Abstract
Astrocytes are essential gatekeepers of brain homeostasis, and the disruption of their functions can contribute to the development of several neurological diseases. Among astrocyte signaling pathways, the intracellular second messenger Ca2+ plays a pivotal role in regulating the release of gliotransmitters, which actively modulate fundamental processes in the brain such as synaptic plasticity and memory function. Several studies over the years support the idea that dysregulated astrocytic Ca2+ homeostasis represents a relevant mechanism in Alzheimer’s disease pathogenesis. Early works in transgenic mice modelling Alzheimer’s disease reported increased Ca2+ activity in astroglial cells, supporting the idea of hyperactivity as a common trait of astrocytes in this pathology. However, recent studies have described astrocyte Ca2+ hypoactivity in various mouse models, revealing a more complex and heterogeneous scenario. In this review, we summarize and critically discuss the main studies addressing the direction(s) of astrocytic Ca2+ signaling dysfunction in mouse models of Alzheimer’s disease. We prioritize investigations performed in ex vivo and in vivo conditions, carefully comparing the different experimental approaches used to measure Ca2+ activity in astrocytes. By integrating results across multiple mouse models and methodological strategies, we aim to provide a more complete picture of astrocyte Ca2+ dysregulation in Alzheimer’s disease.
1. Astrocytes and Ca2+ Signaling Pathways
Astrocytes have been initially acknowledged to play a merely trophic and supportive role in the central nervous system. However, during the past three decades, accumulating evidence has demonstrated that these cells are central regulators of brain homeostasis and actively participate in key physiological processes, including the modulation of synaptic transmission, neurovascular coupling, ion buffering and immune responses [1,2,3,4,5].
Astrocytes express a plethora of receptors that allow them to sense changes in their surrounding environment. As for the other cell types, the activation of these receptors triggers various intracellular signaling pathways, depending on the specific receptor involved and its physiological function [6,7]. Among the various intracellular pathways, Ca2+ signaling in astrocytes has emerged as a crucial mediator of many of their functions, particularly in regulating gliotransmitter release and neuron–glia communication. Indeed, increases in astrocytic Ca2+ concentration can trigger the release of a variety of gliotransmitters, including glutamate, GABA, ATP, D-serine and BDNF, which modulate synaptic transmission and neuronal excitability [8,9,10,11,12,13,14,15]. Through Ca2+-dependent activity, astrocytes influence both pre- and postsynaptic terminals, regulating neurotransmitter release probability, receptor activation and synaptic strength, ultimately shaping memory processes [6]. Consequently, astrocytic Ca2+ signaling emerges as a fundamental mechanism linking cellular activity to circuit level up to behavioral outcomes. Thanks to the development of chemical and genetically encoded Ca2+ indicators, studying astrocytic Ca2+ signaling has historically become a standard method to assess astrocyte activity or functionality [16,17]. Astrocytes rely on several mechanisms to activate intracellular Ca2+ signaling (for a recent review, see [18]). One major pathway involves G protein-coupled receptors (GPCRs) [6], also known as metabotropic receptors. GPCRs can be coupled to three subtypes of G protein (Gq, Gs and Gi), differently linked to the intracellular second messengers Ca2+ and cyclic adenosine monophosphate (cAMP) [19].
In the present review, we will focus on Gq receptors, mentioning metabotropic glutamate receptors (mGluRs), purinergic receptors (P2YRs) and adrenergic receptors (ARs). Upon ligand binding, Gq receptors activate phospholipase C (PLC), which, in turn, converts phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 then binds to IP3 receptors (IP3Rs) on the endoplasmic reticulum (ER), promoting channel opening and the release of stored Ca2+ into the cytosol. This massive release of Ca2+ from the ER triggers a mechanism known as store-operated Ca2+ entry (SOCE): stromal interaction molecules (STIMs) sense ER depletion and recruit plasma membrane Ca2+ release-activated Ca2+ (CRAC) channels, formed by ORAI proteins, allowing Ca2+ influx from the extracellular space. In addition to metabotropic signaling, astrocytes also express ionotropic receptors and various ion channels that permit direct ion flux across the plasma membrane. Some of these, such as TRPA1 and the mechanoreceptor Piezo1, are Ca2+ permeable receptors mediating extracellular Ca2+ influx upon activation. Other mechanisms leading to intracellular Ca2+ increases involve ion exchangers such as the Na+/Ca2+ exchanger (NCX). Activation of the glutamate transporter 1 (GLT-1) leads to Na+ influx into the cytoplasm, which forces NCX into the reverse mode, promoting Ca2+ entry from the extracellular space. On this note, such Ca2+ influx, as well as Ca2+ release from IP3Rs, can trigger a process known as Ca2+-induced Ca2+ release (CICR), in which the rise of the cytosolic Ca2+ concentration activates calcium-binding sites in ryanodine receptors (RyRs) and in activated IP3Rs on the ER, resulting in regenerative Ca2+ release from internal stores [20,21].
2. Alzheimer’s Disease: Overview and Mouse Models
Astrocytic dysfunction has been increasingly implicated in the pathogenesis of numerous neurological and neurodegenerative disorders, including Alzheimer’s disease (AD). AD is a progressive neurodegenerative condition characterized by a cognitive decline and memory issues, representing the most common form of dementia, currently affecting over 40 million individuals worldwide as reported by the Alzheimer’s Disease International (www.alzint.org accessed on 1 March 2026). Histologically, AD is characterized by the extracellular accumulation of amyloid beta (Aβ) plaques and the intracellular formation of neurofibrillary tangles composed by hyperphosphorylated tau protein. These pathological hallmarks are associated with synaptic dysfunction, neuronal loss and neuroinflammation [22]. Despite decades of intensive research, the precise pathogenic mechanisms underlying AD remain poorly understood, significantly hindering the development of effective disease-modifying therapies.
It is estimated that only 1–5% of AD patients carry genetic mutations directly associated with early-onset familial AD (EOFAD). These mutations affect three genes, coding for the amyloid precursor protein (APP) and for presenilin 1 and 2 (PS1 and PS2). Presenilins are part of the catalytic unit of γ-secretase, the enzyme responsible for the C-terminal cleavage of Aβ peptides. Mutations in one or more of these genes in AD patients drive an increase in the production of insoluble Aβ species [23,24,25,26]. The majority of AD cases, conversely, do not present distinctive genetic mutations and are therefore classified as sporadic, indicating a multifactorial etiology that includes environmental, metabolic and lifestyle-related risk factors and underlies late-onset AD (LOAD). Among sporadic AD, the presence of the apolipoprotein E variant ApoE4 represents the major genetic risk factor increasing the probability to develop the pathology [27]. This complexity further hampers the identification of a unified pathogenic pathway suitable as therapeutic target.
To model the disease and explore its mechanisms, various transgenic mouse lines harboring EOFAD mutations have been developed. Although these animal models fail to fully recapitulate the pathological features of the human disease, they can provide valuable insights into the cellular and molecular mechanisms underlying AD. To date, relatively few studies have investigated astrocytic Ca2+ activity in the context of AD. In this review, we summarize all available studies that address this topic using ex vivo or in vivo approaches. Specifically, we find 10 studies examining astrocytic Ca2+ activity in nine different mouse lines. The information on these mouse lines is also available on Alzforum (www.alzforum.org/research-models accessed on 1 March 2026) and graphically summarized in Figure 1.

Figure 1.
Schematic representation of the phenotypes displayed by the Alzheimer’s disease mouse models described in the present review [28,29,30,31,32,33,34,35]. For each mouse line, the onset of gliosis, intracellular neuronal Aβ accumulation, Aβ plaque deposition and cognitive impairment is indicated by a color-coded bar starting in correspondence of the onset age. Arrows mark the age analyzed in the studies and specify the direction of astrocytic Ca2+ dysfunction.
- Tg2576
Tg2576 mice overexpress in heterozygosis a mutant form of human APP (isoform 695) with the Swedish mutation (KM670/671NL), which, per se, increases total Aβ production, under the control of the hamster prion protein (PrP) promoter. The high levels of APPswe (5-fold above the physiological level) lead to elevated levels of Aβ1–40 and Aβ1–42 between 3 and 9 months of age and amyloid plaque deposits at 11 months of age. Regarding cognitive decline, the results are divergent and report memory impairment already at 6 months or starting from 12 months of age. Microglia activation is observed at 10 months of age [28,36].
- 3xTg
3xTg mice, firstly described in 2003, present the following three mutations in homozygosis: the Swedish K670N/M671L mutation in the human APP gene, the P310L mutation in the Microtubule-Associated Protein Tau (MAPT) gene, both under the control of the mouse Thy1.2 promoter, and the M146V mutation in the PSEN1 gene, under the PSEN1 endogenous promoter. Although MAPT mutation is not associated with EOFAD, it was introduced in this model to produce tangle pathology in transgenic animals. Intracellular Aβ accumulation starts at 3–4 months of age, together with cognitive impairment, while extracellular Aβ deposits appear by 6 months in the frontal cortex. Reactive astrogliosis and microglia activation are observed at 7 months of age. Tau pathology occurs later, with aggregates of conformationally altered and hyperphosphorylated tau detected in the hippocampus of 12–15 months old transgenic mice [29,37].
- Tg-SwDI (Dutch/Iowa)
Tg-SwDI mice harbor three mutations in heterozygosis on the human APP gene: the Swedish K670N/M671L (APPswe), the Dutch E693Q and the Iowa D694N mutations, all under the control of the mouse Thy1.2 promoter. These transgenic animals show plaque-like structures associated with increased levels of soluble forms of Aβ species as early as month 3, with fibrillary amyloid deposits in the cerebral microvasculature starting at 6 months of age. Cognitive decline is observed starting from 3 months and gliosis at 6 months of age [30,38].
- APPswe/PSEN1ΔE9 (APP/PS1)
APPswe/PS1ΔE9 mice were created by co-injecting two different vectors expressing human APPswe and PSEN1 lacking exon 9 under the control of mouse prion protein promoter. In hemizygous mice, plaques deposition and reactive astrogliosis start at 6 months of age and worsen with age. Cognitive impairment is significant at 12 months of age, although some groups reported small differences already at 6 months of age [31,39,40,41].
- APPswe/PSEN1L166P (APPPS1-21)
APPswe/PSEN1L166P mice express human APPswe and PSNE1 carrying the L166P mutation, both under the Thy1 promoter. Hemizygous APPswe/PSEN1L166P mice are characterized by overexpression of human APP (3-fold above the endogenous murine gene) and are highly amyloidogenic (due to a high Aβ42/Aβ40 ratio), with plaque deposition and gliosis starting already at 6 weeks. Despite the high levels of Aβ42 and amyloid plaques, cognitive impairment manifests only between 7 and 8 months of age [32,42,43].
- Tg-ArcSwe
Tg-ArcSwe mice harbor human APP carrying the Swedish (KM670/671NL) mutation and the Arctic mutation (E693G) in heterozygosis under the control of the murine Thy1 promoter. The arctic mutation aggravates the amyloid pathology of the Tg2576 mice, leading to earlier plaque deposition (at 5–6 months of age) and to cognitive decline starting at 4 months of age. This model is characterized by very early (month 1) accumulation of intraneuronal Aβ, elevated CSF Aβ levels and detectable CSF tau protofibrils at 4 months of age. Plaque composition and morphology are different compared to the Tg2576 line, with plaques displaying more homogeneity in terms of structure and size [33,44,45].
- APPNL-F
All mouse models based on APP mutations described so far rely on overexpression of human APP accompanying the endogenous murine protein. To overcome the possible artefact deriving from APP overexpression, Saito and colleagues created two different mouse lines expressing the humanized mutated APP gene under the control of the endogenous mouse APP promoter, thus conferring appropriate cell type and temporal specificity to APP expression. Homozygous APPNL-F mice carry the Swedish “NL” and the Iberian “F” (I716F) mutations, which influence the activity of secretases, resulting in an increase in total Aβ production and Aβ42/Aβ40 ratio, respectively. Amyloid plaque deposition and gliosis start from 6 months of age in both the cortex and hippocampus, and notably, hyperphosphorylated tau is found in dystrophic neurites around plaques. Cognitive impairment appears at around 18 months of age, though in the absence of neurodegeneration [34].
- APPNL-G-F
The second mouse line developed was APPNL-G-F. In addition to the Swedish and Iberian mutations, APPNL-G-F mice carry the Arctic mutation, which makes the pathology more severe by promoting Aβ aggregation through enhanced oligomerization and reduced proteolytic degradation. Amyloid plaque deposition begins as early as month 2, accompanied by gliosis, while cognitive deficits typically appear around 6 months of age [34].
- B6.152H (PS2APP)
Two different transgenic mouse lines carry the APPswe mutation under the control of the Thy1.2 promoter and the Volga (N141I) mutation in PSEN2 driven by the mouse prion protein promoter. These lines differ in their method of generation, resulting in different levels of APP expression. One PS2APP model has been obtained by crossing PS2 (N141I) mice with APPswe mice [46], thus expressing the mutations in heterozygosis, while the PS2APP mouse line known as B6.152H, described in this review, has been developed by co-injecting the two mutated constructs into C57BL/6 zygotes and is maintained as a homozygote line, resulting in higher expression of transcripts [35]. B6.152H mice display less variability in pathology expression and develop amyloid plaques at 6 months of age, along with gliosis, while cognitive deficits appear at 8 months of age [35,47].
3. Astrocytic Ca2+ Dysregulation in AD
Nowadays, many studies support the idea that dysregulated astrocytic Ca2+ homeostasis is a relevant feature of AD pathogenesis. However, it is still unclear and under debate whether astrocytes exhibit increased or decreased Ca2+ activity in AD, whether this dysregulation is an early event preceding plaque deposition or a direct consequence of plaque accumulation and what the molecular mechanisms underlying Ca2+ homeostasis dysfunction are. Literature reviews on this topic largely indicate a clear trend toward increased Ca2+ activity, mirroring the neuronal hyperexcitability that characterizes the early stages of pathology. This predominant view mainly stems from early reports on Tg2576 and APP/PS1 transgenic mice (see above for details on strains), which revealed a higher level of spontaneous Ca2+ activity and the occurrence of intercellular cortical Ca2+ waves linked to over-activation of purinergic receptors. Recent studies, though, provided novel information that widens our perspective on the “direction” of signal disruption in astrocytes from AD models.
In this review, we summarize the studies that addressed these questions, focusing primarily on those employing ex vivo and in vivo imaging techniques, aiming to identify a consensus among the different protocols, age and AD mouse models used (Table 1). Astrocyte phenotype has been classified as “hyperactivity”, “normoactivity” or “hypoactivity”, indicating higher, equivalent or lower spontaneous Ca2+ signals in AD mice compared with control mice. “Hyporesponsiveness”, on the other hand, denotes lower stimulus-evoked Ca2+ signals.
3.1. Astrocytic Ca2+ Dysregulation in AD: Hyperactivity
Takano and colleagues from the Nedergaard lab performed seminal work on astrocytic Ca2+ dysregulation in AD in 2007 while investigating brain vasculature involvement in the early stages of AD in living animals [48]. The crucial role of astrocytes in controlling vasodilation and vasoconstriction had been discovered a few years earlier; thus, the group also analyzed astrocyte Ca2+ signals in three different AD mouse models. The study focused on Tg2576, 3xTg and Tg-SwDI mice at 2–4 months of age, before the onset of extracellular amyloid deposition, neuronal degeneration and gliosis. Chemical Ca2+ indicator Fluo-4 AM and two-photon (2P) microscopy were used to image cell bodies and major processes in astrocytes from cortical layer II in ketamine/xylazine-anesthetized animals.
The authors observed a striking elevation in the frequency of spontaneous intracellular Ca2+ activity in Tg2576 mice relative to wild-type (WT) controls (0.4 events/min in WT vs. 2.3 events/min in Tg2576). However, this finding was not consistently observed in the other transgenic mouse models examined. Specifically, 3xTg mice showed mixed effect: while most mice did not differ from WT controls, a few animals displayed increased astrocyte Ca2+ activity (0.4 events/min in WT vs. 0.9 events/min in “3xTg low activity” vs. 3.5 events/min in “3xTg high activity”). The third mouse line (Tg-SwDI) showed no difference in Ca2+ oscillations compared to littermate WT controls (0.4 events/min in WT vs. 0.7 events/min in Tg-SwDI). Interestingly, when these mice were injected intravenously (i.v.) with Aβ40 peptide, astrocytes responded with a significantly greater increase in oscillation frequency compared to glial cells in their littermate controls. Collectively, this was the first in vivo evidence that astrocytic Ca2+ activity can be dysregulated in AD, although with different phenotypes depending on the mouse model.
Dysregulation of astrocyte Ca2+ signals in Tg2576 mice was investigated also in a study by Pirttimaki and colleagues, from the Parri lab, reporting a strong increase in astrocyte spontaneous activity (0.09 events/min in WT vs. 0.36 events/min in Tg2576) also in hippocampal slices from younger animals (PN4-14) [49].
In 2009, Kuchibhotla and colleagues from the Backsai lab conducted an in-depth characterization of Ca2+ homeostasis in cortical astrocytes from APPswe/PS1ΔE9 transgenic mice [50]. To focus on the chronic phase of amyloid pathology, the authors performed their study on isoflurane-anesthetized animals at 6–8 months of age, a time point corresponding to significant Aβ plaque burden in this model. They employed the Ca2+ indicator Oregon Green BAPTA-1 (OGB-1) to combine fluorescence lifetime imaging microscopy (FLIM) with classical 2-photon Ca2+ imaging, allowing for quantification of both the resting intracellular Ca2+ concentration and spontaneous Ca2+ oscillations in astrocyte somata.
The authors reported significantly elevated basal intracellular Ca2+ concentrations in astrocytes from APPswe/PS1ΔE9 mice compared to WT controls (81 nM in WT vs. 149 nM in APPswe/PS1ΔE9). AD mice were also characterized by an increase in both the percentage of astrocytes displaying spontaneous Ca2+ transients (8.1% in WT vs. 27.9% in APPswe/PS1ΔE9) and the amplitude of these signals (23.2 ΔF/F in WT vs. 33.6 ΔF/F in APPswe/PS1ΔE9). The increase in the fraction of active astrocytes was unaffected by the blockade of neuronal firing through TTX application. Notably, spontaneous hyperactivity was absent at an earlier disease stage (3–3.5-month-old AD mice) lacking amyloid plaque deposition, suggesting that Ca2+ dysregulation correlates with disease progression. In addition, the authors observed the occurrence of peculiar intercellular Ca2+ waves that originated from astrocytes located near amyloid plaques (within 25 μm) and propagated radially for distances up to 200 μm. However, higher amplitude Ca2+ signals were observed in individual astrocytes independently of plaque proximity. Based on these findings, the authors proposed a direct modulatory effect of Aβ plaques on global astrocytic function.
The presence of pathological Ca2+ waves was also reported in 2014 in APPswe/PSEN1L166P mice, an AD model characterized by faster progression of amyloid pathology, with plaque deposition occurring at 6 weeks of age. Delekate and colleagues, from the Petzold lab, investigated astrocytic Ca2+ activity in the somatosensory cortex of 5–9-month-old isoflurane/ketamine-anesthetized animals, by employing the Ca2+ indicator OGB-1 and the intravital marker methoxy-XO4 to image amyloid plaques [51]. Consistent with previous findings, this study reported that transgenic astrocytes exhibit elevated somatic Ca2+ activity. The frequency of somatic Ca2+ transients was used to classify astrocytes as inactive (0 events/min), active (0.1–0.4/min) or hyperactive (>0.4/min), revealing that 33.8% of AD astrocytes displayed hyperactivity compared to 4.1% in the littermate controls. Notably, the study investigated spontaneous activity in dye-loaded astrocytic endfeet, showing that hyperactivity similarly involves these perivascular processes. Also in this model, the increase in astrocyte activity was independent of neuronal firing. However, the authors demonstrated that proximity to amyloid plaques (within 50 µm) significantly increased the fraction of hyperactive and active astrocytes. Notably, the previous study [50] analyzed the correlation of plaque proximity with the increase in signal amplitude but not in signal frequency in astrocytes, thus hampering the comparison of these results. It is noteworthy to underline that the term “hyperactivity” is manifestly an umbrella term that can cover different parameters of Ca2+ activity in astrocytes. The Petzold group further investigated the molecular pathways underlying the increased Ca2+ activity and the propagation of Ca2+ waves in space and time, focusing on the role of purinergic signaling and specifically on P2Y1 receptors, highly upregulated in astrocytes surrounding amyloid plaques. Topical application of a general P2Y receptor antagonist (PPADS, 500 µM) or a P2Y1-specific antagonist (MRS2179, 500 µM) was sufficient to restore Ca2+ activity to levels comparable to littermate control and to block Ca2+ wave propagation. Pharmacology experiments revealed the involvement of ER-dependent Ca2+ release and connexin hemichannels in astrocyte hyperactivity.
The same group, years later, published another study [52] demonstrating that chronic (6 weeks) administration of P2Y1R inhibitors (MRS2179 1 mM or BPTU 10 µM), applied through i.c.v. osmotic minipumps, normalized astrocytic activity and reduced neuronal hyperexcitability in the somatosensory cortex. Notably, the authors disclosed astrocyte hyperactivity also in the dorsal hippocampus, using the genetically encoded Ca2+ indicator GCaMP6f. Despite the use of a genetically encoded Ca2+ indicator, the analysis was limited to cell somata and missed checking the recovery of physiological astrocyte activity upon chronic P2Y1R inhibition. On the other hand, the study analyzed the defects of synaptic plasticity in hippocampal slices from APP/PS1 mice and demonstrated that MRS2179 treatment can rescue impaired hippocampal LTP and prevent cognitive decline, suggesting it may be a promising therapeutic candidate.
In a study published by Bosson and colleagues from the Albrieux lab in 2017, a different brain region and an earlier developmental time point were investigated in the same AD mouse model (APPswe/PSEN1L166P) [53]. Specifically, the authors performed Ca2+ imaging of astrocytes in the CA1 stratum radiatum of hippocampal slices from young mice (PN19-28) and their WT littermates. Their analysis thus focused on the early stage of the disease, characterized by increased Aβ1–42 levels but preceding both the deposition of amyloid plaques and the appearance of reactive astrogliosis.
To investigate astrocytic Ca2+ activity, two imaging strategies were employed: (1) bulk loading of Fluo-4 AM across the entire hippocampal slice allowed visualization of a large population of astrocytes, primarily capturing large somatic Ca2+ events, and (2) whole-cell patch-clamp dye loading of individual astrocytes enabled resolution of principal processes and thinner arborizations, manually segmented in subregions that the authors term “microdomains”, although they represent only a minor fraction of the bushy ensemble of astrocyte microdomains. The key focus of the study was the TRPA1 channel, whose expression was found to double between P19 and P30 in APPswe/PSEN1L166P mice, in coincidence with the onset of Aβ secretion. Despite a similar proportion of active astrocytes between APPswe/PS1L166P and WT littermates, bulk-loaded astrocytes from the AD model displayed a significantly higher frequency of Ca2+ events (0.33 events/min in WT vs. 0.67 events/min in APPswe/PS1L166P), shifting the frequency distribution toward a hyperactive (>0.6 events/min) population (6% in WT vs. 21% in APPswe/PS1L166P). Notably, both event frequency and the fraction of hyperactive cells were fully reversed to WT values by pharmacological blockade of TRPA1 with HC030031 (40 μM). Subregion analysis in patch-loaded astrocytes revealed a low but statistically significant increase in event frequency in APPswe/PS1L166P mice. TRPA1 inhibition normalized the frequency increase (0.40 events/min in WT vs. 0.45 events/min in APPswe/PS1L166P vs. 0.43 events/min APPswe/PS1L166P + HC) and reduced the proportion of active microdomains in APPswe/PS1L166P (55.0% in WT vs. 65.7% in APPswe/PS1L166P vs. 29.3% APPswe/PS1L166P + HC).
Interestingly, bath application of Aβ oligomers (100 nM) to WT hippocampal slices increased both the number of active astrocytes and the percentage of the territory engaged in Ca2+ activity; this effect was abolished by HC030031. These experiments suggest that Ca2+ dysregulation may be an early event in AD pathogenesis and that soluble Aβ oligomers play a crucial role in modulating astrocyte Ca2+ activity and in the establishment of an astrocyte phenotype possibly concurring to neurotoxicity.
3.2. The Turning Point: Hyperactivity and Hyporesponsiveness
The study performed in 2021 by Lines and colleagues from the Araque and Kofuji lab [54] aimed to characterize astrocytic Ca2+ dynamics beyond the soma in the APPswe/PS1ΔE9 mouse model, using GCaMP7 and differentiating somatic signals from events within “arborizations”. Although this genetically encoded Ca2+ indicator allows the visualization of processes and microdomains, it is noteworthy that the analysis was performed on astrocyte territories identified by thresholding sulforhodamine 101 (SR101) fluorescence, thus missing a conspicuous part of microdomains, similarly to what obtained in the work of Bosson and colleagues through individual loading of astrocytes with Fluo-4.
The authors crossed APPswe/PS1ΔE9 mice with a mouse line expressing GCaMP7 under the specific astrocyte GLT1 promoter and analyzed 6–9-month-old mice, at a disease stage subsequent to plaque deposition, and control littermates. Complementing previous findings describing higher signal amplitude in astrocytes from the same AD model [50], this study reported an increased frequency of spontaneous Ca2+ signals in both somata and arborizations of astrocytes from the somatosensory cortex of urethane-anesthetized APPswe/PS1ΔE9 mice. The authors also examined the relationship between Ca2+ activity and the presence of amyloid plaques, revealing that Ca2+ events in astrocytic processes, but not in somata, were more frequent in cortical regions with higher plaque density. However, the frequency of Ca2+ events did not correlate with the distance to the nearest plaque, suggesting that astrocytic hyperactivity is not directly influenced by plaque proximity. Rather, it may depend on the concentration of soluble Aβ, the stage of disease progression or other factors within the pathological milieu. In parallel, the authors monitored sensory-evoked astrocytic Ca2+ responses following electrical stimulation of the contralateral hind paw, performing the first assessment of evoked Ca2+ signals in astrocytes from an AD model. In AD mice, the responses exhibited aberrant dynamics characterized by delayed onset, prolonged rise time and slower decay at both somatic and arborization levels. Besides confirming spontaneous astrocyte hyperactivity, this study thus reveals that astrocyte recruitment by sensory stimulation is slower and dysregulated in APPswe/PS1ΔE9 mice. Furthermore, sensory-evoked responses were shown to negatively correlate with local plaque burden and to be influenced by plaque proximity. Specifically, increasing the distance from plaques was associated with higher astrocyte recruitment by sensory stimulation, in terms of the fraction of activated astrocytes and percentage of astrocyte arborization involved in the response. Although a direct comparison of the amplitude of the response between APPswe/PS1ΔE9 mice and WT littermates is not reported, related graphs suggest a lower amplitude of the response in astrocytes from AD mice. The authors conclude that the presence of Aβ plaques is associated with an increased spontaneous Ca2+ activity in astrocyte processes and with a lower response to sensory inputs, providing the first experimental evidence of hypofunction of astrocytes in AD mouse models.
3.3. Astrocytic Ca2+ Dysregulation in AD: Hypoactivity and Hyporesponsiveness
After many years of general agreement on astrocyte hyperactivity in AD mouse models, several publications have now confirmed that AD astrocytes can also exhibit Ca2+ hypoactivity.
In 2022, Åbjørsbråten and colleagues, from the Enger lab, combined 2P microscopy with virally delivered GCaMP6 to image astrocytes in the somatosensory cortex of awake, behaving 15-month-old tg-ArcSwe mice. This age corresponds to an advanced stage of disease, since plaque deposition, gliosis and cognitive impairment are present in 4–6-month-old animals in this AD model. The study investigated possible perturbations of the norepinephrine–astrocyte Ca2+ signaling axis [55]. Astrocytes respond with Ca2+ increases to noradrenaline in different brain regions via the activation of α1-ARs [56,57,58,59], and they have been specifically reported to display fast Ca2+ transients upon sensory and air-puff stimulation [60]. The authors used a sophisticated experimental paradigm in which mice were free to move on a treadmill while the Ca2+ signal and pupil dilation were simultaneously recorded. An air puff directed at the whiskers and nose was exploited to trigger a noradrenergic startle response, while pupil dilation was used as a readout of locomotion and startle. Event-based ROAs (regions of activity) were automatically detected in GCaMP-expressing astrocytes through an originally developed analysis suite and assigned to manually segmented astrocyte territories (soma, processes and endfeet) to discriminate the recruited territories. During quiet periods, the authors found no differences in astrocytic Ca2+ activity between tg-ArcSwe mice and WT littermate controls. Additionally, they observed rare, long-lasting pathological Ca2+ waves similar to those previously reported in APP/PS1 mouse models, occurring in only 10–15% of the recordings. Spontaneous locomotor activity and startle response were accompanied by pupil dilation and increased astrocytic Ca2+ activity, computed as max ROA density and density rise time. During the startle response, the recruitment of Ca2+ activity in WT and tg-ArcSwe astrocytes was similar, but astrocyte Ca2+ response to locomotion was lower in tg-ArcSwe mice. In addition, pupil dilation and astrocyte responses were coupled in WT mice during both startle response and locomotion but uncoupled in AD mice. Notably, the authors found a significant reduction in noradrenaline release in the cortex of AD mice, thus suggesting that a defect in the degree of stimulation contributes the reduced response of astrocytes. Whether a diminished noradrenergic input is the main driver of the lower astrocyte response remains unclear, since the ability of astrocytes to respond to noradrenaline was not directly assessed in these mice.
Conversely, in the same year, Shah and colleagues from the De Strooper lab reported cell-autonomous dysregulation in astrocyte Ca2+ signals in an AD mouse model, investigating in a more translational study the relationship between resting-state functional connectivity (FC, measured via resting-state fMRI) and astrocytic Ca2+ signaling in the APPNL-F mouse model [61]. Firstly, the study monitored and compared FC and amyloid load in a cohort of cognitively healthy individuals scanned at two time points, revealing that an early increase in anterior–posterior cingulate FC characterized patients developing amyloid accumulation several years after, suggesting its potential as a very early functional biomarker. Driven by these results, the authors shifted the research to the APPNL-F mouse line, focusing on an early time point (3 months) preceding plaque deposition by a few months. Resting-state fMRI and 2P GCaMP6f imaging of Ca2+ dynamics in the cingulate cortex (CCx) of anesthetized animals confirmed the early increase in cingulate FC seen in patients and revealed a significant reduction in astrocytic activity in APPNL-F mice compared to APPNL controls (carrying only the APPswe mutation under its endogenous promoter). Specifically, there was a marked decrease in the percentage of active astrocytes (64% in controls vs. 28% in APPNL-F), in the frequency of Ca2+ events (0.12 peaks/min in controls vs. 0.04 peaks/min in APPNL-F) and in event amplitude (1.29 ΔF/F in controls vs. 1.21 ΔF/F in APPNL-F) in regions of interests (ROIs) encompassing both astrocyte soma and proximal processes. The authors report that Ca2+ activity in distal processes remains unaltered, but it is noteworthy that a single ROI comprising all distal territories was used to monitor distal Ca2+ signals of individual astrocytes, thus hampering an accurate assessment of the activity of discrete microdomains. Decreased astrocyte calcium signaling was observed at early stages of amyloid pathology also in a related, more severe AD mouse model, i.e., APPNL-G-F mice. The impairment of Ca2+ homeostasis in APPNL-F mice was associated with a marked reduction in IP3R2 in astrocytes, a finding consistent in both the APPNL-F mouse model and human AD brain tissue. Given the central role of astrocytes in regulating neuronal activity, the authors attempted to modulate intracellular Ca2+ signaling in APPNL-F mice by inducing AAV-mediated expression of Gq DREADDs (Designer Receptors Exclusively Activated by Designer Drugs) in astrocytes. Consistent with the observed reduction in IP3R2, the administration of the designer agonist CNO to activate DREADDs induced a weaker increase in Ca2+ signals in APPNL-F compared to control APPNL mice. Nonetheless, the increase in astrocytic Ca2+ activity triggered by CNO in APPNL-F mice was sufficient to normalize astrocytic Ca2+ signals and dampen neuronal hyperactivity. Of note, this mouse model also displays increased susceptibility to seizure and behavior hyperactivity during the light–dark cycle, and CNO application completely reversed these clinically relevant behaviors. Interestingly, DREADD activation in control APPNL mice induced astrocyte hyperactivity and promoted neuronal hyperactivity, seizure susceptibility and behavioral hyperactivity, demonstrating the importance of a tightly regulated Ca2+ activity in astrocytes.
In 2023, Lia and colleagues from the Carmignoto lab published a study reporting astrocyte hypoactivity in the B6.152H mouse model of AD [47]. The experiments were mainly performed in ex vivo brain slices from the somatosensory cortex of B6.152H mice, analyzing both spontaneous and evoked activity in astrocytes. Spontaneous signal was monitored also in vivo in isoflurane-anesthetized mice. Importantly, astrocyte-specific GCaMP6 expression and 2P microscopy allowed a detailed analysis of Ca2+ signals from all astrocyte territories, from the soma to principal processes to thin microdomains. The study investigated astrocyte activity at two stages of disease progression, before (3 months of age) and after (6 months of age) the onset of gliosis and plaque deposition in brain cortex. At the early stage, SSCx astrocytes exhibited a non-significant trend toward increased spontaneous Ca2+ activity at microdomains. However, this trend was reversed at 6 months of age, revealing a pronounced Ca2+ hypoactivity characterized by reduced active microdomains, frequency of events and event amplitude. This reduction in spontaneous activity was confirmed in vivo through 2P imaging of astrocytes from the somatosensory cortex of anesthetized B6.152H mice. Interestingly, spontaneous Ca2+ hypoactivity after plaque deposition was not influenced by the distance to plaques.
In analogy with the study of Lines, in which astrocyte response to sensory stimulation was probed, this work characterized, besides spontaneous activity, Ca2+ response to metabotropic agonists. In AD mice, after plaque deposition, astrocytes displayed hyporesponsiveness to ATP and noradrenaline (NA) at the level of all their territories, and this hypofunction was associated with both impaired NA-dependent, astrocyte-mediated LTP in the SSCx and defective tactile memory retention. The authors disclosed as a putative underlying molecular mechanism a significant reduction in ER Ca2+ content and astrocyte expression of STIM1, a key regulator of store-operated Ca2+ entry aimed at refilling ER. Interestingly, lower levels of STIM1 have been reported also in post-mortem tissue from sporadic AD patients [62]. Notably, astrocyte-specific overexpression of STIM1 via a lentiviral vector was successful in restoring both spontaneous and evoked Ca2+ activity in brain slices, as well in normalizing the impaired LTP observed in B6.152H mice. These results confirm how a dysregulation of astrocyte Ca2+ signals can affect brain physiology by altering synaptic plasticity.
In the same year, the work of Lee and colleagues from the Kastanenka lab analyzed the slow-wave activity (SWA) disturbances occurring one month before plaque appearance in young APPswe/PS1ΔE9 mice, resembling those commonly reported in patients in early phases of AD [63]. The study disclosed a correlation between slow-wave disruption, sleep disturbances and a reduction of power in astrocyte Ca2+ transients occurring at the slow-wave frequency (0.2–1 Hz) in the SSCx of isoflurane-anesthetized AD mice. The reduction in amplitude was present in all astrocyte territories, but it was particularly significant at the level of microdomains. Optogenetic stimulation of ChR2-expressing astrocytes at the slow-wave frequency (0.6 Hz) was sufficient to restore SWA in transgenic mice, along with improving memory function. Although this study, unfortunately, did not investigate the degree of recovery of astrocyte Ca2+ signals upon optogenetic activation, the obtained results underscore the tight relationship between astrocyte hypoactivity and performance decline in brain function.

Table 1.
Effects of AD-linked mutations on astrocytic Ca2+ activity in different mouse models of Alzheimer’s disease.
Table 1.
Effects of AD-linked mutations on astrocytic Ca2+ activity in different mouse models of Alzheimer’s disease.
| AD Model | Promoters | Age | Brain Region | Approach | Astrocyte Phenotype | Notes | Study |
|---|---|---|---|---|---|---|---|
| Tg-SwDI | Thy1.2 APPmut | 2–4 months | SSX | In vivo anaesthetized (ketamine + xylazine) | Normoactivity | Hyperactivity when injected with Aβ40 i.v. | Takano et al. [48] |
| 3xTg | Thy1.2-APPswe Thy1.2-MAPT endogenous PSEN1 | Normoactivity Hyperactivity | Most astrocytes showed normoactivity, some others hyperactivity | ||||
| Tg2576 | PrP-APPswe | Hyperactivity | Striking increase in event frequency | ||||
| 4–14 days | Hipp | Ex vivo hippocampal slices | Elevated Aβ42 levels in young Tg2576 mice; bath application of Aβ42 reduces hyperactivity | Pirttimaki et al. [49] | |||
| APPswe /PSEN1ΔE9 | PrP-APPswe/PSEN1 | 6–8 months | Cortex | In vivo anaesthetized (0.5–1.0% isoflurane) | Hyperactivity | Normoactivity before plaques (3.5–4 months) | Kuchibhotla et al. [50] |
| 6–9 months | SSCx | In vivo anaesthetized (urethane 1.8 mg/kg) | Hyperactivity Hyporesponsiveness | Hyporesponsivness to n sensory stimulation | Lines et al. [54] | ||
| 4–6 months | SSCx | In vivo anaesthetized (1.75% isoflurane) | Hypoactivity | Reduced power in SWA-related Ca2+ oscillations | Lee et al. [63] | ||
| APPswe /PS1 L166P | Thy1-APPswe/PSEN1 | 5–9 months | SSCx | In vivo anaesthetized (0.4% isoflurane + ketamine) | Hyperactivity | Dependent on purinergic signal (P2YRs) | Delekate et al. [51,52] |
| 19–28 days | Hipp | Ex vivo hippocampal slices | Hyperactivity | Dependent on TRPA1 signaling | Bosson et al. [53] | ||
| tg-ArcSwe | Thy1-APPArcSwe | 15 months | SSCx | In vivo awake | Normoactivity Hyporesponsiveness | Rare occurrence of waves; hyporesponsiveness linked to lower NA release | Åbjørsbråten et al. [55] |
| APPNL-F | endogenous APP | 3 months | CCx | In vivo anaesthetized (medetomidine 0.05 mg/kg + 0.5% isoflurane) | Hypoactivity | Associated to IP3R2 downregulation | Shah et al. [61] |
| APPNL-F-G | 6 weeks | Not further investigated | |||||
| B6.152H | Thy1-APPswe Prp-PSEN2 | 6 months | SSCx | Ex vivo cortical slices In vivo anaesthetized (isoflurane 0.5–0.8% + carprofen 5 mg/kg) | Hypoactivity Hyporesponsiveness | Associated to reduced ER Ca2+ content | Lia et al. [47] |
4. Methodological Limitations
Despite technological advances and significant progress in the field, different methodological limitations can affect the experimental approaches reported in this review and used to monitor astrocyte Ca2+ dynamics. The first limitation concerns the use of chemical Ca2+ indicators, which primarily load cell bodies and principal processes of astrocytes. The majority of reports on astrocyte signals in AD models rely on chemical indicators and therefore do not provide information on the “bushy” arborization, which constitutes the larger fraction of astrocyte volume [64]. The development of GECIs allowed for spatially extending Ca2+ analysis by reliably capturing Ca2+ activity at the level of astrocytic microdomains [65].
Another major limitation of Ca2+ imaging studies is related to the use of anesthetics in the in vivo experiments. It is well known that anesthesia dampens both neuronal and astrocytic activity [66]. Therefore, although AD and control animals are anesthetized using the same protocol within each study, anesthesia-induced alterations in signaling may still represent a confounding factor worth considering. In this regard, lowering the levels of anesthetics may help reduce the differences in astrocyte Ca2+ signals between awake and anesthetized animals.
A further critical issue is that most studies assess astrocytic Ca2+ activity at only a single time point. This makes it difficult to determine whether hyper- or hypoactive phenotypes represent transient phases in disease progression or stable features of specific mouse models. This limitation could be overcome by performing Ca2+ imaging at multiple disease stages, ideally through longitudinal monitoring of Ca2+ activity in awake mice via chronic cranial windows or GRIN lenses coupled to miniaturized microscopes.
5. Discussion
A variety of mechanisms contributing to Ca2+ dysregulation in astrocytes emerges in the studies described in this review. Different works investigated the proximity of astrocytes to plaques as a possible factor contributing to cell impairment. Indeed, the presence of amyloid plaques as a common hallmark across AD models hints at insoluble amyloid aggregates as the major driving stimulus for astrocyte dysregulation. However, controversial results have been obtained on a possible correlation between plaque proximity and astrocyte Ca2+ signal impairment, in both hypo- and hyperactivity [47,50,51,54]. In addition, several studies have reported signal alterations in astrocytes also prior to the onset of plaque deposition [48,49,53,61,63], and a growing body of evidence now supports the hypothesis that soluble Aβ oligomers may be primarily responsible for some distinctive phenotypes observed in AD mouse models. Notably, already in 2012, Busche and colleagues demonstrated that soluble Aβ42 oligomers induce neuronal hyperactivity in the hippocampus of WT mice and are responsible of neuronal hyperactivity in APPswe/PS1G384A mice. Indeed, acute γ-secretase inhibition reduced soluble Aβ levels and restored physiological levels of activity in AD neurons [67]. Other studies suggest that not only neurons but also astrocytes exhibit altered Ca2+ signaling in response to Aβ exposure. For instance, Takano and colleagues revealed an increase in the frequency of Ca2+ oscillations in cortical astrocytes from WT and Tg-SwDI mice after intravenous administration of Aβ1–40 peptide (0.4 mg/kg). Pirttimaki and colleagues showed that both physiological (300 pM) and pathological (10 nM) concentrations of Aβ1–42 oligomers are sufficient to evoke intracellular Ca2+ transients in hippocampal astrocytes in ex vivo brain slices from WT mice. Furthermore, Aβ-induced astrocytic activity was completely abolished by the application of methyllycaconitine (MLA), an antagonist of the α7 nicotinic acetylcholine receptor subunit, highlighting the role of α7 receptors in mediating astrocytic responses to soluble Aβ oligomers [49]. Similar results were reported by Bosson and colleagues, who exposed hippocampal slices from WT mice to Aβ oligomers (100 nM). This treatment increased both the number of active astrocytes and the proportion of astrocytic territory engaged in Ca2+ activity, and this effect was abolished by the TRPA1 inhibitor HC030031 [53]. These data suggest that Ca2+ signaling can be affected by Aβ oligomers present in the early stages of Alzheimer’s disease and that multiple receptors may be involved, converging onto a common signaling pathway.
In addition to acute or chronic effects of Aβ1–42 oligomers on astrocytes, the occurrence of reactive astrogliosis in AD (and in other neurological disorders) has been suggested to influence astrocyte Ca2+ activity [68], for instance, through the upregulation of plasma membrane receptors. Reactive astrocytes re-express mGluR5 in pathophysiological conditions such as AD [69,70], epilepsy [71] and neuropathic pain [72]. Importantly, mGluR5 re-expression significantly enhances astrocytic Ca2+ activity, whereas its genetic silencing or pharmacological inhibition markedly reduces such activity in models of neuropathic pain and status epilepticus [72,73]. Other receptors involved in the inflammatory response, such as P2Y1 and TRPA1 receptors, are similarly upregulated in some AD mouse models, particularly in regions adjacent to amyloid plaques. Furthermore, pharmacological blockade of these receptors using specific antagonists has demonstrated beneficial effects, including improvements in behavioral outcomes [51,52,53,74].
Alongside alterations in the expression of plasma membrane receptors, animal models of AD can also display abnormalities in intracellular key modulators of Ca2+ signaling. Mutations in presenilins, which also localize in the ER membrane, can reduce the activity of the Sarco-Endoplasmic Reticulum Calcium ATPase (SERCA) that physiologically pumps Ca2+ ions from the cytosol to the ER [75,76]. In B6.152H mice, a diminished expression of STIM1, a key protein responsible for ER refilling following depletion, is accompanied by a reduction in ER Ca2+ content in astrocytes [47]. Interestingly, the expression of mutated PSEN2 alone was not sufficient to reduce Ca2+ signals in astrocytes [47], indicating that the concomitant presence of the APPswe mutation in B6.152H mice, which induces an earlier increase in Aβ species and in Aβ42/Aβ40 ratio [77], is a key factor exacerbating the effect of PSEN mutations on astrocyte Ca2+ homeostasis. This observation suggests a difference in acute and chronic effects of Aβ oligomers on astrocyte Ca2+ signals. In another mouse model, based on multiple mutations in APP (APPNL-F), the downregulation of astrocytic IP3R2 expression has been found associated with the reduction of Ca2+ signals in astrocytes from the CCx [61]. Notably, a downregulation in the expression of STIM1 and of IP3R2 has been reported also in post-mortem AD brains [61,62].
Overall, experimental results indicate that the accumulation of amyloid oligomers in the brain parenchyma, the establishment of a neuroinflammatory environment and the presence of pathogenic mutations together induce alterations in the intracellular Ca2+ signaling pathway of astrocytes and contribute to the development of AD phenotypes.
6. Conclusions
While this review aims to offer a comprehensive view of Ca2+ dysregulation in AD astrocytes, we acknowledge that the complexity of this multifactorial pathology cannot be captured by simplistic definitions. Undoubtedly, despite the discrepancies observed among mouse models and brain regions, we show that robust evidence of disrupted astrocyte Ca2+ homeostasis in AD pathology supports the presence of both hypo- and hyper-activity.
The studies reported here comprise a collection of snapshots capturing astrocyte Ca2+ signals in different moments along the lifetime (young or adult mice) and AD progression (early or late pathology). Moreover, these views can focus on various details (brain regions) from diverse subjects (animal models characterized by distinct cellular expression of different AD mutations) and during discrete activity states (spontaneous or evoked signals). This fragmented information hampers the development of an integrative mechanistic model including all the variables and capable of predicting the functional consequences at synaptic and cognitive levels. However, these snapshots allow us to sketch a larger picture in which Ca2+ signalling is dynamically linked to the fundamental homeostatic role of astrocytes, following an inverted U-shape relationship where too little or too much signaling are equally detrimental for brain function (Figure 2).
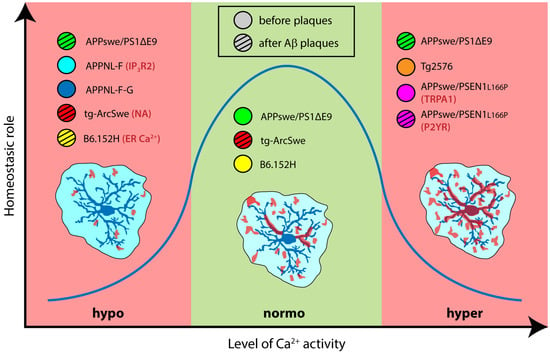
Figure 2.
Schematic representation of the inverted U-shaped relationship (blue trace) showing that both reduced (hypo) and increased (hyper) Ca2+ activity negatively affect the homeostatic function of astrocytes. The position of the circles (color coded to identify the different AD mouse models) in the different sections indicates their Ca2+ phenotype, while their motif fill symbolizes the disease stage (uniform pattern = early stage, i.e., before plaque deposition; striped pattern = late stage, i.e., after plaque deposition). Where investigated, the mechanism underlying signal dysregulation is reported after the AD model designation.
Notably, astrocyte signal dysregulation including both hyper- or hypoexcitability has been reported in animal models of several other disorders affecting the nervous system, including Parkinson’s disease, Huntington’s disease, Down Syndrome, Amyotrophic Lateral Sclerosis and Major Depressive Disorder. These findings urge the scientific community to strengthen the investigation of the diverse molecular mechanisms underlying the disruption of Ca2+ homeostasis in astrocytes, with the aim of identifying therapeutic strategies capable of restoring their central modulatory role.
Author Contributions
A.D.S. and M.Z. contributed equally to the conception, design, writing and revision of this review. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Fondazione Telethon, grant number GMR23T1234 to MZ.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| α1-ARs | Adrenergic receptors |
| AD | Alzheimer’s disease |
| ApoE4 | Apolipoprotein E |
| APP | Amyloid precursor protein |
| ATP | Adenosine triphosphate |
| Aβ | Amyloid beta |
| BDNF | Brain-derived neurotrophic factors |
| cAMP | Cyclic adenosine monophosphate |
| CCX | Cingulate cortex |
| CICR | Ca2+-induced Ca2+ release |
| CNO | Clozapine N-oxide |
| CRAC | Ca2+ release-activated Ca2+ |
| DAG | Diacylglycerol |
| DREADDs | Designer Receptors Exclusively Activated by Designer Drugs |
| EOFAD | Early-onset familial AD |
| ER | Endoplasmic reticulum |
| FC | Functional connectivity |
| FLIM | Lifetime imaging microscopy |
| GABA | Gamma-aminobutyric acid |
| GLT-1 | Glutamate transporter 1 |
| GPCRs | G protein-coupled receptors |
| IP3 | Inositol 1,4,5-trisphosphate |
| IP3Rs | IP3 receptors |
| LOAD | Late-onset AD |
| LTP | Long-term potentiation |
| MAPT | Microtubule-Associated Protein Tau |
| mGluRs | Metabotropic glutamate receptors |
| MLA | Methyllycaconitine |
| NA | Noradrenaline |
| NCX | Na+/Ca2+ exchanger |
| OGB-1 | Oregon Green BAPTA-1 |
| P2YRs | Purinergic receptors |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PLC | Phospholipase C |
| PPADS | Pyridoxalphosphate-6-azophenyl-2′,4′-disulfonic acid |
| PrP | Hamster prion protein |
| PS1 | Presenilin 1 |
| PS2 | Presenilin 2 |
| ROA | Region of activity |
| ROI | Region of interest |
| RyRs | Ryanodine receptors |
| SERCA | Sarco-Endoplasmic Reticulum Calcium ATPase |
| SOCE | Store-operated Ca2+ entry |
| SR101 | Sulforhodamine 101 |
| SSCx | Somatosensory cortex |
| STIM | Stromal interaction molecule |
| SWA | Slow-wave activity |
| WT | Wild type |
References
- Wang, F.; Smith, N.A.; Xu, Q.; Fujita, T.; Baba, A.; Matsuda, T.; Takano, T.; Bekar, L.; Nedergaard, M. Astrocytes Modulate Neural Network Activity by Ca2+-Dependent Uptake of Extracellular K+. Sci. Signal. 2012, 5, ra26. [Google Scholar] [CrossRef]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef]
- Santello, M.; Toni, N.; Volterra, A. Astrocyte Function from Information Processing to Cognition and Cognitive Impairment. Nat. Neurosci. 2019, 22, 154–166. [Google Scholar] [CrossRef]
- Lia, A.; Di Spiezio, A.; Speggiorin, M.; Zonta, M. Two Decades of Astrocytes in Neurovascular Coupling. Front. Netw. Physiol. 2023, 3, 1162757. [Google Scholar] [CrossRef]
- Untiet, V. Astrocytic Chloride Regulates Brain Function in Health and Disease. Cell Calcium 2024, 118, 102855. [Google Scholar] [CrossRef]
- Kofuji, P.; Araque, A. G-Protein-Coupled Receptors in Astrocyte–Neuron Communication. Neuroscience 2021, 456, 71–84. [Google Scholar] [CrossRef]
- Lia, A.; Di Spiezio, A.; Vitalini, L.; Tore, M.; Puja, G.; Losi, G. Ion Channels and Ionotropic Receptors in Astrocytes: Physiological Functions and Alterations in Alzheimer’s Disease and Glioblastoma. Life 2023, 13, 2038. [Google Scholar] [CrossRef]
- Pasti, L.; Zonta, M.; Pozzan, T.; Vicini, S.; Carmignoto, G. Cytosolic Calcium Oscillations in Astrocytes May Regulate Exocytotic Release of Glutamate. J. Neurosci. 2001, 21, 477–484. [Google Scholar] [CrossRef]
- Mothet, J.-P.; Pollegioni, L.; Ouanounou, G.; Martineau, M.; Fossier, P.; Baux, G. Glutamate Receptor Activation Triggers a Calcium-Dependent and SNARE Protein-Dependent Release of the Gliotransmitter D-Serine. Proc. Natl. Acad. Sci. USA 2005, 102, 5606–5611. [Google Scholar] [CrossRef]
- Woo, D.H.; Han, K.-S.; Shim, J.W.; Yoon, B.-E.; Kim, E.; Bae, J.Y.; Oh, S.-J.; Hwang, E.M.; Marmorstein, A.D.; Bae, Y.C.; et al. TREK-1 and Best1 Channels Mediate Fast and Slow Glutamate Release in Astrocytes upon GPCR Activation. Cell 2012, 151, 25–40. [Google Scholar] [CrossRef]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.R.; Robitaille, R.; Volterra, A. Gliotransmitters Travel in Time and Space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef]
- Losi, G.; Vignoli, B.; Granata, R.; Lia, A.; Zonta, M.; Sansevero, G.; Pischedda, F.; Chiavegato, A.; Santi, S.; Zentilin, L.; et al. Spontaneous Activity of Astrocytes Is a Stochastic Functional Signal for Memory Consolidation. Proc. Natl. Acad. Sci. USA 2025, 122, e2500511122. [Google Scholar] [CrossRef]
- de Ceglia, R.; Ledonne, A.; Litvin, D.G.; Lind, B.L.; Carriero, G.; Latagliata, E.C.; Bindocci, E.; Di Castro, M.A.; Savtchouk, I.; Vitali, I.; et al. Specialized Astrocytes Mediate Glutamatergic Gliotransmission in the CNS. Nature 2023, 622, 120–129. [Google Scholar] [CrossRef]
- Li, H.; Zhao, Y.; Dai, R.; Geng, P.; Weng, D.; Wu, W.; Yu, F.; Lin, R.; Wu, Z.; Li, Y.; et al. Astrocytes Release ATP/ADP and Glutamate in Flashes via Vesicular Exocytosis. Mol. Psychiatry 2025, 30, 2475–2489. [Google Scholar] [CrossRef]
- Vignoli, B.; Battistini, G.; Melani, R.; Blum, R.; Santi, S.; Berardi, N.; Canossa, M. Peri-Synaptic Glia Recycles Brain-Derived Neurotrophic Factor for LTP Stabilization and Memory Retention. Neuron 2016, 92, 873–887. [Google Scholar] [CrossRef]
- Shigetomi, E.; Patel, S.; Khakh, B.S. Probing the Complexities of Astrocyte Calcium Signaling. Trends Cell Biol. 2016, 26, 300–312. [Google Scholar] [CrossRef]
- Lohr, C.; Beiersdorfer, A.; Fischer, T.; Hirnet, D.; Rotermund, N.; Sauer, J.; Schulz, K.; Gee, C.E. Using Genetically Encoded Calcium Indicators to Study Astrocyte Physiology: A Field Guide. Front. Cell. Neurosci. 2021, 15, 690147. [Google Scholar] [CrossRef]
- Veiga, A.; Abreu, D.S.; Dias, J.D.; Azenha, P.; Barsanti, S.; Oliveira, J.F. Calcium-Dependent Signaling in Astrocytes: Downstream Mechanisms and Implications for Cognition. J. Neurochem. 2025, 169, e70019. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, T.; Lu, X.; Lan, X.; Chen, Z.; Lu, S. G Protein-Coupled Receptors (GPCRs): Advances in Structures, Mechanisms, and Drug Discovery. Signal Transduct. Target. Ther. 2024, 9, 88. [Google Scholar] [CrossRef]
- Montes de Oca Balderas, P.; Montes de Oca Balderas, H. Synaptic Neuron-Astrocyte Communication Is Supported by an Order of Magnitude Analysis of Inositol Tris-Phosphate Diffusion at the Nanoscale in a Model of Peri-Synaptic Astrocyte Projection. BMC Biophys. 2018, 11, 3. [Google Scholar] [CrossRef]
- Lalo, U.; Pankratov, Y. Astrocyte Ryanodine Receptors Facilitate Gliotransmission and Astroglial Modulation of Synaptic Plasticity. Front. Cell. Neurosci. 2024, 18, 1382010. [Google Scholar] [CrossRef]
- Abdulkhaliq, A.A.; Kim, B.; Almoghrabi, Y.M.; Khan, J.; Ajoolabady, A.; Ren, J.; Bahijri, S.; Tuomilehto, J.; Borai, A.; Pratico, D. Amyloid-β and Tau in Alzheimer’s Disease: Pathogenesis, Mechanisms, and Interplay. Cell Death Dis. 2026, 17, 21. [Google Scholar] [CrossRef]
- Younkin, S.G. The APP and PS1/2 Mutations Linked to Early Onset Familial Alzheimer’s Disease Increase the Extracellular Concentration of Aβ1–42(43). In Presenilins and Alzheimer’s Disease; Springer: Berlin/Heidelberg, Germany, 1998; Volume 37, pp. 27–33. [Google Scholar]
- Weggen, S.; Beher, D. Molecular Consequences of Amyloid Precursor Protein and Presenilin Mutations Causing Autosomal-Dominant Alzheimer’s Disease. Alzheimer’s Res. Ther. 2012, 4, 9. [Google Scholar] [CrossRef]
- Gallwitz, L.; Schmidt, L.; Marques, A.R.A.; Tholey, A.; Cassidy, L.; Ulku, I.; Multhaup, G.; Di Spiezio, A.; Saftig, P. Cathepsin D: Analysis of Its Potential Role as an Amyloid Beta Degrading Protease. Neurobiol. Dis. 2022, 175, 105919. [Google Scholar] [CrossRef]
- Varte, V.; Ralte, L.; Kumar, N.S. Early-Onset Alzheimer’s Disease and Amyloid Precursor Protein Gene Mutations. Discov. Neurosci. 2025, 20, 8. [Google Scholar] [CrossRef]
- Krishnamurthy, H.K.; Jayaraman, V.; Krishna, K.; Wang, T.; Bei, K.; Changalath, C.; Rajasekaran, J.J. An Overview of the Genes and Biomarkers in Alzheimer’s Disease. Ageing Res. Rev. 2025, 104, 102599. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative Memory Deficits, Aβ Elevation, and Amyloid Plaques in Transgenic Mice. Science 1996, 274, 99–103. [Google Scholar] [CrossRef]
- Takahashi, R.H.; Milner, T.A.; Li, F.; Nam, E.E.; Edgar, M.A.; Yamaguchi, H.; Beal, M.F.; Xu, H.; Greengard, P.; Gouras, G.K. Intraneuronal Alzheimer Aβ42 Accumulates in Multivesicular Bodies and Is Associated with Synaptic Pathology. Am. J. Pathol. 2002, 161, 1869–1879. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-Transgenic Model of Alzheimer’s Disease with Plaques and Tangles. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; LaFerla, F.M. Intraneuronal Aβ Causes the Onset of Early Alzheimer’s Disease-Related Cognitive Deficits in Transgenic Mice. Neuron 2005, 45, 675–688. [Google Scholar] [CrossRef]
- Van Nostrand, W.E.; Melchor, J.P.; Cho, H.S.; Greenberg, S.M.; Rebeck, G.W. Pathogenic Effects of D23N Iowa Mutant Amyloid β-Protein. J. Biol. Chem. 2001, 276, 32860–32866. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Xu, F.; Deane, R.; Romanov, G.; Previti, M.L.; Zeigler, K.; Zlokovic, B.V.; Van Nostrand, W.E. Early-Onset and Robust Cerebral Microvascular Accumulation of Amyloid β-Protein in Transgenic Mice Expressing Low Levels of a Vasculotropic Dutch/Iowa Mutant Form of Amyloid β-Protein Precursor. J. Biol. Chem. 2004, 279, 20296–20306. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Slunt, H.H.; Ratovitski, T.; Jenkins, N.A.; Copeland, N.G.; Borchelt, D.R. Co-Expression of Multiple Transgenes in Mouse CNS: A Comparison of Strategies. Biomol. Eng. 2001, 17, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Fadale, D.J.; Anderson, J.; Xu, G.M.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Lee, M.K.; Younkin, L.H.; Wagner, S.L.; et al. Mutant Presenilins Specifically Elevate the Levels of the 42 Residue β-Amyloid Peptide in Vivo: Evidence for Augmentation of a 42-Specific γ Secretase. Hum. Mol. Genet. 2004, 13, 159–170. [Google Scholar] [CrossRef]
- Garcia-Alloza, M.; Robbins, E.M.; Zhang-Nunes, S.X.; Purcell, S.M.; Betensky, R.A.; Raju, S.; Prada, C.; Greenberg, S.M.; Bacskai, B.J.; Frosch, M.P. Characterization of Amyloid Deposition in the APPswe/PS1dE9 Mouse Model of Alzheimer Disease. Neurobiol. Dis. 2006, 24, 516–524. [Google Scholar] [CrossRef]
- Kamphuis, W.; Mamber, C.; Moeton, M.; Kooijman, L.; Sluijs, J.A.; Jansen, A.H.P.; Verveer, M.; de Groot, L.R.; Smith, V.D.; Rangarajan, S.; et al. GFAP Isoforms in Adult Mouse Brain with a Focus on Neurogenic Astrocytes and Reactive Astrogliosis in Mouse Models of Alzheimer Disease. PLoS ONE 2012, 7, e42823. [Google Scholar] [CrossRef]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jäggi, F.; Wolburg, H.; Gengler, S.; et al. Aβ42-driven Cerebral Amyloidosis in Transgenic Mice Reveals Early and Robust Pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef]
- Serneels, L.; Van Biervliet, J.; Craessaerts, K.; Dejaegere, T.; Horré, K.; Van Houtvin, T.; Esselmann, H.; Paul, S.; Schäfer, M.K.; Berezovska, O.; et al. γ-Secretase Heterogeneity in the Aph1 Subunit: Relevance for Alzheimer’s Disease. Science 2009, 324, 639–642. [Google Scholar] [CrossRef]
- Maia, L.F.; Kaeser, S.A.; Reichwald, J.; Hruscha, M.; Martus, P.; Staufenbiel, M.; Jucker, M. Changes in Amyloid-β and Tau in the Cerebrospinal Fluid of Transgenic Mice Overexpressing Amyloid Precursor Protein. Sci. Transl. Med. 2013, 5, 194re2. [Google Scholar] [CrossRef]
- Lord, A.; Kalimo, H.; Eckman, C.; Zhang, X.-Q.; Lannfelt, L.; Nilsson, L.N.G. The Arctic Alzheimer Mutation Facilitates Early Intraneuronal Aβ Aggregation and Senile Plaque Formation in Transgenic Mice. Neurobiol. Aging 2006, 27, 67–77. [Google Scholar] [CrossRef]
- Philipson, O.; Lannfelt, L.; Nilsson, L.N.G. Genetic and Pharmacological Evidence of Intraneuronal Aβ Accumulation in APP Transgenic Mice. FEBS Lett. 2009, 583, 3021–3026. [Google Scholar] [CrossRef] [PubMed]
- Cortese, A.; Delgado-Morales, R.; Almeida, O.F.X.; Romberg, C. The Arctic/Swedish APP Mutation Alters the Impact of Chronic Stress on Cognition in Mice. Eur. J. Neurosci. 2019, 50, 2773–2785. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Matsuba, Y.; Mihira, N.; Takano, J.; Nilsson, P.; Itohara, S.; Iwata, N.; Saido, T.C. Single App Knock-in Mouse Models of Alzheimer’s Disease. Nat. Neurosci. 2014, 17, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Richards, J.G.; Higgins, G.A.; Ouagazzal, A.-M.; Ozmen, L.; Kew, J.N.C.; Bohrmann, B.; Malherbe, P.; Brockhaus, M.; Loetscher, H.; Czech, C.; et al. PS2APP Transgenic Mice, Coexpressing hPS2mut and hAPPswe, Show Age-Related Cognitive Deficits Associated with Discrete Brain Amyloid Deposition and Inflammation. J. Neurosci. 2003, 23, 8989–9003. [Google Scholar] [CrossRef]
- Ozmen, L.; Albientz, A.; Czech, C.; Jacobsen, H. Expression of Transgenic APP MRNA Is the Key Determinant for Beta-Amyloid Deposition in PS2APP Transgenic Mice. Neurodegener. Dis. 2009, 6, 29–36. [Google Scholar] [CrossRef]
- Lia, A.; Sansevero, G.; Chiavegato, A.; Sbrissa, M.; Pendin, D.; Mariotti, L.; Pozzan, T.; Berardi, N.; Carmignoto, G.; Fasolato, C.; et al. Rescue of Astrocyte Activity by the Calcium Sensor STIM1 Restores Long-Term Synaptic Plasticity in Female Mice Modelling Alzheimer’s Disease. Nat. Commun. 2023, 14, 1590. [Google Scholar] [CrossRef]
- Takano, T.; Han, X.; Deane, R.; Zlokovic, B.; Nedergaard, M. Two-Photon Imaging of Astrocytic Ca2+ Signaling and the Microvasculature in Experimental Mice Models of Alzheimer’s Disease. Proc. Ann. N. Y. Acad. Sci. 2007, 1097, 40–50. [Google Scholar] [CrossRef]
- Pirttimaki, T.M.; Codadu, N.K.; Awni, A.; Pratik, P.; Nagel, D.A.; Hill, E.J.; Dineley, K.T.; Parri, H.R. A7 Nicotinic Receptor-Mediated Astrocytic Gliotransmitter Release: Aβ Effects in a Preclinical Alzheimer’s Mouse Model. PLoS ONE 2013, 8, e81828. [Google Scholar] [CrossRef]
- Kuchibhotla, K.V.; Lattarulo, C.R.; Hyman, B.T.; Bacskai, B.J. Synchronous Hyperactivity and Intercellular Calcium Waves in Astrocytes in Alzheimer Mice. Science 2009, 323, 1211–1215. [Google Scholar] [CrossRef]
- Delekate, A.; Füchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 Receptor Signalling Mediates Astrocytic Hyperactivity in Vivo in an Alzheimer’s Disease Mouse Model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef]
- Reichenbach, N.; Delekate, A.; Breithausen, B.; Keppler, K.; Poll, S.; Schulte, T.; Peter, J.; Plescher, M.; Hansen, J.N.; Blank, N.; et al. P2Y1 Receptor Blockade Normalizes Network Dysfunction and Cognition in an Alzheimer’s Disease Model. J. Exp. Med. 2018, 215, 1649–1663. [Google Scholar] [CrossRef]
- Bosson, A.; Paumier, A.; Boisseau, S.; Jacquier-Sarlin, M.; Buisson, A.; Albrieux, M. TRPA1 Channels Promote Astrocytic Ca2+ Hyperactivity and Synaptic Dysfunction Mediated by Oligomeric Forms of Amyloid-β Peptide. Mol. Neurodegener. 2017, 12, 53. [Google Scholar] [CrossRef] [PubMed]
- Lines, J.; Baraibar, A.M.; Fang, C.; Martin, E.D.; Aguilar, J.; Lee, M.K.; Araque, A.; Kofuji, P. Astrocyte-neuronal Network Interplay Is Disrupted in Alzheimer’s Disease Mice. Glia 2022, 70, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Åbjørsbråten, K.S.; Skaaraas, G.H.S.; Cunen, C.; Bjørnstad, D.M.; Binder, K.M.G.G.; Bojarskaite, L.; Jensen, V.; Nilsson, L.N.; Rao, S.B.; Tang, W.; et al. Impaired Astrocytic Ca2+ Signaling in Awake-Behaving Alzheimer’s Disease Transgenic Mice. eLife 2022, 11, e75055. [Google Scholar] [CrossRef] [PubMed]
- Paukert, M.; Agarwal, A.; Cha, J.; Doze, V.A.; Kang, J.U.; Bergles, D.E. Norepinephrine Controls Astroglial Responsiveness to Local Circuit Activity. Neuron 2014, 82, 1263–1270. [Google Scholar] [CrossRef]
- Fischer, T.; Prey, J.; Eschholz, L.; Rotermund, N.; Lohr, C. Norepinephrine-Induced Calcium Signaling and Store-Operated Calcium Entry in Olfactory Bulb Astrocytes. Front. Cell. Neurosci. 2021, 15, 639754. [Google Scholar] [CrossRef]
- Speggiorin, M.; Chiavegato, A.; Zonta, M.; Gómez-Gonzalo, M. Characterization of the Astrocyte Calcium Response to Norepinephrine in the Ventral Tegmental Area. Cells 2024, 14, 24. [Google Scholar] [CrossRef]
- Squires, A.; Park, J. Emerging Roles of Astrocytes in Hippocampal Circuitry and Behavior. Front. Cell. Neurosci. 2025, 19, 1694643. [Google Scholar] [CrossRef]
- Ding, F.; O’Donnell, J.; Thrane, A.S.; Zeppenfeld, D.; Kang, H.; Xie, L.; Wang, F.; Nedergaard, M. A1-Adrenergic Receptors Mediate Coordinated Ca2+ Signaling of Cortical Astrocytes in Awake, Behaving Mice. Cell Calcium 2013, 54, 387–394. [Google Scholar] [CrossRef]
- Shah, D.; Gsell, W.; Wahis, J.; Luckett, E.S.; Jamoulle, T.; Vermaercke, B.; Preman, P.; Moechars, D.; Hendrickx, V.; Jaspers, T.; et al. Astrocyte Calcium Dysfunction Causes Early Network Hyperactivity in Alzheimer’s Disease. Cell Rep. 2022, 40, 111280. [Google Scholar] [CrossRef]
- Pascual-Caro, C.; Espinosa-Bermejo, N.; Pozo-Guisado, E.; Martin-Romero, F.J. Role of STIM1 in Neurodegeneration. World J. Biol. Chem. 2018, 9, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.F.; Russ, A.N.; Zhao, Q.; Perle, S.J.; Maci, M.; Miller, M.R.; Hou, S.S.; Algamal, M.; Zhao, Z.; Li, H.; et al. Optogenetic Targeting of Astrocytes Restores Slow Brain Rhythm Function and Slows Alzheimer’s Disease Pathology. Sci. Rep. 2023, 13, 13075. [Google Scholar] [CrossRef] [PubMed]
- Bindocci, E.; Savtchouk, I.; Liaudet, N.; Becker, D.; Carriero, G.; Volterra, A. Three-Dimensional Ca2+ Imaging Advances Understanding of Astrocyte Biology. Science 2017, 356, eaai8185. [Google Scholar] [CrossRef] [PubMed]
- Shigetomi, E.; Bushong, E.A.; Haustein, M.D.; Tong, X.; Jackson-Weaver, O.; Kracun, S.; Xu, J.; Sofroniew, M.V.; Ellisman, M.H.; Khakh, B.S. Imaging Calcium Microdomains within Entire Astrocyte Territories and Endfeet with GCaMPs Expressed Using Adeno-Associated Viruses. J. Gen. Physiol. 2013, 141, 633–647. [Google Scholar] [CrossRef]
- Thrane, A.S.; Thrane, V.R.; Zeppenfeld, D.; Lou, N.; Xu, Q.; Nagelhus, E.A.; Nedergaard, M. General Anesthesia Selectively Disrupts Astrocyte Calcium Signaling in the Awake Mouse Cortex. Proc. Natl. Acad. Sci. USA 2012, 109, 18974–18979. [Google Scholar] [CrossRef]
- Busche, M.A.; Chen, X.; Henning, H.A.; Reichwald, J.; Staufenbiel, M.; Sakmann, B.; Konnerth, A. Critical Role of Soluble Amyloid-β for Early Hippocampal Hyperactivity in a Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2012, 109, 8740–8745. [Google Scholar] [CrossRef]
- Shigetomi, E.; Saito, K.; Sano, F.; Koizumi, S. Aberrant Calcium Signals in Reactive Astrocytes: A Key Process in Neurological Disorders. Int. J. Mol. Sci. 2019, 20, 996. [Google Scholar] [CrossRef]
- Lim, D.; Iyer, A.; Ronco, V.; Grolla, A.A.; Canonico, P.L.; Aronica, E.; Genazzani, A.A. Amyloid Beta Deregulates Astroglial MGluR5-mediated Calcium Signaling via Calcineurin and Nf-kB. Glia 2013, 61, 1134–1145. [Google Scholar] [CrossRef]
- Shrivastava, A.N.; Kowalewski, J.M.; Renner, M.; Bousset, L.; Koulakoff, A.; Melki, R.; Giaume, C.; Triller, A. β-Amyloid and ATP-Induced Diffusional Trapping of Astrocyte and Neuronal Metabotropic Glutamate Type-5 Receptors. Glia 2013, 61, 1673–1686. [Google Scholar] [CrossRef]
- Aronica, E.; Van Vliet, E.A.; Mayboroda, O.A.; Troost, D.; Da Silva, F.H.L.; Gorter, J.A. Upregulation of Metabotropic Glutamate Receptor Subtype MGluR3 and MGluR5 in Reactive Astrocytes in a Rat Model of Mesial Temporal Lobe Epilepsy. Eur. J. Neurosci. 2000, 12, 2333–2344. [Google Scholar] [CrossRef]
- Kim, S.K.; Hayashi, H.; Ishikawa, T.; Shibata, K.; Shigetomi, E.; Shinozaki, Y.; Inada, H.; Roh, S.E.; Kim, S.J.; Lee, G.; et al. Cortical Astrocytes Rewire Somatosensory Cortical Circuits for Peripheral Neuropathic Pain. J. Clin. Investig. 2016, 126, 1983–1997. [Google Scholar] [CrossRef]
- Danjo, Y.; Shigetomi, E.; Hirayama, Y.J.; Kobayashi, K.; Ishikawa, T.; Fukazawa, Y.; Shibata, K.; Takanashi, K.; Parajuli, B.; Shinozaki, Y.; et al. Transient Astrocytic MGluR5 Expression Drives Synaptic Plasticity and Subsequent Chronic Pain in Mice. J. Exp. Med. 2022, 219, e20210989. [Google Scholar] [CrossRef]
- Paumier, A.; Boisseau, S.; Jacquier-Sarlin, M.; Pernet-Gallay, K.; Buisson, A.; Albrieux, M. Astrocyte–Neuron Interplay Is Critical for Alzheimer’s Disease Pathogenesis and Is Rescued by TRPA1 Channel Blockade. Brain 2022, 145, 388–405. [Google Scholar] [CrossRef]
- Zatti, G.; Burgo, A.; Giacomello, M.; Barbiero, L.; Ghidoni, R.; Sinigaglia, G.; Florean, C.; Bagnoli, S.; Binetti, G.; Sorbi, S.; et al. Presenilin Mutations Linked to Familial Alzheimer’s Disease Reduce Endoplasmic Reticulum and Golgi Apparatus Calcium Levels. Cell Calcium 2006, 39, 539–550. [Google Scholar] [CrossRef]
- Brunello, L.; Zampese, E.; Florean, C.; Pozzan, T.; Pizzo, P.; Fasolato, C. Presenilin-2 Dampens Intracellular Ca2+ Stores by Increasing Ca2+ Leakage and Reducing Ca2+ Uptake. J. Cell. Mol. Med. 2009, 13, 3358–3369. [Google Scholar] [CrossRef]
- Kipanyula, M.J.; Contreras, L.; Zampese, E.; Lazzari, C.; Wong, A.K.C.; Pizzo, P.; Fasolato, C.; Pozzan, T. Ca2+ Dysregulation in Neurons from Transgenic Mice Expressing Mutant Presenilin 2. Aging Cell 2012, 11, 885–893. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.