


OGG1 Preserves Endothelial-Dependent Vasodilation and Regulates the Frequency and Spatial Area of Endothelial Calcium Signals

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Loss of OGG1 Alters Endothelial Calcium Signaling Dynamics
3.2. OGG1 Regulates the Frequency and Spatial Area of Calcium Signals in the Presence of ACh
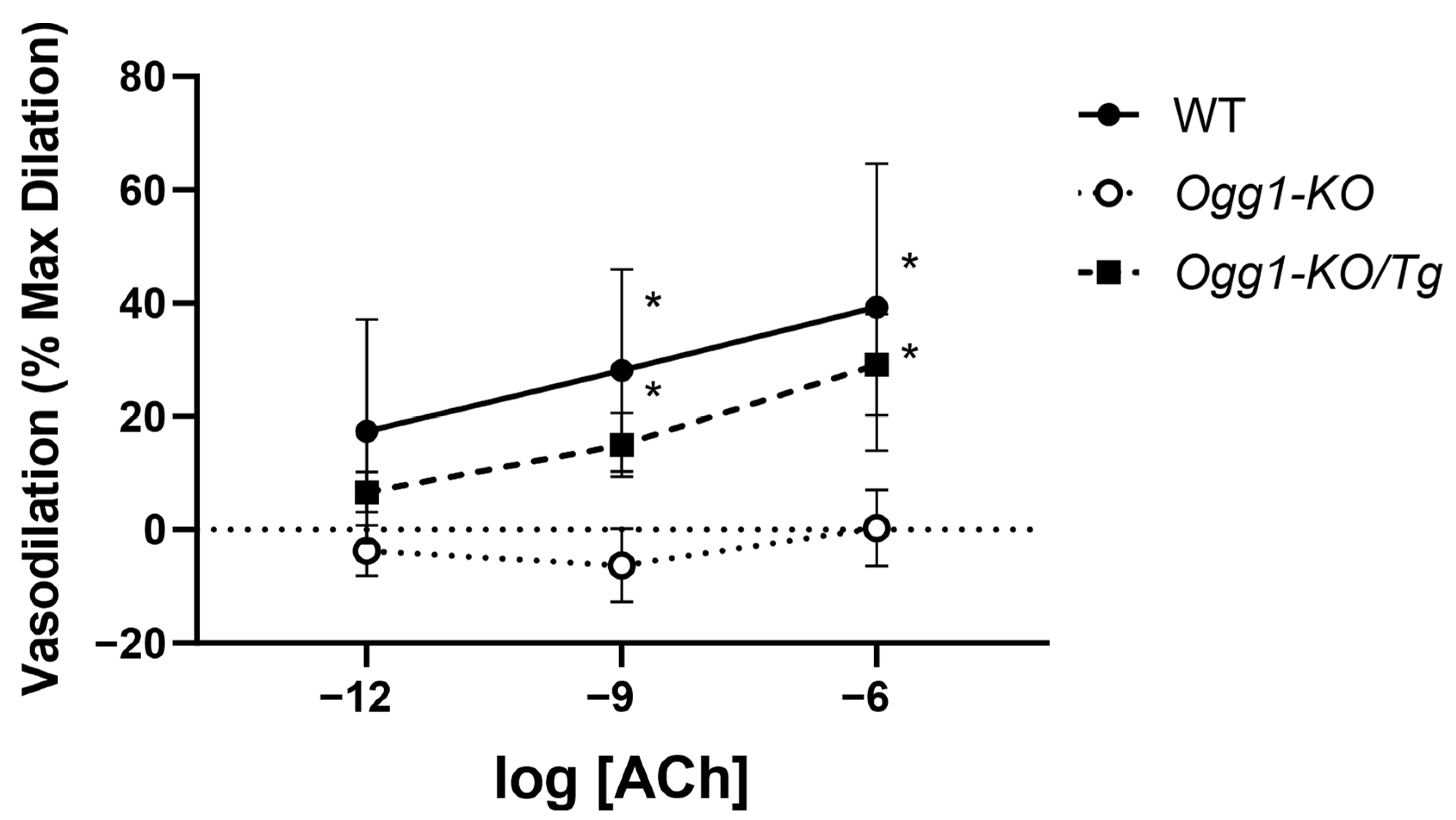
3.3. Endothelial-Dependent Vasodilation Is Impaired by Loss of OGG1 and Rescued by Mitochondrial OGG1 Repletion
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sandoo, A.; van Zanten, J.J.C.S.V.; Metsios, G.S.; Carroll, D.; Kitas, G.D. The Endothelium and Its Role in Regulating Vascular Tone. Open Cardiovasc. Med. J. 2010, 4, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.; Garcia, T.; Aniqa, M.; Ali, S.; Ally, A.; Nauli, S. Endothelial Nitric Oxide Synthase (eNOS) and the Cardiovascular System: In Physiology and in Disease States. Am. J. Biomed. Sci. Res. 2022, 15, 153–177. [Google Scholar]
- Amberg, G.C.; Navedo, M.F. Calcium Dynamics in Vascular Smooth Muscle. Microcirculation 2013, 20, 281–289. [Google Scholar] [CrossRef]
- Ottolini, M.; Sonkusare, S.K. The Calcium Signaling Mechanisms in Arterial Smooth Muscle and Endothelial Cells. Compr. Physiol. 2021, 11, 1831–1869. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Brunetti, V.; Soda, T.; Berra-Romani, R.; Scarpellino, G. Cracking the Endothelial Calcium (Ca2+) Code: A Matter of Timing and Spacing. Int. J. Mol. Sci. 2023, 24, 16765. [Google Scholar] [CrossRef]
- Debir, B.; Meaney, C.; Kohandel, M.; Unlu, M.B. The Role of Calcium Oscillations in the Phenotype Selection in Endothelial Cells. Sci. Rep. 2021, 11, 23781. [Google Scholar] [CrossRef] [PubMed]
- Janaszak-Jasiecka, A.; Płoska, A.; Wierońska, J.M.; Dobrucki, L.W.; Kalinowski, L. Endothelial Dysfunction Due to eNOS Uncoupling: Molecular Mechanisms as Potential Therapeutic Targets. Cell. Mol. Biol. Lett. 2023, 28, 21. [Google Scholar] [CrossRef]
- Davignon, J.; Ganz, P. Role of Endothelial Dysfunction in Atherosclerosis. Circulation 2004, 109, III-27–III-32. [Google Scholar] [CrossRef]
- Ba, X.; Boldogh, I. 8-Oxoguanine DNA Glycosylase 1: Beyond Repair of the Oxidatively Modified Base Lesions. Redox Biol. 2017, 14, 669–678. [Google Scholar] [CrossRef]
- Cadet, J.; Davies, K.J.A. Oxidative DNA Damage & Repair: An Introduction. Free Radic. Biol. Med. 2017, 107, 2–12. [Google Scholar] [CrossRef]
- Shah, A.; Gray, K.; Figg, N.; Finigan, A.; Starks, L.; Bennett, M. Defective Base Excision Repair of Oxidative DNA Damage in Vascular Smooth Muscle Cells Promotes Atherosclerosis. Circulation 2018, 138, 1446–1462. [Google Scholar] [CrossRef] [PubMed]
- Tumurkhuu, G.; Shimada, K.; Dagvadorj, J.; Crother, T.R.; Zhang, W.; Luthringer, D.; Gottlieb, R.A.; Chen, S.; Arditi, M. Ogg1-Dependent DNA Repair Regulates NLRP3 Inflammasome and Prevents Atherosclerosis. Circ. Res. 2016, 119, e76–e90. [Google Scholar] [CrossRef] [PubMed]
- Foote, K.; Rienks, M.; Theofilatos, K.; Shah, A.V.; Figg, N.L.; Finigan, A.; Mayr, M.; Bennett, M. Abstract MP125: Enhancing Base Excision Repair of Oxidative DNA Damage Delays Vascular Aging in Mice. Arterioscler. Thromb. Vasc. Biol. 2020, 40, AMP125. [Google Scholar] [CrossRef]
- Ruchko, M.V.; Gorodnya, O.M.; Zuleta, A.; Pastukh, V.M.; Gillespie, M.N. The DNA Glycosylase Ogg1 Defends against Oxidant-Induced mtDNA Damage and Apoptosis in Pulmonary Artery Endothelial Cells. Free Radic. Biol. Med. 2011, 50, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Torres-Gonzalez, M.; Gawlowski, T.; Kocalis, H.; Scott, B.T.; Dillmann, W.H. Mitochondrial 8-Oxoguanine Glycosylase Decreases Mitochondrial Fragmentation and Improves Mitochondrial Function in H9C2 Cells under Oxidative Stress Conditions. Am. J. Physiol.-Cell Physiol. 2014, 306, C221–C229. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Q.; Watson, L.J.; Jones, S.P.; Epstein, P.N. Cardiac Overexpression of 8-Oxoguanine DNA Glycosylase 1 Protects Mitochondrial DNA and Reduces Cardiac Fibrosis Following Transaortic Constriction. Am. J. Physiol.-Heart Circ. Physiol. 2011, 301, H2073–H2080. [Google Scholar] [CrossRef]
- Okasaka, T.; Matsuo, K.; Suzuki, T.; Ito, H.; Hosono, S.; Kawase, T.; Watanabe, M.; Yatabe, Y.; Hida, T.; Mitsudomi, T.; et al. hOGG1 Ser326Cys Polymorphism and Risk of Lung Cancer by Histological Type. J. Hum. Genet. 2009, 54, 739–745. [Google Scholar] [CrossRef]
- Nock, N.L.; Cicek, M.S.; Li, L.; Liu, X.; Rybicki, B.A.; Moreira, A.; Plummer, S.J.; Casey, G.; Witte, J.S. Polymorphisms in Estrogen Bioactivation, Detoxification and Oxidative DNA Base Excision Repair Genes and Prostate Cancer Risk. Carcinogenesis 2006, 27, 1842–1848. [Google Scholar] [CrossRef]
- Yuzefovych, L.V.; Kahn, A.G.; Schuler, M.A.; Eide, L.; Arora, R.; Wilson, G.L.; Tan, M.; Rachek, L.I. Mitochondrial DNA Repair through OGG1 Activity Attenuates Breast Cancer Progression and Metastasis. Cancer Res. 2016, 76, 30–34. [Google Scholar] [CrossRef]
- Abolhassani, N.; Leon, J.; Sheng, Z.; Oka, S.; Hamasaki, H.; Iwaki, T.; Nakabeppu, Y. Molecular Pathophysiology of Impaired Glucose Metabolism, Mitochondrial Dysfunction, and Oxidative DNA Damage in Alzheimer’s Disease Brain. Mech. Ageing Dev. 2017, 161, 95–104. [Google Scholar] [CrossRef]
- Cardozo-Pelaez, F.; Sanchez-Contreras, M.; Nevin, A.B.C. Ogg1 Null Mice Exhibit Age-Associated Loss of the Nigrostriatal Pathway and Increased Sensitivity to MPTP. Neurochem. Int. 2012, 61, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Simon, H.; Vartanian, V.; Wong, M.H.; Nakabeppu, Y.; Sharma, P.; Lloyd, R.S.; Sampath, H. OGG1 Deficiency Alters the Intestinal Microbiome and Increases Intestinal Inflammation in a Mouse Model. PLoS ONE 2020, 15, e0227501. [Google Scholar] [CrossRef]
- Vartanian, V.; Tumova, J.; Dobrzyn, P.; Dobrzyn, A.; Nakabeppu, Y.; Lloyd, R.S.; Sampath, H. 8-Oxoguanine DNA Glycosylase (OGG1) Deficiency Elicits Coordinated Changes in Lipid and Mitochondrial Metabolism in Muscle. PLoS ONE 2017, 12, e0181687. [Google Scholar] [CrossRef]
- Klungland, A.; Rosewell, I.; Hollenbach, S.; Larsen, E.; Daly, G.; Epe, B.; Seeberg, E.; Lindahl, T.; Barnes, D.E. Accumulation of Premutagenic DNA Lesions in Mice Defective in Removal of Oxidative Base Damage. Proc. Natl. Acad. Sci. USA 1999, 96, 13300–13305. [Google Scholar] [CrossRef]
- Wang, W.; Esbensen, Y.; Kunke, D.; Suganthan, R.; Rachek, L.; Bjørås, M.; Eide, L. Mitochondrial DNA Damage Level Determines Neural Stem Cell Differentiation Fate. J. Neurosci. 2011, 31, 9746–9751. [Google Scholar] [CrossRef] [PubMed]
- Knighten, J.M.; Aziz, T.; Pleshinger, D.J.; Annamdevula, N.; Rich, T.C.; Taylor, M.S.; Andrews, J.F.; Macarilla, C.T.; Francis, C.M. Algorithm for Biological Second Messenger Analysis with Dynamic Regions of Interest. PLoS ONE 2023, 18, e0284394. [Google Scholar] [CrossRef] [PubMed]
- Wenceslau, C.F.; McCarthy, C.G.; Earley, S.; England, S.K.; Filosa, J.A.; Goulopoulou, S.; Gutterman, D.D.; Isakson, B.E.; Kanagy, N.L.; Martinez-Lemus, L.A.; et al. Guidelines for the Measurement of Vascular Function and Structure in Isolated Arteries and Veins. Am. J. Physiol.-Heart Circ. Physiol. 2021, 321, H77–H111. [Google Scholar] [CrossRef]
- Druzhyna, N.M.; Wilson, G.L.; LeDoux, S.P. Mitochondrial DNA Repair in Aging and Disease. Mech. Ageing Dev. 2008, 129, 383–390. [Google Scholar] [CrossRef]
- Francis, M.; Xu, N.; Zhou, C.; Stevens, T. Transient Receptor Potential Channel 4 Encodes a Vascular Permeability Defect and High-Frequency Ca(2+) Transients in Severe Pulmonary Arterial Hypertension. Am. J. Pathol. 2016, 186, 1701–1709. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: A Mitochondrial Love-Hate Triangle. Am. J. Physiol.-Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A Mutual Interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Thomas, W.A.; Florentin, R.A.; Reiner, J.M.; Lee, W.M.; Lee, K.T. Alterations in Population Dynamics of Arterial Smooth Muscle Cells during Atherogenesis: IV. Evidence for a Polyclonal Origin of Hypercholesterolemic Diet-Induced Atherosclerotic Lesions in Young Swine. Exp. Mol. Pathol. 1976, 24, 244–260. [Google Scholar] [CrossRef]
- Bennett, M.R.; Evan, G.I.; Schwartz, S.M. Apoptosis of Human Vascular Smooth Muscle Cells Derived from Normal Vessels and Coronary Atherosclerotic Plaques. J. Clin. Investig. 1995, 95, 2266–2274. [Google Scholar] [CrossRef]
- Geng, Y.J.; Libby, P. Evidence for Apoptosis in Advanced Human Atheroma. Colocalization with Interleukin-1 Beta-Converting Enzyme. Am. J. Pathol. 1995, 147, 251–266. [Google Scholar] [PubMed]
- McFarland, S.J.; Weber, D.S.; Choi, C.-S.; Lin, M.T.; Taylor, M.S. Ablation of Endothelial TRPV4 Channels Alters the Dynamic Ca2+ Signaling Profile in Mouse Carotid Arteries. Int. J. Mol. Sci. 2020, 21, 2179. [Google Scholar] [CrossRef]
- Bacsi, A.; Chodaczek, G.; Hazra, T.K.; Konkel, D.; Boldogh, I. Increased ROS Generation in Subsets of OGG1 Knockout Fibroblast Cells. Mech. Ageing Dev. 2007, 128, 637–649. [Google Scholar] [CrossRef]
- Minowa, O.; Arai, T.; Hirano, M.; Monden, Y.; Nakai, S.; Fukuda, M.; Itoh, M.; Takano, H.; Hippou, Y.; Aburatani, H.; et al. Mmh/Ogg1 Gene Inactivation Results in Accumulation of 8-Hydroxyguanine in Mice. Proc. Natl. Acad. Sci. USA 2000, 97, 4156–4161. [Google Scholar] [CrossRef]
- Osterod, M.; Hollenbach, S.; Hengstler, J.G.; Barnes, D.E.; Lindahl, T.; Epe, B. Age-Related and Tissue-Specific Accumulation of Oxidative DNA Base Damage in 7,8-Dihydro-8-Oxoguanine-DNA Glycosylase (Ogg1) Deficient Mice. Carcinogenesis 2001, 22, 1459–1463. [Google Scholar] [CrossRef]
- Thum, T.; Fraccarollo, D.; Schultheiss, M.; Froese, S.; Galuppo, P.; Widder, J.D.; Tsikas, D.; Ertl, G.; Bauersachs, J. Endothelial Nitric Oxide Synthase Uncoupling Impairs Endothelial Progenitor Cell Mobilization and Function in Diabetes. Diabetes 2007, 56, 666–674. [Google Scholar] [CrossRef]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of Tetrahydrobiopterin Leads to Uncoupling of Endothelial Cell Nitric Oxide Synthase in Hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef]
- Faucher, F.; Doublié, S.; Jia, Z. 8-Oxoguanine DNA Glycosylases: One Lesion, Three Subfamilies. Int. J. Mol. Sci. 2012, 13, 6711–6729. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Fleming, A.M.; Burrows, C.J. Sequencing the Mouse Genome for the Oxidatively Modified Base 8-Oxo-7,8-Dihydroguanine by OG-Seq. J. Am. Chem. Soc. 2017, 139, 2569–2572. [Google Scholar] [CrossRef] [PubMed]
- Brozovich, F.V.; Nicholson, C.J.; Degen, C.V.; Gao, Y.Z.; Aggarwal, M.; Morgan, K.G. Mechanisms of Vascular Smooth Muscle Contraction and the Basis for Pharmacologic Treatment of Smooth Muscle Disorders. Pharmacol. Rev. 2016, 68, 476–532. [Google Scholar] [CrossRef] [PubMed]
- Bir, S.C.; Kelley, R.E. Carotid Atherosclerotic Disease: A Systematic Review of Pathogenesis and Management. Brain Circ. 2022, 8, 127–136. [Google Scholar] [CrossRef]
- Corrado, E.; Rizzo, M.; Coppola, G.; Muratori, I.; Carella, M.; Novo, S. Endothelial Dysfunction and Carotid Lesions Are Strong Predictors of Clinical Events in Patients with Early Stages of Atherosclerosis: A 24-Month Follow-up Study. Coron. Artery Dis. 2008, 19, 139–144. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aziz, T.; Yuzefovych, L.; Rachek, L.; Taylor, M.S.; Francis, C.M. OGG1 Preserves Endothelial-Dependent Vasodilation and Regulates the Frequency and Spatial Area of Endothelial Calcium Signals. Biomolecules 2025, 15, 790. https://doi.org/10.3390/biom15060790
Aziz T, Yuzefovych L, Rachek L, Taylor MS, Francis CM. OGG1 Preserves Endothelial-Dependent Vasodilation and Regulates the Frequency and Spatial Area of Endothelial Calcium Signals. Biomolecules. 2025; 15(6):790. https://doi.org/10.3390/biom15060790
Chicago/Turabian StyleAziz, Takreem, Larysa Yuzefovych, Lyudmila Rachek, Mark S. Taylor, and Christopher M. Francis. 2025. "OGG1 Preserves Endothelial-Dependent Vasodilation and Regulates the Frequency and Spatial Area of Endothelial Calcium Signals" Biomolecules 15, no. 6: 790. https://doi.org/10.3390/biom15060790
APA StyleAziz, T., Yuzefovych, L., Rachek, L., Taylor, M. S., & Francis, C. M. (2025). OGG1 Preserves Endothelial-Dependent Vasodilation and Regulates the Frequency and Spatial Area of Endothelial Calcium Signals. Biomolecules, 15(6), 790. https://doi.org/10.3390/biom15060790