of the chaperone-associated ubiquitin ligase CHIP. Mol. Biol. Cell 2005, 16, 5891–5900. [Google Scholar] [CrossRef]
- Morito, D.; Hirao, K.; Oda, Y.; Hosokawa, N.; Tokunaga, F.; Cyr, D.M.; Tanaka, K.; Iwai, K.; Nagata, K. Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of cftrδF508. Mol. Biol. Cell 2008, 19, 1328–1336. [Google Scholar] [CrossRef]
- Cheng, J.; Guggino, W. Ubiquitination and degradation of CFTR by the E3 ubiquitin ligase MARCH2 through its association with adaptor proteins CAL and STX6. PLoS One 2013, 8, e68001. [Google Scholar] [CrossRef]
- Youker, R.T.; Walsh, P.; Beilharz, T.; Lithgow, T.; Brodsky, J.L. Distinct roles for the Hsp40 and Hsp90 molecular chaperones during cystic fibrosis transmembrane conductance regulator degradation in yeast. Mol. Biol. Cell 2004, 15, 4787–4797. [Google Scholar] [CrossRef]
- Grove, D.E.; Fan, C.-Y.; Ren, H.Y.; Cyr, D.M. The endoplasmic reticulum-associated Hsp40 DnaJB12 and Hsc70 cooperate to facilitate RMA1 E3-dependent degradation of nascent cftrδF508. Mol. Biol. Cell 2011, 22, 301–314. [Google Scholar] [CrossRef]
- Raviol, H.; Sadlish, H.; Rodriguez, F.; Mayer, M.P.; Bukau, B. Chaperone network in the yeast cytosol: Hsp110 is revealed as an Hsp70 nucleotide exchange factor. EMBO J. 2006, 25, 2510–2518. [Google Scholar] [CrossRef]
- Gowda, N.K.C.; Kandasamy, G.; Froehlich, M.S.; Dohmen, R.J.; Andréasson, C. Hsp70 nucleotide exchange factor Fes1 is essential for ubiquitin-dependent degradation of misfolded cytosolic proteins. Proc. Natl. Acad. Sci. USA 2013, 110, 5975–5980. [Google Scholar]
- Eisele, F.; Wolf, D.H. Degradation of misfolded protein in the cytoplasm is mediated by the ubiquitin ligase UBR1. FEBS Lett. 2008, 582, 4143–4146. [Google Scholar] [CrossRef]
- Gardner, R.G.; Nelson, Z.W.; Gottschling, D.E. Degradation-mediated protein quality control in the nucleus. Cell 2005, 120, 803–815. [Google Scholar] [CrossRef]
- Heck, J.W.; Cheung, S.K.; Hampton, R.Y. Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc. Natl. Acad. Sci. USA 2010, 107, 1106–1111. [Google Scholar] [CrossRef]
- Rosenbaum, J.C.; Fredrickson, E.K.; Oeser, M.L.; Garrett-Engele, C.M.; Locke, M.N.; Richardson, L.A.; Nelson, Z.W.; Hetrick, E.D.; Milac, T.I.; Gottschling, D.E.; et al. Disorder targets misorder in nuclear quality control degradation: A disordered ubiquitin ligase directly recognizes its misfolded substrates. Mol. Cell 2011, 41, 93–106. [Google Scholar] [CrossRef]
- Prasad, R.; Kawaguchi, S.; Ng, D.T. A nucleus-based quality control mechanism for cytosolic proteins. Mol. Biol. Cell 2010, 21, 2117–2127. [Google Scholar] [CrossRef]
- Guerriero, C.J.; Weiberth, K.F.; Brodsky, J.L. Hsp70 targets a cytoplasmic quality control substrate to the san1p ubiquitin ligase. J. Biol. Chem. 2013, 288, 18506–18520. [Google Scholar] [CrossRef]
- Shorter, J.; Lindquist, S. Hsp104, Hsp70 and Hsp40 interplay regulates formation, growth and elimination of Sup35 prions. EMBO J. 2008, 27, 2712–2724. [Google Scholar] [CrossRef]
- Summers, D.W.; Douglas, P.M.; Cyr, D.M. Prion propagation by Hsp40 molecular chaperones. Prion 2009, 3, 59–64. [Google Scholar] [CrossRef]
- Park, S.H.; Kukushkin, Y.; Gupta, R.; Chen, T.; Konagai, A.; Hipp, M.S.; Hayer-Hartl, M.; Hartl, F.U. Polyq proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1P chaperone. Cell 2013, 154, 134–145. [Google Scholar] [CrossRef]
- Furth, N.; Gertman, O.; Shiber, A.; Alfassy, O.S.; Cohen, I.; Rosenberg, M.M.; Doron, N.K.; Friedler, A.; Ravid, T. Exposure of bipartite hydrophobic signal triggers nuclear quality control of Ndc10 at the endoplasmic reticulum/nuclear envelope. Mol. Biol. Cell 2011, 22, 4726–4739. [Google Scholar] [CrossRef]
- Oling, D.; Eisele, F.; Kvint, K.; Nystrom, T. Opposing roles of Ubp3-dependent deubiquitination regulate replicative life span and heat resistance. EMBO J. 2014, 33, 747–761. [Google Scholar] [CrossRef]
- Cummings, C.J.; Mancini, M.A.; Antalffy, B.; DeFranco, D.B.; Orr, H.T.; Zoghbi, H.Y. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in Sca1. Nat. Genet. 1998, 19, 148–154. [Google Scholar] [CrossRef]
- Bailey, C.K.; Andriola, I.F.; Kampinga, H.H.; Merry, D.E. Molecular chaperones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2002, 11, 515–523. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Soetandyo, N.; Baek, K.; Hegde, R.; Ye, Y. A ubiquitin ligase-associated chaperone holdase maintains polypeptides in soluble states for proteasome degradation. Mol. Cell 2011, 42, 758–770. [Google Scholar] [CrossRef]
- Takayama, S.; Sato, T.; Krajewski, S.; Kochel, K.; Irie, S.; Milian, J.A.; Reed, J.C. Cloning and functional analysis of BAG-1: A novel Bcl-2-binding protein with anti-cell death activity. Cell 1995, 80, 279–284. [Google Scholar] [CrossRef]
- Lüders, J.; Demand, J.; Höhfeld, J. The ubiquitin-related BAG-1 provides a link between the molecular chaperones Hsc70/Hsp70 and the proteasome. J. Biol. Chem. 2000, 275, 4613–4617. [Google Scholar] [CrossRef]
- Demand, J.; Alberti, S.; Patterson, C.; Höhfeld, J. Cooperation of a ubiquitin domain protein and an E3 ubiquitin ligase during chaperone/proteasome coupling. Curr. Biol. 2001, 11, 1569–1577. [Google Scholar] [CrossRef]
- Alberti, S.; Demand, J.; Esser, C.; Emmerich, N.; Schild, H.; Höhfeld, J. Ubiquitylation of BAG-1 suggests a novel regulatory mechanism during the sorting of chaperone substrates to the proteasome. J. Biol. Chem. 2002, 277, 45920–45927. [Google Scholar] [CrossRef]
- Höhfeld, J.; Jentsch, S. GRPE-like regulation of the Hsc70 chaperone by the anti-apoptotic protein BAG-1. EMBO J. 1997, 16, 6209–6216. [Google Scholar] [CrossRef]
- Kriegenburg, F.; Jakopec, V.; Poulsen, E.G.; Nielsen, S.V.; Roguev, A.; Krogan, N.; Gordon, C.; Fleig, U.; Hartmann-Petersen, R. A chaperone-assisted degradation pathway targets kinetochore proteins to ensure genome stability. PLoS Genet. 2014, 10, e1004140. [Google Scholar] [CrossRef]
- Westhoff, B.; Chapple, J.P.; van der Spuy, J.; Höhfeld, J.; Cheetham, M.E. Hsj1 is a neuronal shuttling factor for the sorting of chaperone clients to the proteasome. Curr. Biol. 2005, 15, 1058–1064. [Google Scholar] [CrossRef]
- Novoselov, S.S.; Mustill, W.J.; Gray, A.L.; Dick, J.R.; Kanuga, N.; Kalmar, B.; Greensmith, L.; Cheetham, M.E. Molecular chaperone mediated late-stage neuroprotection in the SOD1G93A mouse model of amyotrophic lateral sclerosis. PLoS One 2013, 8, e73944. [Google Scholar]
- Gao, X.-C.; Zhou, C.-J.; Zhou, Z.-R.; Zhang, Y.-H.; Zheng, X.-M.; Song, A.-X.; Hu, H.-Y. Co-chaperone HSJ1a dually regulates the proteasomal degradation of ataxin-3. PLoS One 2011, 6, e19763. [Google Scholar]
- Urushitani, M.; Kurisu, J.; Tateno, M.; Hatakeyama, S.; Nakayama, K.-I.; Kato, S.; Takahashi, R. CHIP promotes proteasomal degradation of familial ALS-linked mutant SOD1 by ubiquitinating Hsp/Hsc70. J. Neurochem. 2004, 90, 231–244. [Google Scholar] [CrossRef]
- Kundrat, L.; Regan, L. Identification of residues on Hsp70 and Hsp90 ubiquitinated by the cochaperone CHIP. J. Mol. Biol. 2010, 395, 587–594. [Google Scholar] [CrossRef]
- Peng, J.; Schwartz, D.; Elias, J.E.; Thoreen, C.C.; Cheng, D.; Marsischky, G.; Roelofs, J.; Finley, D.; Gygi, S.P. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 2003, 21, 921–926. [Google Scholar] [CrossRef]
- Muchowski, P.J.; Schaffar, G.; Sittler, A.; Wanker, E.E.; Hayer-Hartl, M.K.; Hartl, F.U. Hsp70 and Hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2000, 97, 7841–7846. [Google Scholar]
- Klucken, J.; Shin, Y.; Masliah, E.; Hyman, B.T.; McLean, P.J. Hsp70 reduces alpha-synuclein aggregation and toxicity. J. Biol. Chem. 2004, 279, 25497–25502. [Google Scholar]
- Evans, C.G.; Wisen, S.; Gestwicki, J.E. Heat shock proteins 70 and 90 inhibit early stages of amyloid beta-(1–42) aggregation in vitro. J. Biol. Chem. 2006, 281, 33182–33191. [Google Scholar] [CrossRef]
- Sørensen, J.G.; Loeschcke, V. Decreased heat-shock resistance and down-regulation of Hsp70 expression with increasing age in adult Drosophila melanogaster. Funct. Ecol. 2002, 16, 379–384. [Google Scholar] [CrossRef]
- Ben-Zvi, A.; Miller, E.A.; Morimoto, R.I. Collapse of proteostasis represents an early molecular event in caenorhabditis elegans aging. Proc. Natl. Acad. Sci. USA 2009, 106, 14914–14919. [Google Scholar] [CrossRef]
- Hebert, L.E.; Scherr, P.A.; Beckett, L.A.; Albert, M.S.; Pilgrim, D.M.; Chown, M.J.; Funkenstein, H.H.; Evans, D.A. Age-specific incidence of Alzheimer’s disease in a community population. JAMA 1995, 273, 1354–1359. [Google Scholar] [CrossRef]
- Cao, K.; Chen-Plotkin, A.S.; Plotkin, J.B.; Wang, L.-S. Age-correlated gene expression in normal and neurodegenerative human brain tissues. PLoS One 2010, 5, e13098. [Google Scholar]
- Ciechanover, A. The ubiquitin proteolytic system and pathogenesis of human diseases: A novel platform for mechanism-based drug targeting. Biochem. Soc. Trans. 2003, 31, 474–481. [Google Scholar] [CrossRef]
- Cipriani, G.; Dolciotti, C.; Picchi, L.; Bonuccelli, U. Alzheimer and his disease: A brief history. Neurol. Sci. 2011, 32, 275–279. [Google Scholar]
- Woulfe, J.M. Abnormalities of the nucleus and nuclear inclusions in neurodegenerative disease: A work in progress. Neuropathol. Appl. Neurobiol. 2007, 33, 2–42. [Google Scholar]
- Chhangani, D.; Jana, N.R.; Mishra, A. Misfolded proteins recognition strategies of E3 ubiquitin ligases and neurodegenerative diseases. Mol. Neurobiol. 2012, 47, 302–312. [Google Scholar] [CrossRef]
- Calise, J.; Powell, S.R. The ubiquitin proteasome system and myocardial Ischemia. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H337–H349. [Google Scholar] [CrossRef]
- Vilchez, D.; Morantte, I.; Liu, Z.; Douglas, P.M.; Merkwirth, C.; Rodrigues, A.P.; Manning, G.; Dillin, A. RPN-6 determines C. Elegans longevity under proteotoxic stress conditions. Nature 2012, 489, 263–268. [Google Scholar] [CrossRef]
- Vilchez, D.; Boyer, L.; Morantte, I.; Lutz, M.; Merkwirth, C.; Joyce, D.; Spencer, B.; Page, L.; Masliah, E.; Berggren, W.T.; et al. Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature 2012, 489, 304–308. [Google Scholar] [CrossRef]
- Xie, Y.; Wolff, D.W.; Wei, T.; Wang, B.; Deng, C.; Kirui, J.K.; Jiang, H.; Qin, J.; Abel, P.W.; Tu, Y. Breast cancer migration and invasion depend on proteasome degradation of regulator of G-protein signaling 4. Cancer Res. 2009, 69, 5743–5751. [Google Scholar] [CrossRef]
- Marber, M.S.; Mestril, R.; Chi, S.H.; Sayen, M.R.; Yellon, D.M.; Dillmann, W.H. Overexpression of the rat inducible 70-kd heat stress protein in a transgenic mouse increases the resistance of the heart to ischemic injury. J. Clin. Investig. 1995, 95, 1446–1456. [Google Scholar] [CrossRef]
- Cummings, C.J.; Sun, Y.; Opal, P.; Antalffy, B.; Mestril, R.; Orr, H.T.; Dillmann, W.H.; Zoghbi, H.Y. Over-expression of inducible Hsp70 chaperone suppresses neuropathology and improves motor function in Sca1 mice. Hum. Mol. Genet. 2001, 10, 1511–1518. [Google Scholar] [CrossRef]
- Nylandsted, J.; Rohde, M.; Brand, K.; Bastholm, L.; Elling, F.; Jaattela, M. Selective depletion of heat shock protein 70 (Hsp70) activates a tumor-specific death program that is independent of caspases and bypasses Bcl-2. Proc. Natl. Acad. Sci. USA 2000, 97, 7871–7876. [Google Scholar] [CrossRef]
- Saluja, A.; Dudeja, V. Heat shock proteins in pancreatic diseases. J. Gastroenterol. Hepatol. 2008, 23, S42–S45. [Google Scholar] [CrossRef]
- Labbadia, J.; Novoselov, S.S.; Bett, J.S.; Weiss, A.; Paganetti, P.; Bates, G.P.; Cheetham, M.E. Suppression of protein aggregation by chaperone modification of high molecular weight complexes. Brain 2012, 135, 1180–1196. [Google Scholar] [CrossRef]
- Grove, D.E.; Rosser, M.F.; Ren, H.Y.; Naren, A.P.; Cyr, D.M. Mechanisms for rescue of correctable folding defects in cftrdelta F508. Mol. Biol. Cell 2009, 20, 4059–4069. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shiber, A.; Ravid, T. Chaperoning Proteins for Destruction: Diverse Roles of Hsp70 Chaperones and their Co-Chaperones in Targeting Misfolded Proteins to the Proteasome. Biomolecules 2014, 4, 704-724. https://doi.org/10.3390/biom4030704
Shiber A, Ravid T. Chaperoning Proteins for Destruction: Diverse Roles of Hsp70 Chaperones and their Co-Chaperones in Targeting Misfolded Proteins to the Proteasome. Biomolecules. 2014; 4(3):704-724. https://doi.org/10.3390/biom4030704
Chicago/Turabian StyleShiber, Ayala, and Tommer Ravid. 2014. "Chaperoning Proteins for Destruction: Diverse Roles of Hsp70 Chaperones and their Co-Chaperones in Targeting Misfolded Proteins to the Proteasome" Biomolecules 4, no. 3: 704-724. https://doi.org/10.3390/biom4030704
APA StyleShiber, A., & Ravid, T. (2014). Chaperoning Proteins for Destruction: Diverse Roles of Hsp70 Chaperones and their Co-Chaperones in Targeting Misfolded Proteins to the Proteasome. Biomolecules, 4(3), 704-724. https://doi.org/10.3390/biom4030704