Abstract
OGG1 and MUTYH are base excision repair (BER) DNA glycosylases (DGs) from the Helix–hairpin–Helix superfamily responsible for initiating and coordinating the repair of 8-oxo-7,8-dihydroguanine (OG), and its replication-derived mispair with adenine (OG:A), respectively. The DNA repair activities of these DGs are pivotal to safeguarding nuclear and mitochondrial genomes. Indeed, DG functional impairment is associated with numerous pathologies, including neurodegenerative diseases, metabolic syndromes, and cancer. The timely and precise localization and processing of oxidized nucleobases carried out by these DGs are modulated by a complex regulatory network at both transcriptional and posttranslational levels, as well as intricate protein–protein interaction networks. In the absence of regulation, inappropriate and imbalanced DG activity may trigger telomeric instability, changes in transcriptional profiles and cell death. This review focuses on summarizing key features of OGG1 and MUTYH function, with a special emphasis on structure, regulation, and novel emerging roles.
1. Introduction
A hallmark of oxidative DNA damage is the oxidation of guanine to 8-oxo-7,8-dihydroguanine (OG), which forms and distributes unevenly within our genome, with basal levels estimated around 10,000 OG per mammalian cell [1,2]. The highly oxidizing environment of mitochondria results in a 10-fold higher accumulation of OG than in the nucleus. The molecular issue with OG pertains to its highly mutagenic nature, which is underscored by the fact that DNA polymerases tend to incorporate an adenine (A) opposite OG, inducing G:C→T:A transversion mutations [3] (Figure 1A).
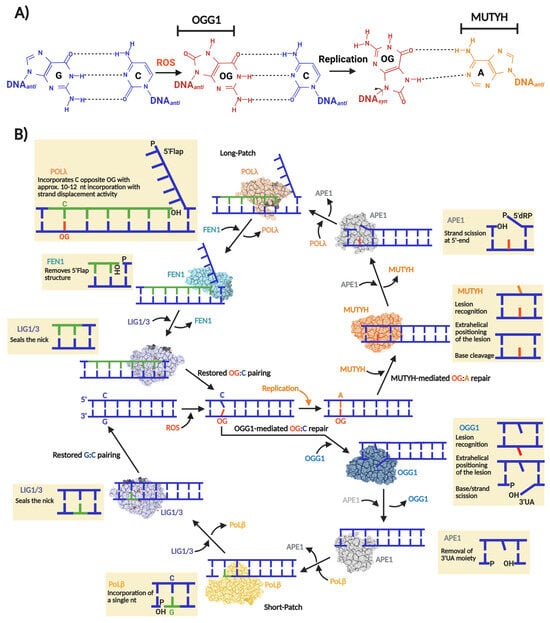
Figure 1.
Base excision repair of OG:C and OG:A in human cells. (A) OG miscoding potential arises from anti versus syn nucleotide conformations that allow for distinct base pairing patterns. The syn conformation of OG presents the Hoogsteen face, instead of the canonical Watson–Crick face, favoring the incorporation of an adenine opposite OG during a replication event initiating a GC→TA transversion. The nucleobases subjected to OGG1 and MUYTH repair are indicated. (B) “One-reaction–one enzyme” scheme representing the flow of enzymatic reactions to repair OG:C and OG:A via OGG1- and MUTYH-mediated BER short- and long-patch, respectively. In the case of OG:C repair, OGG1, as bifunctional DG, carries out OG excision producing an AP site that thereafter is converted to 3′-α,β-unsaturated aldehyde (3′UA) via β-elimination. The 3′UA is processed by APE1 leaving a 3′-OH nick which is used by Polβ and a DNA ligase (LIG1 or LIG3) to incorporate a G and ligate the nick, respectively. In the case of OG:A repair, MUTYH, as monofunctional DG, carries out only the adenine excision opposite OG, producing an AP site which in turn is processed by APE1 leaving a 5′-2-deoxyribose-5-phosphate (5′dRP) and a 3′-OH nick. Then, Polλ incorporate several nucleotides (10–12) generating a 5′Flap structure. To consolidate the repair, the 5′Flap is processed by FEN1 and the remaining nick ligated by LIG1 or LIG3.
The replication-associated miscoding of OG is problematic across organisms in all domains of life. Consequently, a highly conserved enzymatic pathway in archaea, bacteria, and eukaryotes mitigates OG-induced mutagenesis. This pathway, first described by Michaels and Miller as the “Guanine Oxidation” (GO) repair system, consists of three enzymes: MutM, MutY, and MutT [4,5]. MutM, also known as formamidopyrimidine glycosylase (Fpg), is a Helix–2Turn–Helix (H2TH) DNA glycosylase found primarily in bacteria and archaea that excises OG paired with cytosine, generating an a apurinic/apyrimidinic (AP) site subsequently processed by BER to restore the G:C base pair [6,7]. If OG escapes MutM-mediated repair, the pro-mutagenic OG:A mispair is generated after replication. MutY removes adenine opposite OG, producing an AP site that is required to regenerate OG:C, thereby allowing MutM another opportunity for lesion removal [8].
MutT, the third component of the GO system, is not a DNA glycosylase. Instead, it is d(OG)TPase that hydrolyzes oxidized d(OG)TPs, preventing their incorporation into DNA by DNA polymerases [9]. Initially, the GO system was described in Escherichia coli, but there was substantial evidence of its existence in other prokaryotes and eukaryotes [4]. Today, it is well documented that such a mechanism is present, at least partially, in most genera of each domain of life [10,11]. Particularly, in human cells, the GO system is encoded by the MUTYH and MTH1 genes, whose protein products are MutY and MutT homologs, respectively. Regarding MutM, the human genome does not harbor a homolog which acts directly and efficiently on OG:C lesions. Instead, OGG1 is the human OG DNA glycosylase that belongs to the Helix–Hairpin–Helix (HhH) DG superfamily [11,12]. Figure 1B shows a scheme of the human OGG1- and MUTYH-mediated BER.
Woven within the many years of studying of OGG1 and MUTYH are threads indicating functional roles beyond DNA repair. For example, OGG1 is involved in modulating the expression of several genes involved in immunological response and carcinogenesis [13,14,15]. Telomeres undergoing oxidative stress accumulate OG, ultimately leading to rapid cellular senescence [16], as a consequence of replicative stress mediated by OGG1 and MUTYH [17]. Similarly, MUTYH-initiated BER functions have been shown to act as a molecular switch for tumor suppression, inducing apoptosis in cells burdened with excessive oxidative DNA damage [18,19]. Herein, we aim to summarize what is known about OGG1 and MUTYH transcriptional and posttranslational regulation, as well as what is known about how repair activity is modulated by protein–protein interactions (PPIs). We previously reviewed the structural and biochemical properties of these enzymes as members of the HhH DNA glycosylase superfamily [11].
2. OGG1
OGG1 is the main DG in charge of the direct removal of OG opposite C in human cells [11]. OGG1 can also excise the ring-opened lesion 2,6-diamino-4-hydroxy-5-formamidophyrimidine (FapyG) with comparable efficiency [20]. Although both lesions are miscoding, their distinct structures lead to different mutagenic outcomes during DNA replication [21]. OG adopts a rigid planar structure with syn and anti conformers, arising from rotation about the N-glycosidic bond. The syn conformer of OG presents its thymine-mimicking Hoogsteen pairing face to the DNA polymerase to mediate A mis-insertion and ultimately GC→TA transversion (Figure 1A). In contrast, the cleavage of the 5-membered ring in FapyG allows for more varied structural conformations rendering different promutagenic outcomes. This includes GC→AT transitions, as well as GC→TA and GC→CA transversions [21,22,23]. The oxidation of G to OG within the genome is estimated to occur with the high frequency of 500 to 1000 lesions cell−1 day−1 [24] and both OG and FapyG are accumulated at a similar extent when cells are irradiated with UV light [25]. However, FapyG is detected at approximately threefold higher levels than OG in human leukemia cells exposed to ionizing radiation [26,27]. In contrast, hydroxyl radical oxidation under physiological conditions does not produce significant amounts of FapyG [28,29], suggesting that this lesion is unlikely to arise endogenously, but may be a factor to consider in radiation therapy. Several additional DNA glycosylases contribute to the repair of FapyG lesions, including NTHL1, NEIL1, and NEIL3, which process FapyG in distinct DNA contexts [30,31,32,33,34,35]. NEIL1 exhibits negligible activity on OG:C lesions [36,37], underscoring the central role of OGG1 in OG repair.
The importance of OGG1-mediated repair is reflected in its association with multiple pathologies, including metabolic syndromes [38,39], inflammatory diseases [13,40], cancer [41,42], and neurological diseases like Alzheimer’s [43,44] and Parkinson’s [45]. Moreover, OGG1 and MUTYH might play an evolutionary role in maintaining a balanced GC content in eukaryotic genomes [10,46], as defective OG repair imposes strong selective pressure that biases genomes toward increased TA content.
2.1. OGG1 as a Transcriptional Modulator
OG is unevenly distributed throughout the genome [1,47,48], governed in part by chromatin topology and DNA sequence context. GC-rich sequences, such as those localized in cis-regulatory elements of promoters, are the most susceptible to oxidative DNA damage, and therefore, OG accumulation [1,2]. Notably, more than 70% of human promoters are GC-rich regions [49]. Consequently, OG—and by extension OGG1—may exert significant influence over promoter dynamics and gene transcription. Interestingly, OGG1’s influence over promoter activity is mediated by two distinct modus operandi; one relies simply on unproductive OG recognition within promoters, and the other promotes or prevents G-quadruplex formation (Figure 2). Both mechanisms initiate signaling cascades that modulate transcription via transcriptional machinery recruitment.
A series of studies from the Boldogh laboratory established a link between OG formation, OGG1, and inflammatory signaling. Early evidence from the BALB/c mouse models of airway inflammation showed that reduced OGG1 expression attenuates airway hyperresponsiveness, implicating OGG1 in proinflammatory response [50], indicating that OGG1 was participating in proinflammatory responses. OGG1 expression enhances TNFα induced activation of the proinflammatory cytokine CXCL2 [51]. Among the different molecular events triggered by TNFα is an almost immediate (within 30 min) increment of intracellular ROS [52], OG accumulation in GC-rich promoters of proinflammatory cytokine encoding genes [51,53], and release and nuclear translocation of NF-κB, a master transcriptional factor (TF) that regulates inflammation [54]. ChiP analyses revealed enrichment of both OGG1 and NF-κB enrichment at TNF responsive promoters; however, OGG1 binding did not coincide with reduced OG levels, indicating that NF-κB recruitment is independent of OG excision [53,55]. Instead, OGG1 enzymatic activity is transiently suppressed following TNFα exposure through reversible cysteine oxidation, as evidenced by recovery with DTT [53]. These findings support a model in which TNFα -induced ROS inactivates OGG1, enabling nonproductive binding at OG-enriched GC-rich promoters. This inactive OGG1 scaffold facilitates recruitment of transcription factors such as NF-κB, thereby promoting proinflammatory gene expression (Figure 2A) [47]. Of note, it was reported that OG is able to modulate gene expression without need of its repair [56]. Moreover, several innate immune cells are hypersensitive to DNA oxidation due to insufficient downstream BER activity, ultimately leading to apoptosis [57,58,59]. These observations highlight critical roles for both OG and OGG1 in shaping inflammatory transcriptional responses and immune cell fate.

Figure 2.
Role of OGG1 in gene transcription. (A) Transcriptional induction exerted by OGG1 by non-productive mechanism in which OG recognition is required without glycosylase activity. TNFα-induced ROS production inactivates OGG1’s glycosylase activity probably by means of cysteine oxidation without affecting OG recognition. When OGG1 recognizes and binds OG within proinflammatory gene promoters, it recruits the transcriptional factor NF-κB, which in turn recruits the transcriptional machinery to initiate transcription. (B) Transcriptional induction/repression exerted by OGG1 by productive mechanism that implies OG excision and G-quadruplex formation. When OG is formed within the coding strand, its excision by OGG1 forms an AP site causing a concomitant thermodynamically unfavorable form of DNA forcing to adopt a thermodynamically favorable G-quadruplex structure. The AP site in the G-quadruplex is recognized by APE1, which in turn recruits the transcriptional machinery. In the case that the G-quadruplex is formed within the template strand, transcriptional repression results; modified from [60]. (C) G-quadruplex structure of human c-MYC promoter solved by NMR (PDB 2LBY; [61]).
Figure 2.
Role of OGG1 in gene transcription. (A) Transcriptional induction exerted by OGG1 by non-productive mechanism in which OG recognition is required without glycosylase activity. TNFα-induced ROS production inactivates OGG1’s glycosylase activity probably by means of cysteine oxidation without affecting OG recognition. When OGG1 recognizes and binds OG within proinflammatory gene promoters, it recruits the transcriptional factor NF-κB, which in turn recruits the transcriptional machinery to initiate transcription. (B) Transcriptional induction/repression exerted by OGG1 by productive mechanism that implies OG excision and G-quadruplex formation. When OG is formed within the coding strand, its excision by OGG1 forms an AP site causing a concomitant thermodynamically unfavorable form of DNA forcing to adopt a thermodynamically favorable G-quadruplex structure. The AP site in the G-quadruplex is recognized by APE1, which in turn recruits the transcriptional machinery. In the case that the G-quadruplex is formed within the template strand, transcriptional repression results; modified from [60]. (C) G-quadruplex structure of human c-MYC promoter solved by NMR (PDB 2LBY; [61]).

The transcriptional activation mechanism that relies on OGG1’s enzymatic activity is associated with G-quadruplex structures. As mentioned, GC-rich promoters are susceptible to formation of OG. Many of these promoters harbor repeating arrays of guanines (Gn≥3N1–7Gn≥3N1–7 Gn≥3N1–7Gn≥3), which may fold to form three-dimensional DNA structures with stacked quartets of guanines stabilized by Hoogsteen base pairing and potassium ion coordination [62] (Figure 2B). Genome-wide G-quadruplex-seq analyses have identified over 700,000 G-quadruplexes in the human genome [63], of which approximately 10,000 are mapped within promoters and 5′-UTR regions [64]. Moreover, many DNA repair gene promoters are predicted to form G-quadruplex structures, including those from BER genes such as: NTHL1, PCNA, FEN1, NEIL1, and NEIL3 [64,65,66], as well as oncogenes promoters like KRAS and VEGF [67,68]. Therefore, G-quadruplexes can up- or downregulate repair of oxidative DNA damage, and indirectly, their own formation.
Burrows and co-workers proposed that formation of G-quadruplexes within potential G-quadruplex forming sequences (PQS) requires a series of destabilizing steps [69]. Oxidation of a 5′ G to form OG within the PQS and subsequent OG excision by OGG1 and formation of an AP site combine to destabilize local duplex formation, favoring folding into a G-quadruplex structure. APE1 then recognizes the AP site and recruits transcription factors to initiate transcription (Figure 2C). Notably, APE1’s pro-transcriptional function requires AP-site binding, but not endonuclease activity [70], similar to OGG1 induction of proinflammatory genes. The regulatory outcome of this pathway depends on G-quadruplex positioning; structures formed on the coding strand promote transcription, whereas those on the template strand repress gene expression [60]. Beyond this mechanism, additional roles for OGG1 in transcriptional regulation have been reported [69,71,72].
2.2. OGG1 Gene Structure and Regulation
The human OGG1 gene is located in the short arm of chromosome 3 and has a length of 7456 bp. Thus far, 12 distinct functional splice variants have been identified and are produced as a product of the swapping of 8 exons (Figure 3). The most studied isoforms are the α- (1a) and β-isoforms (2a) [73]. Interestingly, all isoforms encode for a mitochondrial localization signal (MLS) at the beginning of the coding region of the mRNA. The α-variants also contain a nuclear localization signal (NLS) at the 3′-end [74]. This means that the NLS is the dominant localization signal for OGG1 intracellular localization.
2.2.1. OGG1 Promoter and Transcriptional Regulation
Initially OGG1 was thought to be a housekeeping gene with relatively consistent expression levels maintained throughout the cell cycle [75]. However, tissue-specific expression data from the Human Protein Atlas reveal substantial variability in OGG1 levels, with particularly high expression in the kidney and lymph nodes (Figure 3, [76]). For instance, in kidney cells, the cell cycle control and cell proliferation processes are under tight regulation, with growth rates less than 1% [77] and a particular arrest in G1/0 of the cell cycle [78]. The organ’s high energetic demand leads to persistent oxidative stress [79] resulting in progressive OG accumulation over the life-span [80]. These observations suggest that OGG1 plays a critical role in removing OG during periods of cell cycle arrest, thereby preventing the accumulation of G:C→T:A transversion mutations prior to DNA replication.
The OGG1 promoter region was initially studied by Dhénaut and colleagues (2000) [75]. They identified that the promoter region up to position −135 is the minimum active component required to maintain transcription. Moreover, they identified several putative transcriptional factor (TF) binding sites, including sites for SP1 and the nuclear respiratory factor (NRF1; Figure 3). The latter is a specialized TF which regulates antioxidant genes and response to oxidative stress [81]. Further studies demonstrated that downregulation of NRF1 reduces OGG1 expression, establishing NRF1 as a key transcriptional regulator of OGG1 [82]. Disruption of this regulation has been linked to impaired handling of oxidative DNA damage in diabetes [82] and estrogen-induced breast cancer [83]. The SP1 binding site was mapped in a segment containing positions −474 to −431 within the OGG1 promoter [84]. SP1 regulates genes that control cell cycle control, apoptosis, hormonal activation, and other processes [85]. Notably, cadmium exposure suppresses SP1 binding at the OGG1 promoter, leading to reduced OGG1 expression [84]. Cadmium is a widespread environmental carcinogen that induces chromosomal aberrations, DNA stand breaks, and OG accumulation [86,87]. Thus, the inhibition of SP1 binding to the OGG1 promoter might play an important role in Cadmium-induced DNA damage and carcinogenesis.
Another set of TFs identified as regulators of OGG1 are the Nuclear Transcription Factor Y-A (NF-YA) [88,89], AP-4 [90], and p53 [91]. Of note, the tumor suppressor p53 regulates different DNA repair pathways, either indirectly or directly [92,93] including several BER genes, such as MUTYH [18], APE1 [94], and Polβ [95]. These transcription factors regulate cell-cycle progression and their dysfunction is linked to colorectal, gastric, and hepatic cancers [96,97,98]. Recently, it has been reported that OGG1 regulates its own expression [99]. Geng et al. demonstrated that OGG1 is recruited to and physically binds its own promoter, a region which spans from position −297 to +3, to increase OGG1 expression and OG repair. This recruitment is enhanced by OGG1’s interaction with SIRT2, an NAD+-dependent sirtuin deacetylase that targets histones H3 and H4 [100,101]. However, the OGG1-SIRT2 interaction and induction of OGG1 transcription are independent of acetylation/deacetylation. Instead, it relies on upstream events such as SIRT2 phosphorylation in an ATM/ATR-dependent manner. Corroborating this finding, the OGG1-SIRT2 interaction is enhanced in cells subjected to oxidative stress [99], suggesting that oxidative DNA damage serves as the initiating signal for OGG1 transcriptional activation.

Figure 3.
Promoter and mRNA structures of OGG1 and its tissue-specific mRNA and protein expression. (Upper Panel). Transcriptional factor binding sites mapped within OGG1 promoter. Transcriptional factors (TFs) or proteins controlling OGG1 expression with their functional annotation are shown. (Lower Panel). GC rich regions are indicated with asterisks. Tissue-specific protein and mRNA expression of OGG1. Expression profiles were retrieved from the Human Protein Atlas database [102]. Degree of protein expression is normalized based on None, Low, Medium, and High, as reported in the database. mRNA expression is reported as normalized expression (nTPM) from a consensus dataset combining transcriptomics from the Human Protein Atlas (HPA) and Genotype-Tissue Expression (GTEx) RNA-seq datasets.
Figure 3.
Promoter and mRNA structures of OGG1 and its tissue-specific mRNA and protein expression. (Upper Panel). Transcriptional factor binding sites mapped within OGG1 promoter. Transcriptional factors (TFs) or proteins controlling OGG1 expression with their functional annotation are shown. (Lower Panel). GC rich regions are indicated with asterisks. Tissue-specific protein and mRNA expression of OGG1. Expression profiles were retrieved from the Human Protein Atlas database [102]. Degree of protein expression is normalized based on None, Low, Medium, and High, as reported in the database. mRNA expression is reported as normalized expression (nTPM) from a consensus dataset combining transcriptomics from the Human Protein Atlas (HPA) and Genotype-Tissue Expression (GTEx) RNA-seq datasets.

2.2.2. Epigenetic Control of OGG1
As a GC rich promoter including several SP1 binding sites [75,84], the OGG1 promoter is epigenetically active and linked to cellular responses to pesticide exposure, arsenic genotoxicity, and several diseases. For instance, methyl parathion, an oxidizing organophosphate pesticide that induces oxidative stress in germ cells [103] causes global hypomethylation and an increase in promoter-specific methylation of OGG1 at two CpG sites [104]. Likewise, Wang et al. (2021) showed that arsenic exposure in human bronchial epithelial (HBE) cells induces oxidative stress with concomitant inhibition of TET-mediated DNA demethylation, resulting in OGG1 hypermethylation and reduced protein expression [105]. Another physiological challenge that affects OGG1 methylation is the prolonged exposure to estrogen 17 beta-estradiol, which is associated with breast cancer [106]. In a prospective longitudinal cohort study including 582 male participants from the Boston, USA, the DNA methylation levels of different genes in blood leukocytes were sampled over the course of nine years. The results indicated that the OGG1 promoter region was methylated, which is associated with certain types of cancer, including prostate [107].
Oxidative DNA damage and OG accumulation are biomarkers for neurodegeneration and aging [80,108,109]. In fact, the low expression of OGG1 and MTH1 is a hallmark of Alzheimer’s disease (AD) [110,111]. These data suggest that the partial dysfunction of the GO system exacerbates the progression of AD. Interestingly, there is evidence that this correlation might have an epigenetic origin. Italian and Chinese late-onset AD cohorts reported modest but detectable changes in OGG1 promoter methylation in peripheral blood cells, including APOE ε4 carriers [112,113]. However, in a Polish population, OGG1 promoter methylation accompanied by reduced mRNA expression in AD patients [114]. Although these differences may reflect ethnic or genetic variability [115], altered methylation of the OGG1 promoter emerges as a recurring feature associated with AD and other diseases.
2.3. OGG1 Protein Structure and Regulation
In 2001, the Verdine laboratory reported the first crystal structure of OGG1 [116]. Distinct from other HhH enzymes, OGG1 is a modular DG composed of two domains; the HhH-GDP superfamily domain and the TATA-binding protein (TBP)-like domain. The latter domain does not make contact with the DNA and has been proposed to serve as a scaffold for PPIs, based on being the locus for binding of the DNA break sensor Poly (ADP-ribose) polymerase 1 (PARP-1) [117]. The HhH-GDP domain carries out OG recognition, DNA engagement, and catalysis. It comprises two α-helical subdomains linked by the featured HhH motif (Figure 4). These subdomains flank the catalytic pocket where catalytic residues Asp268 and Lys249 participate in the excision of OG opposite C, and strand scission at the 3′ end of the produced AP site via β-elimination [116].
Regulation of OGG1 at the protein level occurs via posttranslational modifications (PTM) and PPIs. Particularly, PTMs (phosphorylation, acetylation, ubiquitination) are able to control enzymatic activity, protein stability, intracellular localization, and even PPIs [118] (Table 1). This type of protein regulation has been described as pivotal in maintaining genomic integrity [119].
2.3.1. Ubiquitination
Several studies have indicated that OGG1 may be ubiquitinated. An initial report showed that OGG1 is ubiquitinated in response to hyperthermia, namely in Hela cells at 42 °C [120]. In this study a marked decrease of OGG1 activity was found after 2 h of thermic treatment. The decrease in OGG1 DG activity was explained by two means; (1) heat-induced misfolding, and (2) a diminishment in OGG1 protein levels mediated by proteasomal degradation. The authors identified the C-terminus of HSC70-interacting protein (CHIP) as the E3 ligase involved in the ubiquitination of OGG1 in hyperthermic conditions. However, the residues of OGG1 subjected to ubiquitination were not identified. A study by Parson and co-workers identified NEDD4L as an E3 ubiquitin ligase targeting OGG1 specifically at residue Lys341 [121]. Interestingly, in cellulo studies where NEDDL4 downregulation is carried out in U2O2 cell lines demonstrated that OGG1 protein stability is enhanced under ionizing radiation-induced oxidative stress. Moreover, OGG1 ubiquitination inhibits its glycosylase/lyase activity in vitro. The authors correlated the prolonged OGG1 stability and the enhanced DG activity with poor cell survival post irradiation.

Figure 4.
OGG1 primary and quaternary structures in complex with DNA and their protein–protein interaction network. On top, a scheme of the amino acid sequence is shown highlighting (in different colors) each functional domain. Residues subjected to posttranslational modifications and regions involved in protein–protein interactions are indicated. Ubiquitination, acetylation and phosphorylation are indicated with asterisk in green, clear blue and red colors, respectively. The crystallographic structure of OGG1 (PDB 1FN7 [116]) is shown in different colors for each functional domain. The catalytic cavity is shown in cyan and the DNA in black. The protein–protein interaction network shows protein partners for OGG1 based on the BioGRIG database (Blue, [122]) and based on literature (Red). The protein partners found in BioGRID were filtered based just on physical interactions. The figure of OGG1 structure was generated in PyMOL version 3.0.4.
Figure 4.
OGG1 primary and quaternary structures in complex with DNA and their protein–protein interaction network. On top, a scheme of the amino acid sequence is shown highlighting (in different colors) each functional domain. Residues subjected to posttranslational modifications and regions involved in protein–protein interactions are indicated. Ubiquitination, acetylation and phosphorylation are indicated with asterisk in green, clear blue and red colors, respectively. The crystallographic structure of OGG1 (PDB 1FN7 [116]) is shown in different colors for each functional domain. The catalytic cavity is shown in cyan and the DNA in black. The protein–protein interaction network shows protein partners for OGG1 based on the BioGRIG database (Blue, [122]) and based on literature (Red). The protein partners found in BioGRID were filtered based just on physical interactions. The figure of OGG1 structure was generated in PyMOL version 3.0.4.

Ubiquitination has also been suggested to mediate the repair of OGG1-DNA crosslinks implicated to form in cells [123]. DNA–protein crosslinks are potent contributors to genome instability, aging, and cancer [124]. The OGG1-DNA crosslinks form by Schiff base formation between Lys residues of OGG1 and 3′-phospho-α,β-unsaturated aldehyde (3′UA) intermediates arising from OGG1-mediated β-elimination at the AP site product of OGG1 glycosylase activity (Figure 1) [11,125]. Lys249 can form a Schiff base with the 3′-UA, and in vitro, the Schiff base can be detected via reduction with NaBH4 treatment to form a stable DNA–protein crosslink [12,125]. OGG1-DNA crosslinks can be repaired by two different mechanisms, both mediated by ubiquitination: NER or homologous recombination [126]. Specifically, OGG1 polyubiquitination of Lys341 via Lys48 ubiquitination leads to proteasomal degradation and resolution of the lesion via NER. However, when OGG1 is polyubiquitinated by Lys63 ubiquitination, the lesion is resolved via homologous recombination in a proteasome-independent manner.

Table 1.
Posttranslational modifications (PTM) and protein–protein interaction (PPI) controlling OGG1.
Table 1.
Posttranslational modifications (PTM) and protein–protein interaction (PPI) controlling OGG1.
| PTM | Site | Effect [Reference] |
|---|---|---|
| Ubiquitination | K341 | ↓ glycosylase/lyase activity [121], ↑ proteasomal degradation [126] |
| K289 | Unknown [127] | |
| K298 | Unknown [128] | |
| Phosphorylation | S326 | ↑ glycosylase activity [129] |
| Acetylation | K338/K341 | ↑ turnover [130,131] |
| PPI | Effect [Reference] | |
| Polβ | ↑ turnover [132] | |
| Polλ | Unknown [132] | |
| XPC | ↑ turnover [133] | |
| UV-DDB | ↑ turnover [134] | |
| RAD52 | ↑ turnover [135] | |
| PARP1 | ↓ glycosylase activity [117] | |
| XRCC1 | ↑ glycosylase and lyase activity [136] | |
| RAD9-RAD1-HUS1 | ↑ glycosylase and lyase activity [137] | |
Up-arrow (↑) and down-arrow (↓) symbols indicate an enhancement or diminishment of the referred biochemical parameter.
OGG1’s Lys289 [127] and Lys297 [128] have also been identified as targets for ubiquitination through high-accuracy mass spectrometry approaches. However, there is no characterization of the biochemical and cellular effects of ubiquitination at these residues. Nonetheless, ubiquitination clearly serves as a regulatory mechanism for OGG1 activity, facilitating the removal of potentially lethal intermediates generated during BER. This underscores the importance of these PTMs in fine-tuning repair and preventing futile DNA processing.
2.3.2. Phosphorylation
The first PTM reported for OGG1 was serine phosphorylation, described in 2002 [138]. Although the modified residue was not identified, chromatin-associated OGG1 was shown to co-precipitate with protein kinase C (PKC), and in vitro phosphorylation experiments demonstrated that this PTM was carried out by the α, β, and γ isoforms of PKC. This modification does not impact OGG1 glycosylase activity, but instead regulates its nuclear localization [138]. Phosphorylated OGG1 associates with chromatin, while unphosphorylated protein localizes to the nuclear matrix. OGG1 is found in both compartments during interphase, but association to condensed chromatin is specific to mitosis. These findings suggest that PKC-mediated phosphorylation spatially regulates OGG1, potentially facilitating OG detection and repair across distinct chromatin states and phases of the cell cycle.
Additional kinases have been reported to phosphorylate OGG1, including the serine kinase CDK4 and threonine kinase c-Abl [129]. Although the OGG1 residues these kinases target remain unknown, the in vitro phosphorylation carried out by Cdk4 enhanced OGG1 glycosylase activity 2.5-fold. However, phosphorylation by c-Abl did not cause any changes in biochemical behavior. In silico analysis has identified Ser326 as a potential phosphorylation site. Notably, this residue corresponds with a cancer associated polymorphism (S326C) [139,140,141], whose functional relevance is underscored by defects in nuclear localization [142] and increased genomic instability in homozygous cells [143].
2.3.3. Acetylation
OGG1 acetylation was first reported by the Mitra laboratory in 2006, showing that it was acetylated in HeLa cells [130] with an 2.5-fold increase in response to oxidative stress. The acetyltransferases p300 was identified as being responsible for OGG1 acetylation, mainly at Lys338 and Lys341. In vitro studies demonstrated that this PTM significantly increases OGG1 turnover mediated by APE1. Likewise, an interaction of the deacetylase HDAC1 interaction with OGG1 was detected in immunoprecipitation experiments [130]. Lys338 and Lys341 acetylation have been observed in age-related cataracts (ARCs), a common eye disease in the elderly [144]. Immunoprecipitation and siRNA assays confirmed that p300 was the major acetyltransferase in lens epithelium cells and demonstrated that the deacetylase SIRT1 actively removes acetyl groups from OGG1. OG accumulation in an ARC could result from an imbalance of OGG1 interactions with p300 and SIRT1, favoring deacetylation and reduced OG removal. Similar regulatory patterns have been observed in human skeletal muscle following exercise [130] and in rat hippocampal cells [145].
In contrast, SIRT3 is an NAD+-dependent deacetylase with mitochondrial activity. Indeed, mitochondrial OGG1 physically interacts with and is deacetylated by SIRT3, a modification which stabilizes OGG1, enhances OG excision, and protects genomic integrity by limiting apoptosis during oxidative stress [146]. These findings collectively underscore the importance of acetylation in regulating OGG1 activity.
2.4. OGG1 Regulation by Protein–Protein Interactions
OGG1’s PPIs have been a topic of interest since early 2000 [147,148,149]. In Figure 4, an updated PPI network of OGG1 is displayed, highlighting protein partners involved in different cellular processes including transcriptional regulation, chromatin remodeling, proteasomal degradation, cell cycle control, and cellular differentiation. As expected, OGG1 interacts with AP-site signaling proteins involved in BER (PARP1 and XRCC1), as well as with NER through its interaction with ERCC8 and ERCC6. This complex and broad PPI network strongly suggests that expression and activity of OGG1 is fine-tuned by different cellular processes and vice versa. In this section, we will delineate an updated version of OGG1’s PPI network, focusing on recent reports and newer interpretations.
2.4.1. Regulation of OGG1 Turnover
Most DGs bind their product (AP-containing DNA) more tightly than their substrate in vitro [148]. Particularly, OGG1 forms a stable complex with DNA with a half-life of 18 min [150]. This stable complex is due to OGG1’s 400-fold tighter affinity for AP: C-containing DNA than its substrate, OG:C (Kd < 0.005 vs. 2 nM, respectively) [151]. Such differences in binding renders a markedly slow turnover (k3 = 0.04–0.08 min−1) [34,152]. In stark contrast is APE1’s turnover, with a rate of at least 2.7 min−1 [153]. This suggests that OGG1’s turnover is the rate limiting step in OG processing, and potentially a regulatory point for BER. OGG1’s turnover is stimulated by other DNA repair enzymes like APE1 [154,155,156] and NEIL [34]. Interestingly, despite OGG1 turnover stimulation, there is no physical interaction reported with APE1 nor NEIL1. The mechanism proposed implies a transient release of the AP site by OGG1 that allows its rapid and opportunistic binding and processing by APE1, preventing retrograde OGG1 binding [157]. The Zharkov Lab demonstrated that Polβ and OGG1 interact with an affinity of 580 nM (Kd) and such interaction favors OGG1’s turnover by means of disrupting OGG1-DNA complexes, potentially by displacement of OGG1 from its product by passive diffusion of Polβ [158].
Other PPI reported to promote OGG1 turnover are Xeroderma pigmentosum C (XPC) and the UV-damaged DNA binding (UV-DDB) NER proteins. Both participate in recognizing UV-induced lesions like cyclobutene pyrimidine dimers (CPD) and (6-4) photoproducts, and recruitment of downstream NER proteins to the repair site [133,134]. Far western analysis demonstrated that XPC and OGG1 physically interact, resulting in enhanced OGG1 turnover [133]. More recently, the Van Houten Lab demonstrated that UV-DDB has 2-fold higher affinity for AP sites than OGG1 [134]. Consequently, UV-DDB promotes OGG1’s turnover by competing for the AP site [157]. Similar results were reported for MUTYH (see Section 3.4.3). Another partner for OGG1 is the homologous recombination protein RAD52. This interaction displays contrasting biochemical effects with inhibition of RAD52, while OGG1 turnover is stimulated [135]. The interaction of OGG1 with RAD52 suggests a regulatory mechanism of some sort but the details and implicated remain to be clarified
2.4.2. OGG1 Interactions with DNA Strand Break Signaling Proteins
As stated, OGG1 interacts through its TATA binding-like domain with protein the C-terminal domain of PARP-1 in vitro and in cellulo [117]. PARP-1 plays a role in repairing ssDNA breaks through PARylation of itself and surrounding proteins when bound to ssDNA breaks derived from AP sites processed by APE1. Enhanced PARP-1 PARylation activity leads to OGG1 inhibition. This inhibition only occurs upon incubation with active PARP-1 in the presence of its cofactor NAD+. Although the mechanisms of OGG1 PARylation are not completely understood, its role in modulating OG repair may help explain the etiology of several OGG1-associated pathologies.
The X-ray repair cross-complementing protein 1 (XRCC1), functions as a molecular scaffold that interacts with a variety of DNA repair proteins to signal and accelerate the repair of ssDNA breaks [159]. Marsin et al. (2003) reported a physical and functional interaction between OGG1 and XRCC1 which enhances up to 3-fold and 2-fold the OGG1 glycosylase activity and Schiff base intermediate formation associated to lyase activity, respectively [136]. Interestingly, XRCC1 and APE1 have a synergistic effect on OGG1 glycosylase activity. A structural mapping of the interaction between XRCC1 and OGG1 showed that two regions participate in the interaction, the BRCT1 and a hinge region between the N-terminal domain and BRCT1 of XRCC1. Surprisingly, APE1 binds to both sites, as well. These results suggest that XRCC1 functions as a facilitator of the AP-site hand off between OGG1 and APE1, enhancing the processivity of at least the first two reactions in the coordinated repair of OG.
2.4.3. OGG1 Interaction with Cell-Cycle Control Proteins
Both OGG1 and MUTYH have been reported to interact with the 9-1-1 checkpoint complex [137,160], a heterotrimer comprising Rad9, Rad1, and Hus1 subunits, adopting a donut-like structure similar to PCNA [161]. The 9-1-1 complex senses stalled replications forks produced by ssDNA breaks formed directly by DNA damage or as intermediates of DNA repair events. Once the complex recognizes these abortive replication sites, the DNA damage checkpoint response is to arrest the cell cycle [162].
Park et al. (2009) characterized the in cellulo and in vitro interaction between OGG1 and the 9-1-1 complex [137]. In HEK293 cells treated with H2O2, OGG1 and RAD9 co-localized in the nucleus, suggesting that the OGG1-9-1-1 complex is formed following OG formation. Enzymatic studies demonstrated that OGG1 does not bind a specific subunit of the 9-1-1 complex, but instead, each monomer stimulates OG excision and Schiff base formation to a similar extent. However, the full 9-1-1 complex rendered major OG repair in vitro and in vivo. OGG1, as a bifunctional DG, is able to generate ssDNA breaks, so the non-specific binding of OGG1 to the 9-1-1 complex might be triggered as a product of ssDNA break formation.
3. MUTYH
MUTYH is a peculiar DG, as it excises an undamaged adenine erroneously base pairing with the OG lesion, thereby preventing the formation of stable T:A base pairs. The recombinant MUTYH enzyme also excises A opposite G, although with reduced efficiency compared to the OG:A substrate [163]. However, experiments in Escherichia coli and HEK293FT cells showed that G:A is not efficiently processed by MUTYH in a cellular context [164,165], suggesting that OG:A is the preferable substrate. Human MUTYH displays base excision activity for 2-hydroxyadenine opposite each canonical base and OG [166], as well, but the biological significance of 2-hydroxyad enine repair remains unclear.
Another peculiarity of MUTYH is its narrow substrate specificity. Whereas most DNA glycosylases are promiscuous, MUTYH displays a strict preference for OG:A. For example, NTHL1 and AlkA are each capable of repairing more than nine DNA lesions [11]. Structural analyses of MutY/MUTYH and other HhH-DGs have shown that the strict preference of MUTYH for OG:A is due to the tight fit of adenine inside the active site, as well as a broad H-bond network between residues involved in A and OG recognition [11]. Moreover, most DGs interact primarily with the lesion-containing strand, but MUTYH makes extensive contacts with both the A- and OG-containing DNA strands [11,167].
3.1. MUTYH as Mutagenesis and Tumor Suppressor During Oxidative Stress
The importance of MUTYH in DNA repair is accentuated by an increased risk of polyposis and colorectal cancer (CRC) predisposition in individuals with inherited MUTYH variants, in an autosomal recessive condition known as MUTYH-associated polyposis (MAP) [168,169,170]. Individuals harboring biallelic germline mutations display an increased risk of developing colorectal cancer. Moreover, MAP patients have an increased risk of developing extracolonic cancers such as ovarian, bladder, breast, and others [169]. Although <1% of colorectal cancers are related to biallelic MUTYH mutations, about 1 to 2% of the worldwide population are carriers of monoallelic MUTYH mutations [171]. Of note, reports have been made proposing that monoallelic mutations in MUTYH itself or in combination with other pathologies might be a driver of the carcinogenic process [172,173,174,175,176]. Although the mechanism by which monoallelic mutations in MUTYH become pathologic remains unknown, these studies strongly suggest that the spectrum of diseases and underlying molecular mechanism in which mutated MUTYH participates is beyond MAP.
MAP arises from inherited loss-of-function mutations leading to increased G:C→T:A transversions. The mutational burden elevates the likelihood of oncogenic alterations in genes like APC and KRAS [177]. A role of MUTYH in tumor-suppressive signaling during acute oxidative stress has also been suggested [18,178]. For instance, MUYTH-Knockout mice showed the highest increase (5.3-fold) in tumorigenesis events (particularly in the colon) and up to a 70-fold increase under KBrO3-induced oxidative stress [41,179,180]. This suggests that MUTYH is required to attenuate tumorigenesis.
The Nakabeppu laboratory demonstrated that selective accumulation of nuclear or mitochondrial OG reduced cell viability in response to menadione-induced oxidative stress, indicating that OG accumulation in each organelle triggers cell death distinctly [19]. Nuclear OG-induced cell death requires PARP activity and concomitant nuclear translocation of Apoptosis-Inducing Factor (AIF), which promotes chromatin condensation, DNA fragmentation, and cell death [19,181]. In contrast, mitochondrial OG accumulation leads to functional and morphological degeneration [19]. Consistent with these outcomes, oxidative stress is accompanied by marked activation of calpain, a calcium-dependent protease that contributes to apoptotic cell death [182]. MUTYH downregulation significantly diminishes ssDNA breaks and cell death during oxidative stress, concomitant with abrogation of AIF and calpain activation. These findings suggest that excessive build-up of OG:A in cells might lead to futile MUTYH-mediated repair, which can be cytotoxic. In this context, MUTYH shifts from maintaining the genome to a pro-apoptotic trigger, eliminating cells at high risk of acquiring OG-induced transversions (Figure 5).
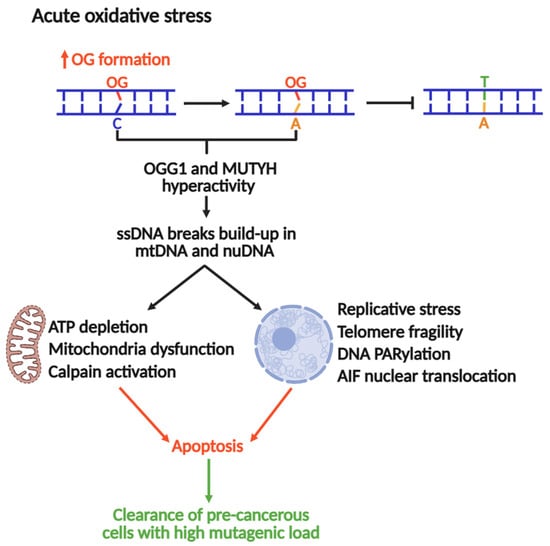
Figure 5.
MUTYH and OGG1 as drivers of genomic instability and apoptosis to suppress tumorigenesis under acute oxidative stress.
Recent work published by the Opresko lab suggests that OG in telomeres causes replicative stress and an excess of ssDNA breaks, inducing telomeric fragility [16]. A follow-up study associated this replicative stress to OGG1 and MUTYH repair activities, where MUTYH and OGG1 knockouts in fibroblasts rescued OG-induced telomere fragility [17]. Therefore, under conditions of acute oxidative stress, the repair of OG produces intermediates such as ssDNA breaks or DG-crosslinks (as reported for OGG1) [126], which stall DNA replication, a major source of genomic instability and cytotoxicity. Consequently, MUTYH-mediated DNA repair and pro-apoptotic activities should be strictly regulated to control the mutagenic load and cellular fate. Moreover, the full understanding of this mechanism might be a potential avenue for the development of therapies against MAP and other types of cancers.
3.2. MUTYH Gene Structure and Regulation
The human MUTYH gene is mapped on the short arm of chromosome 1 and has a length of 11.2 kb with 16 exons (Figure 6). Currently, 18 transcripts have been identified, clustered into three main types: α, β, and γ. The main difference among these transcript variations are the first exons where the 5′ Untranslated Region (UTR) length differs. Namely, in the case of the α transcripts, the first exon also harbors a mitochondrial localization peptide. These transcripts derive from alternative splicing and different transcription start sites modulated by transcription factors [183] (discussed below). A shared feature in most MUTYH mRNA is the presence of exons 4 through 15 and a Nuclear Localization Peptide at the beginning and end of the mRNA. Of note, although mitochondrial isoform α have three protein localization signals (two NLS and an MTL), they are mainly mitochondrial MUTYH isoforms. In contrast with OGG1, MLS is the dominant localization signal in MUTYH [178]. The isoforms α3, β3, β5, and γ3 are the most abundant transcripts of MUTYH [18].
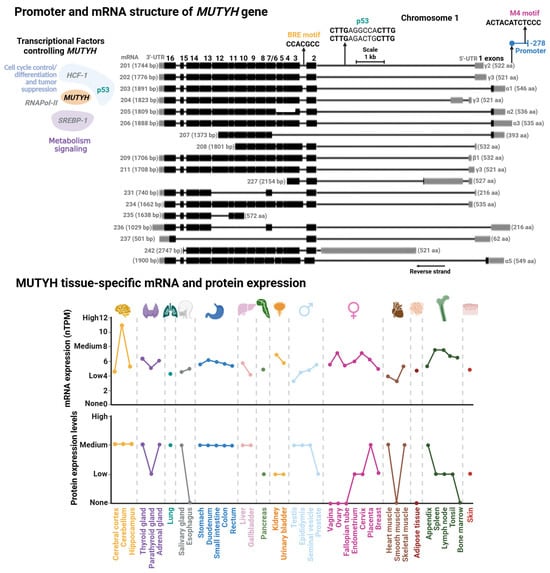
Figure 6.
Promoter and mRNA structures of MUTYH and its tissue-specific mRNA and protein expression. (Upper Panel) Transcriptional factor binding sites mapped within MUTYH promoter. Transcriptional factors (TFs) controlling MUTYH expression with their functional annotation are shown. TFs shown in gray are suggested according to TFs-binding M4 motif [183]; however, there is no direct evidence for their regulation of MUTYH. (Lower Panel) Tissue-specific protein and mRNA expression of MUTYH. Expression profiles were retrieved from the Human Protein Atlas database [102]. Full description of this analysis is shown in Figure 3.
3.2.1. MUTYH Promoter and Transcriptional Regulation
Regulatory elements controlling MUTYH expression were first characterized in 2014. Oka et al. showed that nuclear MUTYH-β transcripts are regulated by the tumor suppressor p53 by two p53 response elements located within the first intron of the MUTYH gene [18] (Figure 6). More recently, the Plotz laboratory identified two regulatory elements within the MUTYH promoter: an M4 and a B-responsive element (BRE) [183]. M4 is the cognate site of SREBP-1 and HCF-1 TFs, which participates in genetic regulation on sterol metabolism [184], and hematopoietic and embryonic stem cells [185]. Mutational studies demonstrated that the M4 motif functions as a global regulator of most MUTYH mRNAs [183]. The BRE motif is a core promoter element associated to RNA polymerase II-dependent transcription. Mutational analysis and location of the transcriptional start site of the BRE motif and GC-box suggest that these elements regulate nuclear MUTYH-β and mitochondrial MUTYH-α mRNAs, respectively.
3.2.2. MUTYH Epigenetic Control
The genetic expression of MUTYH has been explored in different cell lines, as well as in vivo and ex vivo with different genotoxic stressors and pathological conditions [114,186,187,188,189]. A common feature in most of these studies is that MUTYH mRNA expression is tissue- and cell cycle-dependent. MUTYH is highly expressed in the brain, mildly expressed in the respiratory system, skin, and reproductive tissue, and low expression in the gastrointestinal system (Figure 6). It has been reported that organellar MUTYH transcripts are also differentially expressed in different tissues and different stages of the cell cycle [190,191,192]. For instance, the nuclear MUTYH mRNA-β is upregulated in proliferating cells, while variant α is mainly expressed in post mitotic human cells [191]. The association of MUTYH expression and cellular proliferation is consistent with OG:A mismatches as replication-associated DNA lesions. Highly proliferative cells, such as those derived from the reproductive system, are exposed to more replicative stress than low proliferative cells, such as muscle cells, correlating with high metabolic demand and ROS production in highly proliferative cells [193,194].
The variable expression profile of MUTYH suggests tight tissue-specific regulation to ensure appropriate OG:A repair. Any perturbation that leads to expression changes might trigger adverse consequences for cellular function, as discussed above. This suggests that epigenetics might be a strong regulatory element of MUTYH expression. However, contrary to OGG1, the regulatory regions of MUTYH characterized so far are not particularly GC-rich, and the few sites identified are not regulatorily active [183]. Additionally, to our knowledge, epigenetic control of MUTYH has not been explored. Nonetheless, several studies have implicated dysregulation of MUTYH epigenetic control in pathologies like sporadic colorectal cancer [195,196,197].
3.3. MUTYH Protein Structure and Regulation
OG recognition and adenine excision by MUTYH are highly coordinated [198]. The enzyme possesses two distinct domains: the catalytic domain and the OG recognition domain (Figure 7). The OG recognition domain finds the OG moiety using a region known as the FSH loop, which contains highly conserved Phe, Ser, and His residues (F446, S447, and H448 in relation to MUTYH human isoform α5). The OG within the OG:A mismatch adopts the syn-conformation, in contrast to OG or G in OG:C/G:C base pairs that adopt canonical anti-conformation of Watson–Crick base pairs [199,200,201,202]. Several studies have shown that bacterial and human MUTYH detect 2-amino group of OG that is exposed in the major groove only in OG-syn:A-anti base pairs [164,203]. Upon OG:A detection, MutY rotates the OG to the anti-conformation, where the 8-oxo and 7-amino moieties are located in the major groove, stabilized with H-bond contacts with the S447 within the FSH loop [198].
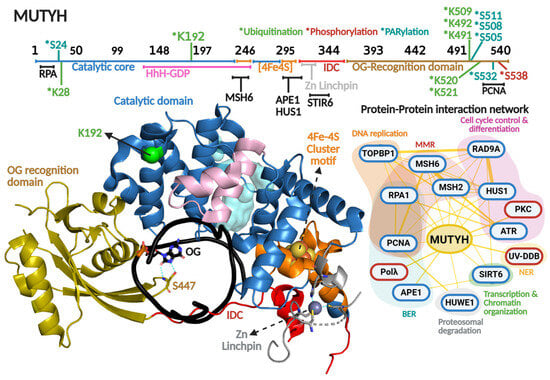
Figure 7.
MUTYH primary and quaternary structures in complex with DNA and their protein–protein interaction network. On top, a scheme of the amino acid sequence is shown highlighting (in different colors) each functional domain. Residues subjected to posttranslational modifications and regions involved in protein–protein interactions are indicated. Ubiquitination, PARylation and phosphorylation are indicated with asterisk in green, clear blue and red colors, respectively Crystallographic structure of MUTYH (8FAY [167]) is shown in different colors for each functional domain. Catalytic cavity is shown in cyan and the DNA in black. Zn linchpin motif of MUTYH is superimposed from mouse MUTYH structure (PDB 7EF8 [166]). Protein–protein interactions show protein partners for MUTYH based on the BioGRIG database [122] (Blue edge) and based on the literature (Red edge). Protein partners found in BioGRID were filtered based just on physical interactions. Figure of MUTYH structure was generated in PyMOL version 3.0.4.
Other important structural and functional components of MutY/MUTYH homologs are the coordination of a [4Fe-4S] cluster and zinc ion. The [4Fe-4S] cluster is a cofactor present in bacterial, archaeal, and eukaryotic MutY homologs, while the Zn coordination is exclusively conserved amongst mammalian MUTYH homologs [11]. Both cofactors have been described as important elements in DNA repair, despite not being involved directly in catalysis [167,204,205,206,207]. The [4Fe-4S] is coordinated by four cysteines within the [4Fe-4S] cluster motif, which is part of the catalytic domain, and is involved in DNA lesion localization/recognition and protein stability. One of the most outstanding functional roles of this cofactor is its DNA-mediated redox activity, which modulates MUTYH affinity for DNA and has been proposed to direct the quest for DNA lesions within genomes of MUTYH and other [4Fe-4S] cluster-containing DGs such as NTHL1 [208,209,210]. More recently, we solved the X-ray crystallographic structure of the human MUTYH-DNA complex, and along with biochemical profiling of cancer associated variants, coevolutionary and molecular dynamic analyses, we discovered that the [4Fe-4S] cluster allosterically regulates the positioning and protonation of the catalytic Asp236 residue required for adenine excision [167,211]. Therefore, the [4Fe-4S] cluster is a multifaceted cofactor in MUTYH. The Zn is coordinated by three cysteines and one histidine to form the zinc linchpin motif within the interdomain connector (IDC) that structurally connects the catalytic domain and OG recognition domain of MUTYH [166]. The presence of the Zn linchpin, along with a longer IDC region (up to 6-fold longer than bacterial MutY homologs) has been described as a scaffold for PPIs, as detailed below (Figure 7 and Table 2).
3.3.1. Phosphorylation
The first reports of MUTYH phosphorylation were by Gu and Lu in 2001. They reported that cell extracts from H2009 cell lines showed a marked reduction in MUTYH activity after dephosphorylation treatment [212]. Interestingly, the dephosphorylation effect on MUTYH activity was greater with G:A vs. OG:A-containing DNA substrates (50- vs. 4-fold reduction), suggesting that MUTYH activity and specificity is regulated by this type of PTM. In 2003, Parker et al. showed that defects in MUTYH phosphorylation are responsible, at least partially, for diminished OG:A repair in cell extracts of colorectal cancer cell lines [213]. Through the use of co-immunoprecipitation and in vitro phosphorylation assays, the protein kinase PKC was identified as the kinase responsible for MUTYH phosphorylation (Figure 7). In 2010, the David Lab identified that residue Ser538 is phosphorylated within the PCNA binding region [214]. Mutation to Alanine at this position renders lower protein stability than the WT and S538D phosphomimic mutation. The mutants exhibited a 10-fold reduction in binding affinity for an uncleavable adenine analog opposite OG. Because the phosphorylation site maps to the PCNA-binding region, these biochemical defects suggest that Ser538 phosphorylation is critical for OG:A recognition and coupling MUTYH to DNA replication.
Table 2.
Posttranslational modifications (PTM) and protein–protein interaction (PPI) controlling MUTYH.
Table 2.
Posttranslational modifications (PTM) and protein–protein interaction (PPI) controlling MUTYH.
| PTM | Site | Effect [Reference] |
|---|---|---|
| Phosphorylation | S538 | ↑ protein stability [214] |
| Ubiquitination | K28/K192 | Unknown [215,216] |
| K491/K492/K509/K520/ K521 | ↑ proteasomal degradation [217] ↓ nuclear translocation [217] | |
| PARylation | S24/S511/S505/S508/S532 | Unknown [218] |
| PPI | Effect [Reference] | |
| MutSα | ↑ glycosylase activity (at low [MutSα]) [219] ↓ glycosylase activity (at high [MutSα]) [219] | |
| PCNA | Couples MUTYH repair to DNA replication [220] | |
| RAD9-RAD1-HUS1 | ↑ glycosylase activity [160,221] | |
| APE1 | ↑ protein:DNA complex formation [222] | |
| SIRT6 | ↑ glycosylase activity [223] | |
| UV-DDB | ↑ turnover [224] | |
Up-arrow (↑) and down-arrow (↓) symbols indicate an enhancement or diminishment of the referred biochemical parameter.
3.3.2. Ubiquitination
MUTYH harbors seven Lys residues mapped as ubiquitination sites: Lys28, Lys192, Lys491, Lys492, Lys509, Lys520, and Lys521 (Figure 7 and Table 2). Namely, ubiquitination of Lys28 and Lys192 were identified through high-throughput proteomic analyses [215,216], but the biological significance of the ubiquitination of those Lys residues in MUTYH is unknown. On the contrary, Dorn et al. (2014) demonstrated that the E3 ubiquitin ligase Mule/HUWE1 interacts with and ubiquitinates MUTYH’s C-terminal region (amino acids 475–535), resulting in downregulation of MUTYH [217]. Within that region, the mutation of five Lys resides (Lys491, Lys492, Lys509, Lys520, and Lys521) to Arg resulted in augmented MUTYH in HEK293T cells. Ubiquitination regulates MUTYH’s cellular localization and repair capacity during oxidative stress. Cellular fractionation and immunofluorescence assays showed that ubiquitinated MUTYH is retained mainly in the cytoplasm, while ubiquitin-free localizes to nuclei and associates with chromatin.
DNA damage is a common feature of Acute Kidney Injury (AKI), induced by ischemia and genotoxic agents such as cisplatin [225]. Recently, Yang and colleagues (2025) showed that patients and mice models of AKI have decreased levels of MUTYH [226]. Furthermore, MUTYH knockout mice had higher expression of markers of Cisplatin-induced AKI compared to WT, and overexpression of nuclear MUTYH isoforms ameliorated AKI and Cisplatin-induced DNA damage. Proteomic analysis of cisplatin-treated cells showed upregulation of Mule/HUWE1 ubiquitin ligase and Mule/HUWE1-MUTYH enrichment in cisplatin-treated cells [226]. These results suggest that Mule/HUWE1 tightly controls MUTYH protein turnover.
3.3.3. PARylation
A feature of MUTYH-mediated DNA repair is the rapid activation of PARP-1 upon oxidative DNA damage [19,227]. As mentioned above, accumulation of nuclear OG drives PARP1-dependent nuclear translocation of AIF, DNA fragmentation, and ultimately, apoptosis [19] (Figure 5). PARP1 association with MUTYH was initially proposed as a DNA repair-triggered signaling event. In 2019, Hendriks et al. identified multiple PARylation sites in MUTYH (Ser24, Ser511, Ser505, Ser508, and Ser532) [218] (Figure 7). The functional consequences of this remain unknown. PARylation is known to regulate key aspects of DNA repair, including DNA association [228], recruitment of downstream BER proteins [229], and deubiquitination to prevent degradation [230]. As such, although the role of PARylation of MUTYH is still unclear, it is likely that is represents an additional regulatory role controlling MUTYH-mediated repair.
3.4. MUTYH’s Regulation by Protein–Protein Interactions
MUTYH is subjected to a wide network of PPIs linking MUTYH function to cellular process beyond BER, including interactions with other DNA repair pathways (MMR and NER), as well as DNA replication, cell cycle control and differentiation, proteasomal degradation, transcription, and chromatin organization (Figure 5). The effects of some PPI on MUTYH function are detailed in the following sections.
3.4.1. MUTYH-MutSα: Avoidance of Pro-Mutagenic Activity of MUTYH
MUTYH might channel BER toward pro-mutagenic repair if it removes A from the template strand after misincorporation of G across A by DNA polymerases. Such misincorporation might derive from replicative DNA polymerases (α, δ and ε) [231,232,233], or even from DNA repair polymerases (Polβ) [234]. Thus far, it is not known how such a pro-mutagenic scenario is avoided. Although MutY homologs exhibit adenine excision from A:G mismatches in vitro, such activity is negligible in a cellular context [164,165]. This suggests that MutY/MUTYH-mediated repair activity on A:G mismatches might be negatively regulated in cells. A potential regulatory mechanism involves MUTYH and MMR interacting, specifically through the MSH2 and MSH6 proteins that form the MutSα heterodimeric complex of MMR (Figure 7). MutSα recruits downstream MMR factors via MutL homologs [235], and its interaction with MUTYH maps to the [4Fe-4S] cluster motif [219]. At low concentrations of MutSα (0.25 to 4 nM), MUTYH affinity for A:OG-containing DNA duplexes increases up to 8-fold, with a 2-fold enhancement in adenine excision activity [219]. Higher concentrations of MutSα (>6 nM) inhibit MUTYH activity by 15%. Additionally, MutSα binds A:G and A:OG mispairs comparably (58 and 78 nM, respectively) [236], while MutY shows a 50-fold preference for an uncleavable analog of A opposite OG vs. G [237].
These observations support a model in which MutSα negatively regulates MUTYH toward A:G mismatches. At low stoichiometric ratios, interaction with MutSα enhances intrinsic preference for A:OG substrates. However, upon OG-independent replicative stress, where A is mis-incorporated across G, enriched MutSα recruitment and its stoichiometric excess over MUTYH may competitively inhibit MUTYH activity, preserving specificity for OG:A. Genomic OG accumulation in MMR-deficient mouse embryonic fibroblasts [238,239] and the downregulation of MSH2 and MSH6 in cells treated with non-cytotoxic concentration of H2O2 [240] might favor MUTYH-mediated OG:A repair.
3.4.2. PCNA and 9-1-1 Complex as Structural and Functional Scaffolds for MUTYH-Associated BERosome Assembly
BER has been proposed to proceed through the assembly of a macromolecular repair complex, dubbed the “BERosome,” at sites requiring repair [241]. Given the critical nature of MUTYH-derived repair intermediates [16,17,19], the formation of a MUTYH-associated BERosome likely ensures that enzymes required for efficient and faithful OG:A repair are co-localized at the repair site, protecting and processing genotoxic intermediates. This is particularly important because OG is on the opposite strand and the associated repair polymerase Polλ, which preferentially incorporates A over C (kcat/KM; 42 vs. 11 min−1 μM−1, respectively) [242]. Indeed, MUTYH and APE1 enhance Polλ fidelity in vitro by biasing towards C incorporation [243], Thus, the formation of a BERosome comprised of all these enzymes could function as a proofreading hub, allowing rapid re-initiation of repair by MUTYH if adenine is mis-incorporated.
PCNA is a homotrimer, ring-shaped DNA sliding clamp that encircles DNA and plays a central role in DNA replication and repair [244]. PCNA’s PPI are mediated by its interdomain connector loop (IDCL) and PCNA-interacting protein (PIP) box. The Lu-Chang Lab identified a C-terminal PIP box (QXX(I/L/M)XXFF) in MUTYH [245] (Figure 7), establishing MUTYH as a PCNA-interacting protein, supporting its role in replication-associated repair [220]. Indeed, the disruption of the PCNA-MUTYH binding interface abrogates OG:A repair in vivo [246]. PCNA interacts with downstream proteins like MUTYH [247], APE1 [248], and Polλ [249]. Likewise, MUTYH interacts with APE1 [250] and Polλ [250]. During oxidative stress, MUTYH is recruited to damaged DNA along with PCNA and Polλ [249]. Since PCNA contains three PPI IDCL motifs, it can bind to three different protein partners, suggesting a scaffolding role for a MUTYH-associated BERosome during S phase.
The Rad9-Rad1-Hus1 (9-1-1) complex is another sliding clamp which also has a donut-like structure composed of the Rad9, Rad1, and Hus1 subunits [251]. The 9-1-1 complex is loaded onto the lesion site, activating the cell cycle checkpoint and distinct DNA repair pathways [252]. It physically interacts with and modulates the activity of MUTYH [221,247] and other BER enzymes [137,253,254,255,256,257,258]. MUTYH physically interacts with the 9-1-1 complex through its Rad9 and Hus1 subunits [221,247]. These interactions stimulate MUTYH activity and are enriched upon ionizing radiation and oxidative stress [160,221]. Interestingly, it is reported that MUTYH, APE1, and Hus1 form a stable complex in vitro and in cells following oxidative stress [250]. This suggests that the 9-1-1 complex might assist with and regulate the assembly of the MUTYH-associated BERosome at the cell cycle checkpoint, as is the proposed function of PCNA during DNA replication. However, there is no direct evidence of the formation of the BERosome for OG:A repair. It is possible that multi-BER enzyme complexes may be transient and therefore hard to isolate, but their existence could explain the exquisite regulation of BER and substrate-product hand-off.
3.4.3. Regulation of MUTYH Turnover
MUTYH, like other DGs, including OGG1, has the biochemical feature of limited turnover in vitro. For instance MUTYH has at least 3200-fold higher affinity for its product (AP-site analog [THF]:OG; KD < 10 pM) than its substrate (A analog [fluorinated-A]:OG; KD = 32 nM), resulting in a markedly slow turnover (k3 of 0.02 min−1) [167]. This could be explained by MUTYH embracing the AP site:OG-containing DNA after base excision. In a cellular context, this could be a regulatory step, with turnover being mediated through PTMs or PPIs. Protection of AP:OG sites would be vital, given that resulting ssDNA and dsDNA breaks are more cytotoxic than AP:OG [259].
MUTYH activity is impacted by its protein partners. For instance, human APE1 stimulates mouse MUTYH activity on OG:A-containing DNA 60-fold when APE1 is in excess (80:1 APE1:MUTYH ratio). Other proteins have shown similar effects on MUTYH, such as the 9-1-1 complex [260] and SIRT6 [223]. However, there is no direct kinetic evidence for enhancement of MUTYH’s turnover. The increase in activity might result from favoring the formation of enzyme-substrate complexes, as evidenced by the MUTYH-APE1 interaction [222], or increasing the fraction of active protein, where the protein partner might function as a chaperone favoring the proper folding of the inactive protein population [261].
The Van Houten and David laboratories, along with other collaborators, carried out detailed studies to evaluate the impact of NER protein UV-DDB on MUTYH, OGG1, and APE1 [134]. They showed that UV-DDB, with an inherently robust ability to recognize and bind AP-sites, stimulates OGG1, APE1, and MUTYH activities [134]. In fact, UV-DDB stimulates MUTYH, increasing its turnover up to 5-fold [224]. EMSA, atomic force microscopy, and single molecule analysis demonstrated that UV-DDB interacts transiently with MUTYH, inducing its dissociation from OG:AP-site-containing DNA, reducing the half-life of the MUTYH-DNA complex from 8800 to 590 s. Hence, UV-DDB directly regulates MUTYH OG:A repair by favoring the release of its product, the OG:AP site, thus indirectly regulating the subsequent downstream BER reactions. Mutations in the genes encoding UV-DDB or MUTYH that disrupt their interaction, and consequently full repair of OG:A, might contribute in part to the build-up of DNA repair intermediates, compromising cell viability and contributing to MAP etiology.
4. Concluding Remarks
Over 30 years of amazing work led by numerous laboratories around the globe have demonstrated that the DNA repair properties of OGG1 and MUTYH are not limited to BER, but also transcriptional regulation and complex signaling events which determine genomic and cellular fate. The interplay these DGs have through their PTMs and PPIs arm OGG1 and MUTYH with the potential to impact distinct cellular aspects beyond BER. Therefore, understanding their regulation at the transcriptional and protein level is pivotal to understanding the functions of these enzymes, as well as their implication in carcinogenesis and disease. Leveraging this knowledge can aid the development of potential drugs to enhance or hijack repair. Finally, there are still questions regarding the mechanisms these enzymes utilize. For example, understanding a general mechanism of product turnover may shed light on the implication of OG repair in telomeric instability and apoptosis. Additionally, understanding BERosome formation could present novel opportunities for therapeutic intervention.
Author Contributions
Conceptualization and funding acquisition: C.H.T.-A., S.S.D. and L.G.B.; figure design and elaboration: C.H.T.-A., A.P.G.-R. and E.G.G.-G.; writing—original draft: All authors; writing—review and editing: M.M., S.S.D. and C.H.T.-A. All authors have read and agreed to the published version of the manuscript.
Funding
This work is funded by CONAHCYT-Ciencia de Frontera (México) grant CF-2023-G-1168 (CHTA and LGB). The work in the David laboratory on MUTYH was funded by National Cancer Institutes of the National Institutes of Health (CA067985). A.P.G.R was supported by the CONAHCYT/SECIHTI master’s degree fellowship (4002329) and M.M. was supported by a National Institutes of Environmental Health Sciences (NIEHS)-funded predoctoral fellowship (T32 ES007059). The APC was partially funded by CONAHCYT-Ciencia de Frontera (México) grant CF-2023-G-1168 (C.H.T.-A. and L.G.B.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ding, Y.; Fleming, A.M.; Burrows, C.J. Sequencing the mouse genome for the oxidatively modified base 8-oxo-7, 8-dihydroguanine by OG-Seq. J. Am. Chem. Soc. 2017, 139, 2569–2572. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M.; Miura, T.; Furuichi, M.; Tominaga, Y.; Tsuchimoto, D.; Sakumi, K.; Nakabeppu, Y. A genome-wide distribution of 8-oxoguanine correlates with the preferred regions for recombination and single nucleotide polymorphism in the human genome. Genome Res. 2006, 16, 567–575. [Google Scholar] [CrossRef] [PubMed]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef]
- Michaels, M.; Miller, J.H. The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7, 8-dihydro-8-oxoguanine). J. Bacteriol. 1992, 174, 6321. [Google Scholar] [CrossRef] [PubMed]
- Michaels, M.L.; Cruz, C.; Grollman, A.P.; Miller, J.H. Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc. Natl. Acad. Sci. USA 1992, 89, 7022–7025. [Google Scholar] [CrossRef]
- Boiteux, S.; O’Connor, T.R.; Laval, J. Formamidopyrimidine-DNA glycosylase of Escherichia coli: Cloning and sequencing of the fpg structural gene and overproduction of the protein. EMBO J. 1987, 6, 3177–3183. [Google Scholar] [CrossRef]
- Fromme, J.C.; Verdine, G.L. Structural insights into lesion recognition and repair by the bacterial 8-oxoguanine DNA glycosylase MutM. Nat. Struct. Biol. 2002, 9, 544–552. [Google Scholar] [CrossRef]
- Fromme, J.C.; Banerjee, A.; Huang, S.J.; Verdine, G.L. Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature 2004, 427, 652–656. [Google Scholar] [CrossRef]
- Maki, H.; Sekiguchi, M. MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature 1992, 355, 273–275. [Google Scholar] [CrossRef]
- Trasviña-Arenas, C.H.; David, S.S.; Delaye, L.; Azuara-Liceaga, E.; Brieba, L.G. Evolution of Base Excision Repair in Entamoeba histolytica is shaped by gene loss, gene duplication, and lateral gene transfer. DNA Repair 2019, 76, 76–88. [Google Scholar] [CrossRef]
- Trasvina-Arenas, C.; Demir, M.; Lin, W.-J.; David, S.S. Structure, function and evolution of the Helix-hairpin-Helix DNA glycosylase superfamily: Piecing together the evolutionary puzzle of DNA base damage repair mechanisms. DNA Repair 2021, 108, 103231. [Google Scholar] [CrossRef]
- Fromme, J.C.; Bruner, S.D.; Yang, W.; Karplus, M.; Verdine, G.L. Product-assisted catalysis in base-excision DNA repair. Nat. Struct. Mol. Biol. 2003, 10, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Visnes, T.; Cázares-Körner, A.; Hao, W.; Wallner, O.; Masuyer, G.; Loseva, O.; Mortusewicz, O.; Wiita, E.; Sarno, A.; Manoilov, A. Small-molecule inhibitor of OGG1 suppresses proinflammatory gene expression and inflammation. Science 2018, 362, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Vlahopoulos, S.; Adamaki, M.; Khoury, N.; Zoumpourlis, V.; Boldogh, I. Roles of DNA repair enzyme OGG1 in innate immunity and its significance for lung cancer. Pharmacol. Ther. 2019, 194, 59–72. [Google Scholar] [CrossRef]
- Fleming, A.M.; Burrows, C.J. Oxidative stress-mediated epigenetic regulation by G-quadruplexes. NAR Cancer 2021, 3, zcab038. [Google Scholar] [CrossRef]
- Barnes, R.P.; de Rosa, M.; Thosar, S.A.; Detwiler, A.C.; Roginskaya, V.; Van Houten, B.; Bruchez, M.P.; Stewart-Ornstein, J.; Opresko, P.L. Telomeric 8-oxo-guanine drives rapid premature senescence in the absence of telomere shortening. Nat. Struct. Mol. Biol. 2022, 29, 639–652. [Google Scholar] [CrossRef]
- De Rosa, M.; Barnes, R.P.; Detwiler, A.C.; Nyalapatla, P.R.; Wipf, P.; Opresko, P.L. OGG1 and MUTYH repair activities promote telomeric 8-oxoguanine induced senescence in human fibroblasts. Nat. Commun. 2025, 16, 893. [Google Scholar] [CrossRef]
- Oka, S.; Leon, J.; Tsuchimoto, D.; Sakumi, K.; Nakabeppu, Y. MUTYH, an adenine DNA glycosylase, mediates p53 tumor suppression via PARP-dependent cell death. Oncogenesis 2014, 3, e121, Erratum in Oncogenesis 2015, 4, e142. [Google Scholar] [CrossRef]
- Oka, S.; Ohno, M.; Tsuchimoto, D.; Sakumi, K.; Furuichi, M.; Nakabeppu, Y. Two distinct pathways of cell death triggered by oxidative damage to nuclear and mitochondrial DNAs. EMBO J. 2008, 27, 421–432. [Google Scholar] [CrossRef]
- Krishnamurthy, N.; Haraguchi, K.; Greenberg, M.M.; David, S.S. Efficient removal of formamidopyrimidines by 8-oxoguanine glycosylases. Biochemistry 2008, 47, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Oden, P.N.; Ryan, B.J.; Yang, H.; Freudenthal, B.D.; Greenberg, M.M. Biochemical and structural characterization of Fapy• dG replication by Human DNA polymerase β. Nucleic Acids Res. 2024, 52, 5392–5405. [Google Scholar] [CrossRef]
- Tomar, R.; Minko, I.G.; Sharma, P.; Kellum, A.H.; Lei, L.; Harp, J.M.; Iverson, T.; Lloyd, R.S.; Egli, M.; Stone, M.P. Base excision repair of the N-(2-deoxy-d-erythro-pentofuranosyl)-urea lesion by the hNEIL1 glycosylase. Nucleic Acids Res. 2023, 51, 3754–3769. [Google Scholar] [CrossRef]
- Bacurio, J.H.T.; Yang, H.; Naldiga, S.; Powell, B.V.; Ryan, B.J.; Freudenthal, B.D.; Greenberg, M.M.; Basu, A.K. Sequence context effects of replication of Fapy• dG in three mutational hot spot sequences of the p53 gene in human cells. DNA Repair 2021, 108, 103213. [Google Scholar] [CrossRef]
- Lindahl, T.; Barnes, D. Repair of endogenous DNA damage. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Woodbury, NY, USA, 2000; pp. 127–134. [Google Scholar]
- Doetsch, P.W.; Zastawny, T.H.; Martin, A.M.; Dizdaroglu, M. Monomeric base damage products from guanine, adenine, and thymine induced by exposure of DNA to ultraviolet radiation. Biochemistry 1995, 34, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M.; Coskun, E.; Jaruga, P. Measurement of oxidatively induced DNA damage and its repair, by mass spectrometric techniques. Free. Radic. Res. 2015, 49, 525–548. [Google Scholar] [CrossRef]
- Delaney, S.; Jarem, D.A.; Volle, C.B.; Yennie, C.J. Chemical and biological consequences of oxidatively damaged guanine in DNA. Free. Radic. Res. 2012, 46, 420–441. [Google Scholar] [CrossRef]
- Alshykhly, O.R.; Fleming, A.M.; Burrows, C.J. Carboxamido-5-formamido-2-iminohydantoin, in addition to 8-oxo-7, 8-dihydroguanine, is the major product of the iron-Fenton or X-ray radiation-induced oxidation of guanine under aerobic reducing conditions in nucleoside and DNA contexts. J. Org. Chem. 2015, 80, 6996–7007. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Dingman, J.C.; Burrows, C.J. CO2 protects cells from iron-Fenton oxidative DNA damage in Escherichia coli and humans. Proc. Natl. Acad. Sci. USA 2024, 121, e2419175121. [Google Scholar] [CrossRef] [PubMed]
- Luna, L.; Bjørås, M.; Hoff, E.; Rognes, T.; Seeberg, E. Cell-cycle regulation, intracellular sorting and induced overexpression of the human NTH1 DNA glycosylase involved in removal of formamidopyrimidine residues from DNA. Mutat. Res. DNA Repair 2000, 460, 95–104. [Google Scholar] [CrossRef]
- Hazra, T.K.; Izumi, T.; Kow, Y.W.; Mitra, S. The discovery of a new family of mammalian enzymes for repair of oxidatively damaged DNA, and its physiological implications. Carcinogenesis 2003, 24, 155–157. [Google Scholar] [CrossRef][Green Version]
- Liu, M.; Doublié, S.; Wallace, S.S. Neil3, the final frontier for the DNA glycosylases that recognize oxidative damage. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2013, 743, 4–11. [Google Scholar] [CrossRef]
- Liu, M.; Bandaru, V.; Bond, J.P.; Jaruga, P.; Zhao, X.; Christov, P.P.; Burrows, C.J.; Rizzo, C.J.; Dizdaroglu, M.; Wallace, S.S. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 4925–4930. [Google Scholar] [CrossRef] [PubMed]
- Mokkapati, S.K.; Wiederhold, L.; Hazra, T.K.; Mitra, S. Stimulation of DNA glycosylase activity of OGG1 by NEIL1: Functional collaboration between two human DNA glycosylases. Biochemistry 2004, 43, 11596–11604. [Google Scholar] [CrossRef] [PubMed]
- Minko, I.G.; Vartanian, V.L.; Tozaki, N.N.; Coskun, E.; Coskun, S.H.; Jaruga, P.; Yeo, J.; David, S.S.; Stone, M.P.; Egli, M. Recognition of DNA adducts by edited and unedited forms of DNA glycosylase NEIL1. DNA Repair 2020, 85, 102741. [Google Scholar] [CrossRef]
- Bandaru, V.; Zhao, X.; Newton, M.R.; Burrows, C.J.; Wallace, S.S. Human endonuclease VIII-like (NEIL) proteins in the giant DNA Mimivirus. DNA Repair 2007, 6, 1629–1641. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bandaru, V.; Sunkara, S.; Wallace, S.S.; Bond, J.P. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair 2002, 1, 517–529. [Google Scholar] [CrossRef]
- Sampath, H.; Vartanian, V.; Rollins, M.R.; Sakumi, K.; Nakabeppu, Y.; Lloyd, R.S. 8-Oxoguanine DNA glycosylase (OGG1) deficiency increases susceptibility to obesity and metabolic dysfunction. PLoS ONE 2012, 7, e51697. [Google Scholar] [CrossRef]
- Vartanian, V.; Tumova, J.; Dobrzyn, P.; Dobrzyn, A.; Nakabeppu, Y.; Lloyd, R.S.; Sampath, H. 8-oxoguanine DNA glycosylase (OGG1) deficiency elicits coordinated changes in lipid and mitochondrial metabolism in muscle. PLoS ONE 2017, 12, e0181687. [Google Scholar] [CrossRef]
- Mabley, J.G.; Pacher, P.; Deb, A.; Wallace, R.; Elder, R.H.; Szabó, C. Potential role for 8-oxoguanine DNA glycosylase in regulating inflammation. FASEB J. 2005, 19, 1–18. [Google Scholar] [CrossRef]
- Xie, Y.; Yang, H.; Cunanan, C.; Okamoto, K.; Shibata, D.; Pan, J.; Barnes, D.E.; Lindahl, T.; McIlhatton, M.; Fishel, R. Deficiencies in mouse Myh and Ogg1 result in tumor predisposition and G to T mutations in codon 12 of the K-ras oncogene in lung tumors. Cancer Res. 2004, 64, 3096–3102. [Google Scholar] [CrossRef]
- Zhou, P.-T.; Li, B.; Ji, J.; Wang, M.-M.; Gao, C.-F. A systematic review and meta-analysis of the association between OGG1 Ser326Cys polymorphism and cancers. Med. Oncol. 2015, 32, 31. [Google Scholar] [CrossRef]
- Mao, G.; Pan, X.; Zhu, B.-B.; Zhang, Y.; Yuan, F.; Huang, J.; Lovell, M.A.; Lee, M.P.; Markesbery, W.R.; Li, G.-M. Identification and characterization of OGG1 mutations in patients with Alzheimer’s disease. Nucleic Acids Res. 2007, 35, 2759–2766. [Google Scholar] [CrossRef]
- Pao, P.-C.; Patnaik, D.; Watson, L.A.; Gao, F.; Pan, L.; Wang, J.; Adaikkan, C.; Penney, J.; Cam, H.P.; Huang, W.-C. HDAC1 modulates OGG1-initiated oxidative DNA damage repair in the aging brain and Alzheimer’s disease. Nat. Commun. 2020, 11, 2484. [Google Scholar] [CrossRef]
- Cardozo-Pelaez, F.; Sanchez-Contreras, M.; Nevin, A.B. Ogg1 null mice exhibit age-associated loss of the nigrostriatal pathway and increased sensitivity to MPTP. Neurochem. Int. 2012, 61, 721–730. [Google Scholar] [CrossRef]
- Lind, P.A.; Andersson, D.I. Whole-genome mutational biases in bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 17878–17883. [Google Scholar] [CrossRef]
- Fang, Y.; Zou, P. Genome-wide mapping of oxidative DNA damage via engineering of 8-oxoguanine DNA glycosylase. Biochemistry 2019, 59, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Amente, S.; Di Palo, G.; Scala, G.; Castrignanò, T.; Gorini, F.; Cocozza, S.; Moresano, A.; Pucci, P.; Ma, B.; Stepanov, I. Genome-wide mapping of 8-oxo-7, 8-dihydro-2′-deoxyguanosine reveals accumulation of oxidatively-generated damage at DNA replication origins within transcribed long genes of mammalian cells. Nucleic Acids Res. 2019, 47, 221–236. [Google Scholar] [CrossRef]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Bacsi, A.; Aguilera-Aguirre, L.; Szczesny, B.; Radak, Z.; Hazra, T.K.; Sur, S.; Ba, X.; Boldogh, I. Down-regulation of 8-oxoguanine DNA glycosylase 1 expression in the airway epithelium ameliorates allergic lung inflammation. DNA Repair 2013, 12, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Ba, X.; Bacsi, A.; Luo, J.; Aguilera-Aguirre, L.; Zeng, X.; Radak, Z.; Brasier, A.R.; Boldogh, I. 8-oxoguanine DNA glycosylase-1 augments proinflammatory gene expression by facilitating the recruitment of site-specific transcription factors. J. Immunol. 2014, 192, 2384–2394. [Google Scholar] [CrossRef]
- Jamaluddin, M.; Wang, S.; Boldogh, I.; Tian, B.; Brasier, A.R. TNF-α-induced NF-κB/RelA Ser276 phosphorylation and enhanceosome formation is mediated by an ROS-dependent PKAc pathway. Cell. Signal. 2007, 19, 1419–1433. [Google Scholar] [CrossRef]
- Pan, L.; Zhu, B.; Hao, W.; Zeng, X.; Vlahopoulos, S.A.; Hazra, T.K.; Hegde, M.L.; Radak, Z.; Bacsi, A.; Brasier, A.R. Oxidized guanine base lesions function in 8-oxoguanine DNA glycosylase-1-mediated epigenetic regulation of nuclear factor κB-driven gene expression. J. Biol. Chem. 2016, 291, 25553–25566. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Regulation of NF-κB by TNF family cytokines. In Seminars in Immunology; Elsevier: Amsterdam, The Netherlands, 2014; pp. 253–266. [Google Scholar]
- Pan, L.; Hao, W.; Zheng, X.; Zeng, X.; Abbasi, A.A.; Boldogh, I.; Ba, X. OGG1-DNA interactions facilitate NF-κB binding to DNA targets. Sci. Rep. 2017, 7, 43297. [Google Scholar] [CrossRef] [PubMed]
- Obermann, T.; Sakshaug, T.; Kanagaraj, V.V.; Abentung, A.; Sarno, A.; Bjørås, M.; Scheffler, K. Genomic 8-oxoguanine modulates gene transcription independent of its repair by DNA glycosylases OGG1 and MUTYH. Redox Biol. 2024, 79, 103461. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Goldstein, M.; Heylmann, D.; Kaina, B. Human monocytes undergo excessive apoptosis following temozolomide activating the ATM/ATR pathway while dendritic cells and macrophages are resistant. PLoS ONE 2012, 7, e39956. [Google Scholar] [CrossRef]
- Zhao, S.; Klattenhoff, A.W.; Thakur, M.; Sebastian, M.; Kidane, D. Mutation in DNA polymerase beta causes spontaneous chromosomal instability and inflammation-associated carcinogenesis in mice. Cancers 2019, 11, 1160. [Google Scholar] [CrossRef]
- Zhao, S.; Habib, S.L.; Senejani, A.G.; Sebastian, M.; Kidane, D. Role of base excision repair in innate immune cells and its relevance for cancer therapy. Biomedicines 2022, 10, 557. [Google Scholar] [CrossRef]
- Fleming, A.M.; Zhu, J.; Ding, Y.; Burrows, C.J. Oxo-7,8-dihydroguanine in the context of a gene promoter G-quadruplex is an on–off switch for transcription. ACS Chem. Biol. 2017, 12, 2417–2426. [Google Scholar] [CrossRef]
- Mathad, R.I.; Hatzakis, E.; Dai, J.; Yang, D. c-MYC promoter G-quadruplex formed at the 5′-end of NHE III 1 element: Insights into biological relevance and parallel-stranded G-quadruplex stability. Nucleic Acids Res. 2011, 39, 9023–9033. [Google Scholar] [CrossRef]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA secondary structures: Stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef]
- Chambers, V.S.; Marsico, G.; Boutell, J.M.; Di Antonio, M.; Smith, G.P.; Balasubramanian, S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015, 33, 877–881. [Google Scholar] [CrossRef]
- Hänsel-Hertsch, R.; Beraldi, D.; Lensing, S.V.; Marsico, G.; Zyner, K.; Parry, A.; Di Antonio, M.; Pike, J.; Kimura, H.; Narita, M. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016, 48, 1267–1272. [Google Scholar] [CrossRef]
- Fleming, A.M.; Zhu, J.; Ding, Y.; Visser, J.A.; Zhu, J.; Burrows, C.J. Human DNA repair genes possess potential G-quadruplex sequences in their promoters and 5′-untranslated regions. Biochemistry 2018, 57, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.T.; Vallur, A.C.; Eddy, J.; Maizels, N. G quadruplexes are genomewide targets of transcriptional helicases XPB and XPD. Nat. Chem. Biol. 2014, 10, 313–318. [Google Scholar] [CrossRef]
- Fleming, A.M.; Zhou, J.; Wallace, S.S.; Burrows, C.J. A role for the fifth G-track in G-quadruplex forming oncogene promoter sequences during oxidative stress: Do these “spare tires” have an evolved function? ACS Cent. Sci. 2015, 1, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Cogoi, S.; Ferino, A.; Miglietta, G.; Pedersen, E.B.; Xodo, L.E. The regulatory G4 motif of the Kirsten ras (KRAS) gene is sensitive to guanine oxidation: Implications on transcription. Nucleic Acids Res. 2018, 46, 661–676. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Burrows, C.J. Interplay of guanine oxidation and G-quadruplex folding in gene promoters. J. Am. Chem. Soc. 2019, 142, 1115–1136. [Google Scholar] [CrossRef]
- Fleming, A.M.; Ding, Y.; Burrows, C.J. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc. Natl. Acad. Sci. USA 2017, 114, 2604–2609. [Google Scholar] [CrossRef]
- Wang, R.; Hao, W.; Pan, L.; Boldogh, I.; Ba, X. The roles of base excision repair enzyme OGG1 in gene expression. Cell. Mol. Life Sci. 2018, 75, 3741–3750. [Google Scholar] [CrossRef]
- Sampath, H.; Lloyd, R.S. Roles of OGG1 in transcriptional regulation and maintenance of metabolic homeostasis. DNA Repair 2019, 81, 102667. [Google Scholar] [CrossRef]
- Hashiguchi, K.; Stuart, J.; de Souza-Pinto, N.; Bohr, V. The C-terminal αO helix of human Ogg1 is essential for 8-oxoguanine DNA glycosylase activity: The mitochondrial β-Ogg1 lacks this domain and does not have glycosylase activity. Nucleic Acids Res. 2004, 32, 5596–5608. [Google Scholar] [CrossRef]
- Nishioka, K.; Ohtsubo, T.; Oda, H.; Fujiwara, T.; Kang, D.; Sugimachi, K.; Nakabeppu, Y. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell 1999, 10, 1637–1652. [Google Scholar] [CrossRef]
- Dhénaut, A.; Boiteux, S.; Radicella, J.P. Characterization of the hOGG1 promoter and its expression during the cell cycle. Mutat. Res. DNA Repair 2000, 461, 109–118. [Google Scholar] [CrossRef]
- Sjöstedt, E.; Zhong, W.; Fagerberg, L.; Karlsson, M.; Mitsios, N.; Adori, C.; Oksvold, P.; Edfors, F.; Limiszewska, A.; Hikmet, F. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 2020, 367, eaay5947. [Google Scholar] [CrossRef]
- Cove-Smith, A.; Hendry, B.M. The regulation of mesangial cell proliferation. Nephron Exp. Nephrol. 2008, 108, e74–e79. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Sureshbabu, A.; Ryter, S.W.; Choi, M.E. Oxidative stress and autophagy: Crucial modulators of kidney injury. Redox Biol. 2015, 4, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Nie, B.; Gan, W.; Shi, F.; Hu, G.-X.; Chen, L.-G.; Hayakawa, H.; Sekiguchi, M.; Cai, J.-P. Age-dependent accumulation of 8-oxoguanine in the DNA and RNA in various rat tissues. Oxidative Med. Cell. Longev. 2013, 2013, 303181. [Google Scholar] [CrossRef]
- Biswas, M.; Chan, J.Y. Role of Nrf1 in antioxidant response element-mediated gene expression and beyond. Toxicol. Appl. Pharmacol. 2010, 244, 16–20. [Google Scholar] [CrossRef]
- Habib, S.L.; Yadav, A.; Kidane, D.; Weiss, R.H.; Liang, S. Novel protective mechanism of reducing renal cell damage in diabetes: Activation AMPK by AICAR increased NRF2/OGG1 proteins and reduced oxidative DNA damage. Cell Cycle 2016, 15, 3048–3059. [Google Scholar] [CrossRef]
- Singh, B.; Chatterjee, A.; Ronghe, A.M.; Bhat, N.K.; Bhat, H.K. Antioxidant-mediated up-regulation of OGG1 via NRF2 induction is associated with inhibition of oxidative DNA damage in estrogen-induced breast cancer. BMC Cancer 2013, 13, 1–9. [Google Scholar] [CrossRef]
- Youn, C.-K.; Kim, S.-H.; Song, S.H.; Chang, I.-Y.; Hyun, J.-W.; Chung, M.-H.; You, H.J. Cadmium down-regulates human OGG1 through suppression of Sp1 activity. J. Biol. Chem. 2005, 280, 25185–25195. [Google Scholar] [CrossRef]
- Chu, S.; Ferro, T.J. Sp1: Regulation of gene expression by phosphorylation. Gene 2005, 348, 1–11. [Google Scholar] [CrossRef]
- Misra, R.R.; Smith, G.T.; Waalkes, M.P. Evaluation of the direct genotoxic potential of cadmium in four different rodent cell lines. Toxicology 1998, 126, 103–114. [Google Scholar] [CrossRef]
- Ochi, T.; Ohsawa, M. Participation of active oxygen species in the induction of chromosomal aberrations by cadmium chloride in cultured Chinese hamster cells. Mutat. Res. Lett. 1985, 143, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Habib, S.L.; Riley, D.J.; Mahimainathan, L.; Bhandari, B.; Choudhury, G.G.; Abboud, H.E. Tuberin regulates the DNA repair enzyme OGG1. Am. J. Physiol. Ren. Physiol. 2008, 294, F281–F290. [Google Scholar] [CrossRef][Green Version]
- Lee, M.-R.; Kim, S.-H.; Cho, H.-J.; Lee, K.-Y.; Moon, A.R.; Jeong, H.G.; Lee, J.-S.; Hyun, J.-W.; Chung, M.-H.; You, H.J. Transcription factors NF-YA regulate the induction of human OGG1 following DNA-alkylating agent methylmethane sulfonate (MMS) treatment. J. Biol. Chem. 2004, 279, 9857–9866. [Google Scholar] [CrossRef]
- Habib, S.L.; Bhandari, B.K.; Sadek, N.; Abboud-Werner, S.L.; Abboud, H.E. Novel mechanism of regulation of the DNA repair enzyme OGG1 in tuberin-deficient cells. Carcinogenesis 2010, 31, 2022–2030. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chatterjee, A.; Mambo, E.; Osada, M.; Upadhyay, S.; Sidransky, D. The effect of p53-RNAi and p53 knockout on human 8-oxoguanine DNA glycosylase (hOgg1) activity. FASEB J. 2006, 20, 112–114. [Google Scholar] [CrossRef]
- Seo, Y.R.; Jung, H.J. The potential roles of p53 tumor suppressor in nucleotide excision repair (NER) and base excision repair (BER). Exp. Mol. Med. 2004, 36, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.L.; Seo, Y.R. p53 regulation of DNA excision repair pathways. Mutagenesis 2002, 17, 149–156. [Google Scholar] [CrossRef]
- Poletto, M.; Legrand, A.J.; Fletcher, S.C.; Dianov, G.L. p53 coordinates base excision repair to prevent genomic instability. Nucleic Acids Res. 2016, 44, 3165–3175. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ahn, J.; Wilson, S.H.; Prives, C. A role for p53 in base excision repair. EMBO J. 2001, 20, 914–923. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, C.; Liu, X.; Zheng, J.; Zhang, F.; Wang, D.; Xue, Y.; Li, X.; Shen, S.; Shao, L. Transcription factor AP-4 (TFAP4)-upstream ORF coding 66 aa inhibits the malignant behaviors of glioma cells by suppressing the TFAP4/long noncoding RNA 00520/microRNA-520f-3p feedback loop. Cancer Sci. 2020, 111, 891–906. [Google Scholar] [CrossRef]
- Song, J.; Xie, C.; Jiang, L.; Wu, G.; Zhu, J.; Zhang, S.; Tang, M.; Song, L.; Li, J. Transcription factor AP-4 promotes tumorigenic capability and activates the Wnt/β-catenin pathway in hepatocellular carcinoma. Theranostics 2018, 8, 3571. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakagawara, A. Role of p53 in cell death and human cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef]
- Geng, A.; Sun, J.; Tang, H.; Yu, Y.; Wang, X.; Zhang, J.; Wang, X.; Sun, X.; Zhou, X.; Gao, N. SIRT2 promotes base excision repair by transcriptionally activating OGG1 in an ATM/ATR-dependent manner. Nucleic Acids Res. 2024, 52, 5107–5120. [Google Scholar] [CrossRef] [PubMed]
- Minten, E.V.; Kapoor-Vazirani, P.; Li, C.; Zhang, H.; Balakrishnan, K.; David, S.Y. SIRT2 promotes BRCA1-BARD1 heterodimerization through deacetylation. Cell Rep. 2021, 34, 108921. [Google Scholar] [CrossRef]
- Ma, L.; Liang, B.; Yang, Y.; Chen, L.; Liu, Q.; Zhang, A. hOGG1 promoter methylation, hOGG1 genetic variants and their interactions for risk of coal-borne arsenicosis: A case-control study. Environ. Toxicol. Pharmacol. 2020, 75, 103330. [Google Scholar] [CrossRef]
- Thul, P.J.; Lindskog, C. The human protein atlas: A spatial map of the human proteome. Protein Sci. 2018, 27, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Piña-Guzmán, B.; Solís-Heredia, M.; Rojas-García, A.; Urióstegui-Acosta, M.; Quintanilla-Vega, B. Genetic damage caused by methyl–parathion in mouse spermatozoa is related to oxidative stress. Toxicol. Appl. Pharmacol. 2006, 216, 216–224. [Google Scholar] [CrossRef]
- Hernandez-Cortes, D.; Alvarado-Cruz, I.; Solís-Heredia, M.; Quintanilla-Vega, B. Epigenetic modulation of Nrf2 and Ogg1 gene expression in testicular germ cells by methyl parathion exposure. Toxicol. Appl. Pharmacol. 2018, 346, 19–27. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, W.; Zhang, A. TET-mediated DNA demethylation plays an important role in arsenic-induced HBE cells oxidative stress via regulating promoter methylation of OGG1 and GSTP1. Toxicol. Vitr. 2021, 72, 105075. [Google Scholar] [CrossRef] [PubMed]
- Platet, N.; Cathiard, A.M.; Gleizes, M.; Garcia, M. Estrogens and their receptors in breast cancer progression: A dual role in cancer proliferation and invasion. Crit. Rev. Oncol. Hematol. 2004, 51, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.P.; Treas, J.; Tyagi, T.; Gao, W. DNA demethylation by 5-aza-2-deoxycytidine treatment abrogates 17 beta-estradiol-induced cell growth and restores expression of DNA repair genes in human breast cancer cells. Cancer Lett. 2012, 316, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Sliwinska, A.; Kwiatkowski, D.; Czarny, P.; Toma, M.; Wigner, P.; Drzewoski, J.; Fabianowska-Majewska, K.; Szemraj, J.; Maes, M.; Galecki, P. The levels of 7, 8-dihydrodeoxyguanosine (8-oxoG) and 8-oxoguanine DNA glycosylase 1 (OGG1)–A potential diagnostic biomarkers of Alzheimer’s disease. J. Neurol. Sci. 2016, 368, 155–159. [Google Scholar] [CrossRef]
- Fukae, J.; Takanashi, M.; Kubo, S.-I.; Nishioka, K.-I.; Nakabeppu, Y.; Mori, H.; Mizuno, Y.; Hattori, N. Expression of 8-oxoguanine DNA glycosylase (OGG1) in Parkinson’s disease and related neurodegenerative disorders. Acta Neuropathol. 2005, 109, 256–262. [Google Scholar] [CrossRef]
- Furuta, A.; Iida, T.; Nakabeppu, Y.; Iwaki, T. Expression of hMTH1 in the hippocampi of control and Alzheimer’s disease. Neuroreport 2001, 12, 2895–2899. [Google Scholar] [CrossRef]
- Iida, T.; Furuta, A.; Nishioka, K.; Nakabeppu, Y.; Iwaki, T. Expression of 8-oxoguanine DNA glycosylase is reduced and associated with neurofibrillary tangles in Alzheimer’s disease brain. Acta Neuropathol. 2002, 103, 20–25. [Google Scholar] [CrossRef]
- Coppedè, F.; Tannorella, P.; Stoccoro, A.; Chico, L.; Siciliano, G.; Bonuccelli, U.; Migliore, L. Methylation analysis of DNA repair genes in Alzheimer’s disease. Mech. Ageing Dev. 2017, 161, 105–111. [Google Scholar] [CrossRef]
- Chen, W.; Zhou, X.; Duan, Y.; Zou, T.; Liu, G.; Ying, X.; Wang, Q.; Duan, S. Association of OGG1 and DLST promoter methylation with Alzheimer’s disease in Xinjiang population. Exp. Ther. Med. 2018, 16, 3135–3142. [Google Scholar] [CrossRef] [PubMed]
- Sliwinska, A.; Sitarek, P.; Toma, M.; Czarny, P.; Synowiec, E.; Krupa, R.; Wigner, P.; Bialek, K.; Kwiatkowski, D.; Korycinska, A. Decreased expression level of BER genes in Alzheimer’s disease patients is not derivative of their DNA methylation status. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2017, 79, 311–316. [Google Scholar] [CrossRef]
- Fraser, H.B.; Lam, L.L.; Neumann, S.M.; Kobor, M.S. Population-specificity of human DNA methylation. Genome Biol. 2012, 13, R8. [Google Scholar] [CrossRef]
- Norman, D.P.; Bruner, S.D.; Verdine, G.L. Coupling of substrate recognition and catalysis by a human base-excision DNA repair protein. J. Am. Chem. Soc. 2001, 123, 359–360. [Google Scholar] [CrossRef]
- Hooten, N.N.; Kompaniez, K.; Barnes, J.; Lohani, A.; Evans, M.K. Poly (ADP-ribose) polymerase. J. Biol. Chem. 2011, 286, 44679–44690. [Google Scholar] [CrossRef]
- Carter, R.J.; Parsons, J.L. Base excision repair, a pathway regulated by posttranslational modifications. Mol. Cell. Biol. 2016, 36, 1426–1437. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.; Ti, D.; Mei, Q.; Liu, J.; Yan, X.; Chen, D.; Li, X.; Wu, Z.; Han, W. The role of posttranslational modifications in DNA repair. BioMed Res. Int. 2020, 2020, 7493902. [Google Scholar] [CrossRef]
- Fantini, D.; Moritz, E.; Auvré, F.; Amouroux, R.; Campalans, A.; Epe, B.; Bravard, A.; Radicella, J.P. Rapid inactivation and proteasome-mediated degradation of OGG1 contribute to the synergistic effect of hyperthermia on genotoxic treatments. DNA Repair 2013, 12, 227–237. [Google Scholar] [CrossRef]
- Hughes, J.R.; Parsons, J.L. The E3 ubiquitin ligase NEDD4L targets OGG1 for ubiquitylation and modulates the cellular DNA damage response. Front. Cell Dev. Biol. 2020, 8, 607060. [Google Scholar] [CrossRef] [PubMed]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Quiñones, J.L.; Thapar, U.; Wilson, S.H.; Ramsden, D.A.; Demple, B. Oxidative DNA-protein crosslinks formed in mammalian cells by abasic site lyases involved in DNA repair. DNA Repair 2020, 87, 102773. [Google Scholar] [CrossRef]
- Tretyakova, N.Y.; IV, A.G.; Ji, S. DNA–protein cross-links: Formation; structural identities; biological outcomes. Acc. Chem. Res. 2015, 48, 1631–1644. [Google Scholar] [CrossRef] [PubMed]
- David, S.S.; Williams, S.D. Chemistry of glycosylases and endonucleases involved in base-excision repair. Chem. Rev. 1998, 98, 1221–1262. [Google Scholar] [CrossRef]
- Essawy, M.; Chesner, L.; Alshareef, D.; Ji, S.; Tretyakova, N.; Campbell, C. Ubiquitin signaling and the proteasome drive human DNA–protein crosslink repair. Nucleic Acids Res. 2023, 51, 12174–12184. [Google Scholar] [CrossRef] [PubMed]
- Akimov, V.; Barrio-Hernandez, I.; Hansen, S.V.; Hallenborg, P.; Pedersen, A.-K.; Bekker-Jensen, D.B.; Puglia, M.; Christensen, S.D.; Vanselow, J.T.; Nielsen, M.M. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 2018, 25, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.A.; Hammerle, L.P.; Andrews, P.S.; Stokes, M.P.; Mustelin, T.; Silva, J.C.; Black, R.A.; Doedens, J.R. Ubiquitin ligase substrate identification through quantitative proteomics at both the protein and peptide levels. J. Biol. Chem. 2011, 286, 41530–41538. [Google Scholar] [CrossRef]
- Hu, J.; Imam, S.Z.; Hashiguchi, K.; de Souza-Pinto, N.C.; Bohr, V.A. Phosphorylation of human oxoguanine DNA glycosylase (α-OGG1) modulates its function. Nucleic Acids Res. 2005, 33, 3271–3282. [Google Scholar] [CrossRef]
- Bhakat, K.K.; Mokkapati, S.K.; Boldogh, I.; Hazra, T.K.; Mitra, S. Acetylation of human 8-oxoguanine-DNA glycosylase by p300 and its role in 8-oxoguanine repair in vivo. Mol. Cell. Biol. 2006, 26, 1654–1665. [Google Scholar] [CrossRef]
- Li, S.; Shi, B.; Liu, X.; An, H.-X. Acetylation and deacetylation of DNA repair proteins in cancers. Front. Oncol. 2020, 10, 573502. [Google Scholar] [CrossRef]
- Braithwaite, E.K.; Kedar, P.S.; Stumpo, D.J.; Bertocci, B.; Freedman, J.H.; Samson, L.D.; Wilson, S.H. DNA polymerases β and λ mediate overlapping and independent roles in base excision repair in mouse embryonic fibroblasts. PLoS ONE 2010, 5, e12229. [Google Scholar] [CrossRef]
- d’Errico, M.; Parlanti, E.; Teson, M.; de Jesus, B.M.B.; Degan, P.; Calcagnile, A.; Jaruga, P.; Bjørås, M.; Crescenzi, M.; Pedrini, A.M. New functions of XPC in the protection of human skin cells from oxidative damage. EMBO J. 2006, 25, 4305–4315. [Google Scholar] [CrossRef]
- Jang, S.; Kumar, N.; Beckwitt, E.C.; Kong, M.; Fouquerel, E.; Rapić-Otrin, V.; Prasad, R.; Watkins, S.C.; Khuu, C.; Majumdar, C.; et al. Damage sensor role of UV-DDB during base excision repair. Nat. Struct. Mol. Biol. 2019, 26, 695–703. [Google Scholar] [CrossRef]
- de Souza-Pinto, N.C.; Maynard, S.; Hashiguchi, K.; Hu, J.; Muftuoglu, M.; Bohr, V.A. The recombination protein RAD52 cooperates with the excision repair protein OGG1 for the repair of oxidative lesions in mammalian cells. Mol. Cell. Biol. 2009, 29, 4441–4454. [Google Scholar] [CrossRef]
- Marsin, S.; Vidal, A.E.; Sossou, M.; Murcia, J.M.-D.; Le Page, F.; Boiteux, S.; De Murcia, G.; Radicella, J.P. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. J. Biol. Chem. 2003, 278, 44068–44074. [Google Scholar] [CrossRef] [PubMed]
- Park, M.J.; Park, J.-H.; Hahm, S.-H.; Ko, S.I.; Lee, Y.R.; Chung, J.H.; Sohn, S.Y.; Cho, Y.; Kang, L.-W.; Han, Y.S. Repair activities of human 8-oxoguanine DNA glycosylase are stimulated by the interaction with human checkpoint sensor Rad9–Rad1–Hus1 complex. DNA Repair 2009, 8, 1190–1200. [Google Scholar] [CrossRef]
- Dantzer, F.; Luna, L.; Bjørås, M.; Seeberg, E. Human OGG1 undergoes serine phosphorylation and associates with the nuclear matrix and mitotic chromatin in vivo. Nucleic Acids Res. 2002, 30, 2349–2357. [Google Scholar] [CrossRef] [PubMed]
- Le Marchand, L.; Donlon, T.; Lum-Jones, A.; Seifried, A.; Wilkens, L.R. Association of the hOGG1 Ser326Cys polymorphism with lung cancer risk. Cancer Epidemiol. Biomark. Prev. 2002, 11, 409–412. [Google Scholar]
- Xu, J.; Zheng, S.L.; Turner, A.; Isaacs, S.D.; Wiley, K.E.; Hawkins, G.A.; Chang, B.-L.; Bleecker, E.R.; Walsh, P.C.; Meyers, D.A. Associations between hOGG1 sequence variants and prostate cancer susceptibility. Cancer Res. 2002, 62, 2253–2257. [Google Scholar]
- Elahi, A.; Zheng, Z.; Park, J.; Eyring, K.; McCaffrey, T.; Lazarus, P. The human OGG1 DNA repair enzyme and its association with orolaryngeal cancer risk. Carcinogenesis 2002, 23, 1229–1234. [Google Scholar] [CrossRef]
- Luna, L.; Rolseth, V.; Hildrestrand, G.A.; Otterlei, M.; Dantzer, F.; Bjørås, M.; Seeberg, E. Dynamic relocalization of hOGG1 during the cell cycle is disrupted in cells harbouring the hOGG1-Cys 326 polymorphic variant. Nucleic Acids Res. 2005, 33, 1813–1824. [Google Scholar] [CrossRef] [PubMed]
- Bravard, A.; Vacher, M.; Moritz, E.; Vaslin, L.; Hall, J.; Epe, B.; Radicella, J.P. Oxidation status of human OGG1-S326C polymorphic variant determines cellular DNA repair capacity. Cancer Res. 2009, 69, 3642–3649. [Google Scholar] [CrossRef]
- Kang, L.; Zhao, W.; Zhang, G.; Wu, J.; Guan, H. Acetylated 8-oxoguanine DNA glycosylase 1 and its relationship with p300 and SIRT1 in lens epithelium cells from age-related cataract. Exp. Eye Res. 2015, 135, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Sarga, L.; Hart, N.; Koch, L.; Britton, S.; Hajas, G.; Boldogh, I.; Ba, X.; Radak, Z. Aerobic endurance capacity affects spatial memory and SIRT1 is a potent modulator of 8-oxoguanine repair. Neuroscience 2013, 252, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Ren, X.; Gowda, A.S.; Shan, Y.; Zhang, L.; Yuan, Y.; Patel, R.; Wu, H.; Huber-Keener, K.; Yang, J. Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death Dis. 2013, 4, e731. [Google Scholar] [CrossRef]
- Fan, J.; Wilson, D.M., II. Protein–protein interactions and posttranslational modifications in mammalian base excision repair. Free. Radic. Biol. Med. 2005, 38, 1121–1138. [Google Scholar] [CrossRef]
- Hang, B.; Singer, B. Protein− Protein Interactions Involving DNA Glycosylases. Chem. Res. Toxicol. 2003, 16, 1181–1195. [Google Scholar] [CrossRef]
- Limpose, K.L.; Corbett, A.H.; Doetsch, P.W. BERing the burden of damage: Pathway crosstalk and posttranslational modification of base excision repair proteins regulate DNA damage management. DNA Repair 2017, 56, 51–64. [Google Scholar] [CrossRef]
- Zharkov, D.O.; Rosenquist, T.A.; Gerchman, S.E.; Grollman, A.P. Substrate specificity and reaction mechanism of murine 8-oxoguanine-DNA glycosylase. J. Biol. Chem. 2000, 275, 28607–28617. [Google Scholar] [CrossRef]
- Yuen, P.K.; Green, S.A.; Ashby, J.; Lay, K.T.; Santra, A.; Chen, X.; Horvath, M.P.; David, S.S. Targeting base excision repair glycosylases with DNA containing transition state mimics prepared via click chemistry. ACS Chem. Biol. 2018, 14, 27–36. [Google Scholar] [CrossRef]
- Leipold, M.D.; Workman, H.; Muller, J.G.; Burrows, C.J.; David, S.S. Recognition and removal of oxidized guanines in duplex DNA by the base excision repair enzymes hOGG1, yOGG1, and yOGG2. Biochemistry 2003, 42, 11373–11381. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, M.R.; O’Brien, P.J. Human AP endonuclease 1 stimulates multiple-turnover base excision by alkyladenine DNA glycosylase. Biochemistry 2009, 48, 6022–6033. [Google Scholar] [CrossRef]
- Saitoh, T.; Shinmura, K.; Yamaguchi, S.; Tani, M.; Seki, S.; Murakami, H.; Nojima, Y.; Yokota, J. Enhancement of OGG1 protein AP lyase activity by increase of APEX protein. Mutat. Res. DNA Repair 2001, 486, 31–40. [Google Scholar] [CrossRef]
- Hill, J.W.; Hazra, T.K.; Izumi, T.; Mitra, S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: Potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001, 29, 430–438. [Google Scholar] [CrossRef]
- Vidal, A.E.; Hickson, I.D.; Boiteux, S.; Radicella, J.P. Mechanism of stimulation of the DNA glycosylase activity of hOGG1 by the major human AP endonuclease: Bypass of the AP lyase activity step. Nucleic Acids Res. 2001, 29, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Esadze, A.; Rodriguez, G.; Cravens, S.L.; Stivers, J.T. AP-endonuclease 1 accelerates turnover of human 8-oxoguanine DNA glycosylase by preventing retrograde binding to the abasic-site product. Biochemistry 2017, 56, 1974–1986. [Google Scholar] [CrossRef] [PubMed]
- Yudkina, A.V.; Endutkin, A.V.; Diatlova, E.A.; Moor, N.A.; Vokhtantsev, I.P.; Grin, I.R.; Zharkov, D.O. Displacement of slow-turnover DNA glycosylases by molecular traffic on DNA. Genes 2020, 11, 866. [Google Scholar] [CrossRef]
- Caldecott, K.W. XRCC1 protein; Form; function. DNA Repair 2019, 81, 102664. [Google Scholar] [CrossRef]
- Shi, G.; Chang, D.-Y.; Cheng, C.-C.; Guan, X.; Venclovas, Č.; Lu, A.-L. Physical and functional interactions between MutY glycosylase homologue (MYH) and checkpoint proteins Rad9–Rad1–Hus1. Biochem. J. 2006, 400, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Day, M.; Oliver, A.W.; Pearl, L.H. Structure of the human RAD17–RFC clamp loader and 9–1–1 checkpoint clamp bound to a dsDNA–ssDNA junction. Nucleic Acids Res. 2022, 50, 8279–8289. [Google Scholar] [CrossRef]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef]
- Porello, S.L.; Leyes, A.E.; David, S.S. Single-turnover and pre-steady-state kinetics of the reaction of the adenine glycosylase MutY with mismatch-containing DNA substrates. Biochemistry 1998, 37, 14756–14764. [Google Scholar] [CrossRef]
- Manlove, A.H.; McKibbin, P.L.; Doyle, E.L.; Majumdar, C.; Hamm, M.L.; David, S.S. Structure–activity relationships reveal key features of 8-oxoguanine: A mismatch detection by the MutY glycosylase. ACS Chem. Biol. 2017, 12, 2335–2344. [Google Scholar] [CrossRef]
- Conlon, S.G.; Khuu, C.; Trasviña-Arenas, C.H.; Xia, T.; Hamm, M.L.; Raetz, A.G.; David, S.S. Cellular Repair of Synthetic Analogs of Oxidative DNA Damage Reveals a Key Structure–Activity Relationship of the Cancer-Associated MUTYH DNA Repair Glycosylase. ACS Cent. Sci. 2024, 10, 291–301. [Google Scholar] [CrossRef]
- Nakamura, T.; Okabe, K.; Hirayama, S.; Chirifu, M.; Ikemizu, S.; Morioka, H.; Nakabeppu, Y.; Yamagata, Y. Structure of the mammalian adenine DNA glycosylase MUTYH: Insights into the base excision repair pathway and cancer. Nucleic Acids Res. 2021, 49, 7154–7163. [Google Scholar] [CrossRef]
- Trasviña-Arenas, C.H.; Dissanayake, U.C.; Tamayo, N.; Hashemian, M.; Lin, W.-J.; Demir, M.; Hoyos-Gonzalez, N.; Fisher, A.J.; Cisneros, G.A.; Horvath, M.P. Structure of human MUTYH and functional profiling of cancer-associated variants reveal an allosteric network between its cluster cofactor and active site required for DNA repair. Nat. Commun. 2025, 16, 3596. [Google Scholar] [CrossRef]
- Al-Tassan, N.; Chmiel, N.H.; Maynard, J.; Fleming, N.; Livingston, A.L.; Williams, G.T.; Hodges, A.K.; Davies, D.R.; David, S.S.; Sampson, J.R. Inherited variants of MYH associated with somatic G: C→ T: A mutations in colorectal tumors. Nat. Genet. 2002, 30, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Magrin, L.; Fanale, D.; Brando, C.; Corsini, L.R.; Randazzo, U.; Di Piazza, M.; Gurrera, V.; Pedone, E.; Russo, T.D.B.; Vieni, S. MUTYH-associated tumor syndrome: The other face of MAP. Oncogene 2022, 41, 2531–2539. [Google Scholar] [CrossRef] [PubMed]
- Raetz, A.G.; David, S.S. When you’re strange: Unusual features of the MUTYH glycosylase and implications in cancer. DNA Repair 2019, 80, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Cleary, S.P.; Cotterchio, M.; Jenkins, M.A.; Kim, H.; Bristow, R.; Green, R.; Haile, R.; Hopper, J.L.; LeMarchand, L.; Lindor, N. Germline MutY human homologue mutations and colorectal cancer: A multisite case-control study. Gastroenterology 2009, 136, 1251–1260. [Google Scholar] [CrossRef]
- Barreiro, R.A.S.; Sabbaga, J.; Rossi, B.M.; Achatz, M.I.W.; Bettoni, F.; Camargo, A.A.; Asprino, P.F.; Galante, P.A.F. Monoallelic deleterious MUTYH germline variants as a driver for tumorigenesis. J. Pathol. 2022, 256, 214–222. [Google Scholar] [CrossRef]
- Win, A.K.; Cleary, S.P.; Dowty, J.G.; Baron, J.A.; Young, J.P.; Buchanan, D.D.; Southey, M.C.; Burnett, T.; Parfrey, P.S.; Green, R.C. Cancer risks for monoallelic MUTYH mutation carriers with a family history of colorectal cancer. Int. J. Cancer 2011, 129, 2256–2262. [Google Scholar] [CrossRef]
- Chan, J.Y.; Toh, M.R.; Chong, S.T.; Ishak, N.D.B.; Kolinjivadi, A.M.; Chan, S.H.; Lee, E.; Boot, A.; Shao-Tzu, L.; Chew, M.-H. Multiple neoplasia in a patient with Gitelman syndrome harboring germline monoallelic MUTYH mutation. NPJ Genom. Med. 2020, 5, 39. [Google Scholar] [CrossRef]
- Zhao, B.; Sun, W.; Wang, Y.; Wu, X.; Li, Y.; Wang, W.; Ni, M.; Yan, P.; Dou, X.; Wang, L. Monoallelic deleterious MUTYH mutations generate colorectal cancer: A case report. Clin. Case Rep. 2023, 11, e8229. [Google Scholar] [CrossRef]
- Grasso, F.; Giacomini, E.; Sanchez, M.; Degan, P.; Gismondi, V.; Mazzei, F.; Varesco, L.; Viel, A.; Bignami, M. Genetic instability in lymphoblastoid cell lines expressing biallelic and monoallelic variants in the human MUTYH gene. Hum. Mol. Genet. 2014, 23, 3843–3852. [Google Scholar] [CrossRef] [PubMed]
- Venesio, T.; Balsamo, A.; Errichiello, E.; Ranzani, G.N.; Risio, M. Oxidative DNA damage drives carcinogenesis in MUTYH-associated-polyposis by specific mutations of mitochondrial and MAPK genes. Mod. Pathol. 2013, 26, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Nakabeppu, Y. DNA glycosylase encoded by MUTYH functions as a molecular switch for programmed cell death under oxidative stress to suppress tumorigenesis. Cancer Sci. 2011, 102, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Tominaga, Y.; Yamauchi, K.; Nakatsu, Y.; Sakumi, K.; Yoshiyama, K.; Egashira, A.; Kura, S.; Yao, T.; Tsuneyoshi, M. MUTYH-null mice are susceptible to spontaneous and oxidative stress–induced intestinal tumorigenesis. Cancer Res. 2007, 67, 6599–6604. [Google Scholar] [CrossRef]
- Sakumi, K.; Tominaga, Y.; Furuichi, M.; Xu, P.; Tsuzuki, T.; Sekiguchi, M.; Nakabeppu, Y. Ogg1 knockout-associated lung tumorigenesis and its suppression by Mth1 gene disruption. Cancer Res. 2003, 63, 902–905. [Google Scholar]
- Cande, C.; Vahsen, N.; Garrido, C.; Kroemer, G. Apoptosis-inducing factor (AIF): Caspase-independent after all. Cell Death Differ. 2004, 11, 591–595. [Google Scholar] [CrossRef]
- Momeni, H.R. Role of calpain in apoptosis. Cell J. 2011, 13, 65. [Google Scholar]
- Köger, N.; Brieger, A.; Hinrichsen, I.M.; Zeuzem, S.; Plotz, G. Analysis of MUTYH alternative transcript expression, promoter function, and the effect of human genetic variants. Hum. Mutat. 2019, 40, 472–482. [Google Scholar] [CrossRef]
- Seo, Y.-K.; Chong, H.K.; Infante, A.M.; Im, S.-S.; Xie, X.; Osborne, T.F. Genome-wide analysis of SREBP-1 binding in mouse liver chromatin reveals a preference for promoter proximal binding to a new motif. Proc. Natl. Acad. Sci. USA 2009, 106, 13765–13769. [Google Scholar] [CrossRef]
- Trung, N.T.; Kremmer, E.; Mittler, G. Biochemical and cellular characterization of transcription factors binding to the hyperconserved core promoter-associated M4 motif. BMC Genom. 2016, 17, 693. [Google Scholar] [CrossRef]
- Chen, J.; Wu, X.; Wang, Y.; Pan, Y.; Ren, Y.; Nakabeppu, Y.; Fan, Y.; Wang, Y. Mutyh deficiency downregulates mitochondrial fusion proteins and causes cardiac dysfunction via α-ketoglutaric acid reduction with oxidative stress. Free. Radic. Res. 2022, 56, 129–142. [Google Scholar] [CrossRef]
- Gao, D.; Wei, C.; Chen, L.; Huang, J.; Yang, S.; Diehl, A.M. Oxidative DNA damage and DNA repair enzyme expression are inversely related in murine models of fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1070–G1077. [Google Scholar] [CrossRef]
- Bjørge, M.D.; Hildrestrand, G.A.; Scheffler, K.; Suganthan, R.; Rolseth, V.; Kuśnierczyk, A.; Rowe, A.D.; Vågbø, C.B.; Vetlesen, S.; Eide, L. Synergistic actions of Ogg1 and mutyh DNA glycosylases modulate anxiety-like behavior in mice. Cell Rep. 2015, 13, 2671–2678. [Google Scholar] [CrossRef] [PubMed]
- Cilli, P.; Ventura, I.; Minoprio, A.; Meccia, E.; Martire, A.; Wilson, S.H.; Bignami, M.; Mazzei, F. Oxidized dNTPs and the OGG1 and MUTYH DNA glycosylases combine to induce CAG/CTG repeat instability. Nucleic Acids Res. 2016, 44, 5190–5203. [Google Scholar] [CrossRef] [PubMed]
- Ichinoe, A.; Behmanesh, M.; Tominaga, Y.; Ushijima, Y.; Hirano, S.; Sakai, Y.; Tsuchimoto, D.; Sakumi, K.; Wake, N.; Nakabeppu, Y. Identification and characterization of two forms of mouse MUTYH proteins encoded by alternatively spliced transcripts. Nucleic Acids Res. 2004, 32, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Plotz, G.; Casper, M.; Raedle, J.; Hinrichsen, I.; Heckel, V.; Brieger, A.; Trojan, J.; Zeuzem, S. MUTYH gene expression and alternative splicing in controls and polyposis patients. Hum. Mutat. 2012, 33, 1067–1074. [Google Scholar] [CrossRef]
- Englander, E.W.; Hu, Z.; Sharma, A.; Lee, H.M.; Wu, Z.H.; Greeley, G.H.; MYH, R. a glycosylase for repair of oxidatively damaged DNA, has brain-specific isoforms that localize to neuronal mitochondria. J. Neurochem. 2002, 83, 1471–1480. [Google Scholar] [CrossRef]
- Gharib, A.; De Paulis, D.; Li, B.; Augeul, L.; Couture-Lepetit, E.; Gomez, L.; Angoulvant, D.; Ovize, M. Opposite and tissue-specific effects of coenzyme Q2 on mPTP opening and ROS production between heart and liver mitochondria: Role of complex I. J. Mol. Cell. Cardiol. 2012, 52, 1091–1095. [Google Scholar] [CrossRef]
- Panov, A.; Dikalov, S.; Shalbuyeva, N.; Hemendinger, R.; Greenamyre, J.T.; Rosenfeld, J. Species-and tissue-specific relationships between mitochondrial permeability transition and generation of ROS in brain and liver mitochondria of rats and mice. Am. J. Physiol. Cell Physiol. 2007, 292, C708–C718. [Google Scholar] [CrossRef]
- Kuno, T.; Matsubara, N.; Tsuda, S.; Kobayashi, M.; Hamanaka, M.; Yamagishi, D.; Tsukamoto, K.; Yamano, T.; Noda, M.; Ikeuchi, H. Alterations of the base excision repair gene MUTYH in sporadic colorectal cancer. Oncol. Rep. 2012, 28, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, E.F.R.; Ribeiro, M.L.; Magro, D.O.; Carvalho, J.; Kanno, D.T.; Martinez, C.A.R.; Coy, C.S.R. Tissue expresion of the genes mutyh and ogg1 in patients with sporadic colorectal cancer. ABCD. Arq. Bras. Cir. Dig. 2017, 30, 98–102. [Google Scholar] [CrossRef]
- Thet, M.; Plazzer, J.-P.; Capella, G.; Latchford, A.; Nadeau, E.A.; Greenblatt, M.S.; Macrae, F. Phenotype Correlations With Pathogenic DNA Variants in the MUTYH Gene: A Review of Over 2000 Cases. Hum. Mutat. 2024, 2024, 8520275. [Google Scholar] [CrossRef]
- Russelburg, L.P.; Murray, V.L.O.; Demir, M.; Knutsen, K.R.; Sehgal, S.L.; Cao, S.; David, S.S.; Horvath, M.P. Structural Basis for Finding OG Lesions and Avoiding Undamaged G by the DNA Glycosylase MutY. ACS Chem. Biol. 2019, 15, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Batra, V.K.; Shock, D.D.; Beard, W.A.; McKenna, C.E.; Wilson, S.H. Binary complex crystal structure of DNA polymerase β reveals multiple conformations of the templating 8-oxoguanine lesion. Proc. Natl. Acad. Sci. USA 2012, 109, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, T.D.; Jain, R.; Johnson, R.E.; Prakash, L.; Prakash, S.; Aggarwal, A.K. Structural basis for error-free replication of oxidatively damaged DNA by yeast DNA polymerase η. Structure 2010, 18, 1463–1470. [Google Scholar] [CrossRef]
- Benítez, B.S.; Barbati, Z.R.; Arora, K.; Bogdanovic, J.; Schlick, T. How DNA polymerase X preferentially accommodates incoming dATP opposite 8-oxoguanine on the template. Biophys. J. 2013, 105, 2559–2568. [Google Scholar] [CrossRef][Green Version]
- Brieba, L.G.; Eichman, B.F.; Kokoska, R.J.; Doublié, S.; Kunkel, T.A.; Ellenberger, T. Structural basis for the dual coding potential of 8-oxoguanosine by a high-fidelity DNA polymerase. EMBO J. 2004, 23, 3452–3461. [Google Scholar] [CrossRef]
- Lee, A.J.; Majumdar, C.; Kathe, S.D.; Van Ostrand, R.P.; Vickery, H.R.; Averill, A.M.; Nelson, S.R.; Manlove, A.H.; McCord, M.A.; David, S.S. Detection of OG: A lesion mispairs by MutY relies on a single His residue and the 2-amino group of 8-oxoguanine. J. Am. Chem. Soc. 2020, 142, 13283–13287. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, N.N.; Khuu, C.; Babu, C.S.; Bertolani, S.J.; Rajavel, A.N.; Spear, J.E.; Armas, J.A.; Wright, J.D.; Siegel, J.B.; Lim, C. The Zinc Linchpin Motif in the DNA Repair Glycosylase MUTYH: Identifying the Zn2+ Ligands and Roles in Damage Recognition and Repair. J. Am. Chem. Soc. 2018, 140, 13260–13271. [Google Scholar] [CrossRef]
- Engstrom, L.M.; Brinkmeyer, M.K.; Ha, Y.; Raetz, A.G.; Hedman, B.; Hodgson, K.O.; Solomon, E.I.; David, S.S. A zinc linchpin motif in the MUTYH glycosylase interdomain connector is required for efficient repair of DNA damage. J. Am. Chem. Soc. 2014, 136, 7829–7832. [Google Scholar] [CrossRef] [PubMed]
- Porello, S.L.; Cannon, M.J.; David, S.S. A substrate recognition role for the 2+ cluster of the DNA repair glycosylase MutY. Biochemistry 1998, 37, 6465–6475. [Google Scholar] [CrossRef] [PubMed]
- Chepanoske, C.L.; Golinelli, M.-P.; Williams, S.D.; David, S.S. Positively Charged Residues within the Iron–Sulfur Cluster Loop of E. coli MutY Participate in Damage Recognition and Removal. Arch. Biochem. Biophys. 2000, 380, 11–19. [Google Scholar] [CrossRef]
- Arnold, A.R.; Grodick, M.A.; Barton, J.K. DNA charge transport: From chemical principles to the cell. Cell Chem. Biol. 2016, 23, 183–197. [Google Scholar] [CrossRef]
- Boal, A.K.; Yavin, E.; Lukianova, O.A.; O’Shea, V.L.; David, S.S.; Barton, J.K. DNA-bound redox activity of DNA repair glycosylases containing clusters. Biochemistry 2005, 44, 8397–8407. [Google Scholar] [CrossRef]
- Sontz, P.A.; Mui, T.P.; Fuss, J.O.; Tainer, J.A.; Barton, J.K. DNA charge transport as a first step in coordinating the detection of lesions by repair proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 1856–1861. [Google Scholar] [CrossRef]
- Demir, M.; Russelburg, L.P.; Lin, W.-J.; Trasviña-Arenas, C.H.; Huang, B.; Yuen, P.K.; Horvath, M.P.; David, S.S. Structural snapshots of base excision by the cancer-associated variant MutY N146S reveal a retaining mechanism. Nucleic Acids Res. 2023, 51, 1034–1049. [Google Scholar] [CrossRef]
- Gu, Y.; Lu, A.-L. Differential DNA recognition and glycosylase activity of the native human MutY homolog (hMYH) and recombinant hMYH expressed in bacteria. Nucleic Acids Res. 2001, 29, 2666–2674. [Google Scholar] [CrossRef]
- Parker, A.R.; O’Meally, R.N.; Sahin, F.; Su, G.H.; Racke, F.K.; Nelson, W.G.; DeWeese, T.L.; Eshleman, J.R. Defective human MutY phosphorylation exists in colorectal cancer cell lines with wild-type MutY alleles. J. Biol. Chem. 2003, 278, 47937–47945. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Brinkmeyer, M.K.; Eigenheer, R.A.; David, S.S. Ser 524 is a phosphorylation site in MUTYH and Ser 524 mutations alter 8-oxoguanine (OG): A mismatch recognition. DNA Repair 2010, 9, 1026–1037. [Google Scholar] [CrossRef]
- Stes, E.; Laga, M.; Walton, A.; Samyn, N.; Timmerman, E.; De Smet, I.; Goormachtig, S.; Gevaert, K. A COFRADIC protocol to study protein ubiquitination. J. Proteome Res. 2014, 13, 3107–3113. [Google Scholar] [CrossRef]
- Oshikawa, K.; Matsumoto, M.; Oyamada, K.; Nakayama, K.I. Proteome-wide identification of ubiquitylation sites by conjugation of engineered lysine-less ubiquitin. J. Proteome Res. 2012, 11, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Dorn, J.; Ferrari, E.; Imhof, R.; Ziegler, N.; Hübscher, U. Regulation of human MutYH DNA glycosylase by the E3 ubiquitin ligase mule. J. Biol. Chem. 2014, 289, 7049–7058. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, I.A.; Larsen, S.C.; Nielsen, M.L. An advanced strategy for comprehensive profiling of ADP-ribosylation sites using mass spectrometry-based proteomics. Mol. Cell. Proteom. 2019, 18, 1010–1026. [Google Scholar] [CrossRef]
- Gu, Y.; Parker, A.; Wilson, T.M.; Bai, H.; Chang, D.-Y.; Lu, A.-L. Human MutY homolog, a DNA glycosylase involved in base excision repair, physically and functionally interacts with mismatch repair proteins human MutS homolog 2/human MutS homolog 6. J. Biol. Chem. 2002, 277, 11135–11142. [Google Scholar] [CrossRef]
- Boldogh, I.; Milligan, D.; Lee, M.S.; Bassett, H.; Lloyd, R.S.; McCullough, A.K. hMYH cell cycle-dependent expression, subcellular localization and association with replication foci: Evidence suggesting replication-coupled repair of adenine: 8-oxoguanine mispairs. Nucleic Acids Res. 2001, 29, 2802–2809. [Google Scholar] [CrossRef]
- Chang, D.-Y.; Lu, A.-L. Interaction of checkpoint proteins Hus1/Rad1/Rad9 with DNA base excision repair enzyme MutY homolog in fission yeast, Schizosaccharomyces pombe. J. Biol. Chem. 2005, 280, 408–417. [Google Scholar] [CrossRef]
- Yang, H.; Clendenin, W.M.; Wong, D.; Demple, B.; Slupska, M.M.; Chiang, J.-H.; Miller, J.H. Enhanced activity of adenine-DNA glycosylase (Myh) by apurinic/apyrimidinic endonuclease (Ape1) in mammalian base excision repair of an A/GO mismatch. Nucleic Acids Res. 2001, 29, 743–752. [Google Scholar] [CrossRef]
- Hwang, B.-J.; Jin, J.; Gao, Y.; Shi, G.; Madabushi, A.; Yan, A.; Guan, X.; Zalzman, M.; Nakajima, S.; Lan, L. SIRT6 protein deacetylase interacts with MYH DNA glycosylase, APE1 endonuclease, and Rad9–Rad1–Hus1 checkpoint clamp. BMC Mol. Biol. 2015, 16, 1–16. [Google Scholar] [CrossRef]
- Jang, S.; Schaich, M.A.; Khuu, C.; Schnable, B.L.; Majumdar, C.; Watkins, S.C.; David, S.S.; Van Houten, B. Single molecule analysis indicates stimulation of MUTYH by UV-DDB through enzyme turnover. Nucleic Acids Res. 2021, 49, 8177–8188. [Google Scholar] [CrossRef] [PubMed]
- McSweeney, K.R.; Gadanec, L.K.; Qaradakhi, T.; Ali, B.A.; Zulli, A.; Apostolopoulos, V. Mechanisms of cisplatin-induced acute kidney injury: Pathological mechanisms, pharmacological interventions, and genetic mitigations. Cancers 2021, 13, 1572. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, P.; Zhou, K.; Zhang, W.; Liu, S.; Ouyang, J.; Bai, M.; Ding, G.; Huang, S.; Jia, Z. HUWE1-Mediated Degradation of MUTYH Facilitates DNA Damage and Mitochondrial Dysfunction to Promote Acute Kidney Injury. Adv. Sci. 2025, 12, 2412250. [Google Scholar] [CrossRef] [PubMed]
- Raetz, A.G.; Banda, D.M.; Ma, X.; Xu, G.; Rajavel, A.N.; McKibbin, P.L.; Lebrilla, C.B.; David, S.S. The DNA repair enzyme MUTYH potentiates cytotoxicity of the alkylating agent MNNG by interacting with abasic sites. J. Biol. Chem. 2020, 295, 3692–3707. [Google Scholar] [CrossRef]
- Schwarz, S.D.; Xu, J.; Gunasekera, K.; Schürmann, D.; Vågbø, C.B.; Ferrari, E.; Slupphaug, G.; Hottiger, M.O.; Schär, P.; Steinacher, R. Covalent PARylation of DNA base excision repair proteins regulates DNA demethylation. Nat. Commun. 2024, 15, 184. [Google Scholar] [CrossRef]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chen, J.; Huang, S.-Y.N.; Su, Y.P.; Wang, W.; Agama, K.; Saha, S.; Jenkins, L.M.; Pascal, J.M.; Pommier, Y. PARylation prevents the proteasomal degradation of topoisomerase I DNA-protein crosslinks and induces their deubiquitylation. Nat. Commun. 2021, 12, 5010. [Google Scholar] [CrossRef]
- Baranovskiy, A.G.; Duong, V.N.; Babayeva, N.D.; Zhang, Y.; Pavlov, Y.I.; Anderson, K.S.; Tahirov, T.H. Activity and fidelity of human DNA polymerase α depend on primer structure. J. Biol. Chem. 2018, 293, 6824–6843. [Google Scholar] [CrossRef]
- Schmitt, M.W.; Matsumoto, Y.; Loeb, L.A. High fidelity and lesion bypass capability of human DNA polymerase δ. Biochimie 2009, 91, 1163–1172. [Google Scholar] [CrossRef]
- Korona, D.A.; LeCompte, K.G.; Pursell, Z.F. The high fidelity and unique error signature of human DNA polymerase ε. Nucleic Acids Res. 2011, 39, 1763–1773. [Google Scholar] [CrossRef]
- Osheroff, W.P.; Jung, H.K.; Beard, W.A.; Wilson, S.H.; Kunkel, T.A. The fidelity of DNA polymerase β during distributive and processive DNA synthesis. J. Biol. Chem. 1999, 274, 3642–3650. [Google Scholar] [CrossRef] [PubMed]
- Kolodner, R.D.; Marsischky, G.T. Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 1999, 9, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, A.; Berardini, M.; Fishel, R. Activation of human MutS homologs by 8-oxo-guanine DNA damage. J. Biol. Chem. 2002, 277, 8260–8266. [Google Scholar] [CrossRef]
- Chmiel, N.H.; Golinelli, M.-P.; Francis, A.W.; David, S.S. Efficient recognition of substrates and substrate analogs by the adenine glycosylase MutY requires the C-terminal domain. Nucleic Acids Res. 2001, 29, 553–564. [Google Scholar] [CrossRef][Green Version]
- Colussi, C.; Parlanti, E.; Degan, P.; Aquilina, G.; Barnes, D.; Macpherson, P.; Karran, P.; Crescenzi, M.; Dogliotti, E.; Bignami, M. The mammalian mismatch repair pathway removes DNA 8-oxodGMP incorporated from the oxidized dNTP pool. Curr. Biol. 2002, 12, 912–918. [Google Scholar] [CrossRef] [PubMed]
- DeWeese, T.L.; Shipman, J.M.; Larrier, N.A.; Buckley, N.M.; Kidd, L.R.; Groopman, J.D.; Cutler, R.G.; Riele, H.T.; Nelson, W.G. Mouse embryonic stem cells carrying one or two defective Msh2 alleles respond abnormally to oxidative stress inflicted by low-level radiation. Proc. Natl. Acad. Sci. USA 1998, 95, 11915–11920. [Google Scholar] [CrossRef]
- Chang, C.L.; Marra, G.; Chauhan, D.P.; Ha, H.T.; Chang, D.K.; Ricciardiello, L.; Randolph, A.; Carethers, J.M.; Boland, C.R. Oxidative stress inactivates the human DNA mismatch repair system. Am. J. Physiol. Cell Physiol. 2002, 283, C148–C154. [Google Scholar] [CrossRef]
- Hegde, M.L.; Hazra, T.K.; Mitra, S. Functions of disordered regions in mammalian early base excision repair proteins. Cell. Mol. Life Sci. 2010, 67, 3573–3587. [Google Scholar] [CrossRef]
- Burak, M.J.; Guja, K.E.; Hambardjieva, E.; Derkunt, B.; Garcia-Diaz, M. A fidelity mechanism in DNA polymerase lambda promotes error-free bypass of 8-oxo-dG. EMBO J. 2016, 35, 2045–2059. [Google Scholar] [CrossRef] [PubMed]
- van Loon, B.; Hübscher, U. An 8-oxo-guanine repair pathway coordinated by MUTYH glycosylase and DNA polymerase λ. Proc. Natl. Acad. Sci. USA 2009, 106, 18201–18206. [Google Scholar] [CrossRef]
- Moldovan, G.-L.; Pfander, B.; Jentsch, S. PCNA, the maestro of the replication fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.; Gu, Y.; Mahoney, W.; Lee, S.-H.; Singh, K.K.; Lu, A.-L. Human homolog of the MutY repair protein (hMYH) physically interacts with proteins involved in long patch DNA base excision repair. J. Biol. Chem. 2001, 276, 5547–5555. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Tominaga, Y.; Hirano, S.; McKenna, A.E.; Nakabeppu, Y.; Matsumoto, Y. Replication-associated repair of adenine: 8-oxoguanine mispairs by MYH. Curr. Biol. 2002, 12, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Brinkmeyer, M.K.; David, S.S. Distinct functional consequences of MUTYH variants associated with colorectal cancer: Damaged DNA affinity, glycosylase activity and interaction with PCNA and Hus1. DNA Repair 2015, 34, 39–51. [Google Scholar] [CrossRef]
- Dianova, I.I.; Bohr, V.A.; Dianov, G.L. Interaction of human AP endonuclease 1 with flap endonuclease 1 and proliferating cell nuclear antigen involved in long-patch base excision repair. Biochemistry 2001, 40, 12639–12644. [Google Scholar] [CrossRef]
- Maga, G.; Villani, G.; Ramadan, K.; Shevelev, I.; Le Gac, N.T.; Blanco, L.; Blanca, G.; Spadari, S.; Hübscher, U. Human DNA polymerase λ functionally and physically interacts with proliferating cell nuclear antigen in normal and translesion DNA synthesis. J. Biol. Chem. 2002, 277, 48434–48440. [Google Scholar] [CrossRef]
- Luncsford, P.J.; Manvilla, B.A.; Patterson, D.N.; Malik, S.S.; Jin, J.; Hwang, B.-J.; Gunther, R.; Kalvakolanu, S.; Lipinski, L.J.; Yuan, W. Coordination of MYH DNA glycosylase and APE1 endonuclease activities via physical interactions. DNA Repair 2013, 12, 1043–1052. [Google Scholar] [CrossRef]
- Xu, M.; Bai, L.; Gong, Y.; Xie, W.; Hang, H.; Jiang, T. Structure and functional implications of the human rad9-hus1-rad1 cell cycle checkpoint complex. J. Biol. Chem. 2009, 284, 20457–20461. [Google Scholar] [CrossRef]
- Helt, C.E.; Wang, W.; Keng, P.C.; Bambara, R.A. 9-1-1 Complex involvement in DNA repair: Evidence that DNA damage detection machinery participates in DNA repair. Cell Cycle 2005, 4, 529–532. [Google Scholar] [CrossRef]
- Gembka, A.; Toueille, M.; Smirnova, E.; Poltz, R.; Ferrari, E.; Villani, G.; Hübscher, U. The checkpoint clamp, Rad9-Rad1-Hus1 complex, preferentially stimulates the activity of apurinic/apyrimidinic endonuclease 1 and DNA polymerase β in long patch base excision repair. Nucleic Acids Res. 2007, 35, 2596–2608. [Google Scholar] [CrossRef] [PubMed]
- Toueille, M.; El-Andaloussi, N.; Frouin, I.; Freire, R.; Funk, D.; Shevelev, I.; Friedrich-Heineken, E.; Villani, G.; Hottiger, M.O.; Hübscher, U. The human Rad9/Rad1/Hus1 damage sensor clamp interacts with DNA polymerase β and increases its DNA substrate utilisation efficiency: Implications for DNA repair. Nucleic Acids Res. 2004, 32, 3316–3324. [Google Scholar] [CrossRef]
- Guan, X.; Madabushi, A.; Chang, D.-Y.; Fitzgerald, M.E.; Shi, G.; Drohat, A.C.; Lu, A.-L. The human checkpoint sensor Rad9–Rad1–Hus1 interacts with and stimulates DNA repair enzyme TDG glycosylase. Nucleic Acids Res. 2007, 35, 6207–6218. [Google Scholar] [CrossRef]
- Guan, X.; Bai, H.; Shi, G.; Theriot, C.A.; Hazra, T.K.; Mitra, S.; Lu, A.-L. The human checkpoint sensor Rad9–Rad1–Hus1 interacts with and stimulates NEIL1 glycosylase. Nucleic Acids Res. 2007, 35, 2463–2472. [Google Scholar] [CrossRef]
- Wang, W.; Brandt, P.; Rossi, M.L.; Lindsey-Boltz, L.; Podust, V.; Fanning, E.; Sancar, A.; Bambara, R.A. The human Rad9–Rad1–Hus1 checkpoint complex stimulates flap endonuclease 1. Proc. Natl. Acad. Sci. USA 2004, 101, 16762–16767. [Google Scholar] [CrossRef]
- Smirnova, E.; Toueille, M.; Markkanen, E.; Hübscher, U. The human checkpoint sensor and alternative DNA clamp Rad9–Rad1–Hus1 modulates the activity of DNA ligase I, a component of the long-patch base excision repair machinery. Biochem. J. 2005, 389, 13. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, Y.; Ushijima, Y.; Tsuchimoto, D.; Mishima, M.; Shirakawa, M.; Hirano, S.; Sakumi, K.; Nakabeppu, Y. MUTYH prevents OGG1 or APEX1 from inappropriately processing its substrate or reaction product with its C-terminal domain. Nucleic Acids Res. 2004, 32, 3198–3211. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.-J.; Jin, J.; Gunther, R.; Madabushi, A.; Shi, G.; Wilson, G.M.; Lu, A.-L. Association of the Rad9–Rad1–Hus1 checkpoint clamp with MYH DNA glycosylase and DNA. DNA Repair 2015, 31, 80–90. [Google Scholar] [CrossRef]
- Kwon, S.B.; Ryu, K.; Son, A.; Jeong, H.; Lim, K.-H.; Kim, K.-H.; Seong, B.L.; Choi, S.I. Conversion of a soluble protein into a potent chaperone in vivo. Sci. Rep. 2019, 9, 2735, Erratum in Sci. Rep. 2019, 9, 12812. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.