1. Introduction
Acid-sensing ion channels (ASICs) are a family of ion channels that are primarily activated by extracellular acidification [
1]. These channels are highly expressed in the central nervous system and play a pivotal role in maintaining cellular homeostasis, regulating neuronal excitability, and contributing to sensory processes such as pain and touch [
2,
3]. ASICs are particularly important for synaptic function, where they influence synaptic plasticity and contribute to neurophysiological processes like learning and memory [
4,
5]. They are also involved in the detection of environmental pH changes, such as those that occur during ischemic events, where the blood supply to brain tissue is compromised, leading to local acidification [
6]. ASICs have been implicated in a variety of neurological and psychological conditions. These include ischemic stroke [
6,
7], traumatic brain injury [
8,
9], epilepsy [
10,
11], chronic pain [
12,
13], and drug addiction [
4,
14]. During pathological conditions, such as cerebral ischemia, the acidification of the extracellular environment leads to the activation of ASICs, which contributes to neuronal depolarization, excitotoxicity, and subsequent neuronal damage [
6,
7]. Thus, while ASICs have vital roles in normal physiological processes, their dysregulation or overactivation during disease conditions can lead to detrimental effects on brain function and structure [
15].
Cyanide (CN) is a potent, rapid-acting toxin that has significant neurotoxic effects on the brain [
16]. It primarily exerts its toxicity through the inhibition of cytochrome c oxidase in the mitochondrial electron transport chain, leading to a disruption of cellular respiration and adenosine triphosphate (ATP) production [
17]. This results in cellular hypoxia, metabolic acidosis, and the accumulation of lactate. CN is particularly dangerous in situations where oxygen delivery to tissues is already compromised, such as during ischemic events. In the brain, CN exposure can lead to a cascade of neurophysiological events, including neuronal depolarization, excitotoxicity, and, potentially, irreversible neuronal damage [
18,
19]. CN-induced neuronal injury has been linked to disturbances in ion homeostasis, including the dysregulation of ion channels [
20]. CN’s ability to influence ion channels, such as those involved in neurotransmission and cellular signaling, could significantly alter brain function and exacerbate damage in neurodegenerative conditions or during acute ischemic injury. However, while the mechanisms of CN toxicity are well understood in terms of mitochondrial inhibition and metabolic disruption, its effects on specific ion channels in the brain remain less clear and warrant further investigation.
Previous studies have provided evidence that CN can modulate the activity of ion channels [
21], such as N-Methyl-D-Aspartate (NMDA) receptors, which are crucial for synaptic plasticity and memory [
22,
23]. NMDA receptors are a subtype of glutamate receptors that mediate excitatory neurotransmission and play a key role in synaptic plasticity processes such as long-term potentiation and long-term depression. CN has been shown to enhance NMDA receptor-mediated currents [
24], likely through a zinc-dependent mechanism, although the precise pathways remain a subject of ongoing research [
25,
26,
27]. This modulation of NMDA receptors suggests that CN can influence neuronal excitability and synaptic plasticity, which may contribute to its neurotoxic effects.
Given the central role of NMDA receptors in brain function, it is plausible that CN’s action on other ion channels, particularly those involved in pH sensing and neuronal excitability, could also have significant effects on brain activity. One such class of channels, the ASICs, remains understudied in the context of CN exposure. Given that ASICs are activated by extracellular protons and involved in neuronal excitability and acid-base homeostasis, it is important to investigate how CN may influence their function and whether this contributes to its neurotoxic effects. ASICs are integral to the regulation of neuronal excitability, especially in pathological conditions that involve changes in extracellular pH, such as ischemic stroke and traumatic brain injury. Since CN exposure results in metabolic acidosis and a decrease in cellular ATP, it is likely that this could affect the extracellular pH environment and, in turn, influence the activity of ASICs. In particular, the potentiation or inhibition of ASICs by CN could modulate neuronal excitability and exacerbate or mitigate the effects of ischemia or other forms of neurotoxicity.
Although previous studies have demonstrated that CN interacts with other ion channels, such as NMDA receptors [
21,
24,
25,
26,
27], the potential effects of CN on ASIC channels remain largely unexplored. ASICs, particularly ASIC1a, play a role in excitotoxicity and could be critically involved in CN-induced neuronal injury. Investigating how CN modulates ASIC currents, especially in cortical neurons, is therefore essential for understanding the broader neurotoxic effects of CN and how it influences ion channel function in the brain. The primary hypothesis of the current study is that CN potentiates ASIC currents in cortical neurons through a zinc-dependent mechanism. We hypothesize that CN enhances the sensitivity of ASIC channels, particularly ASIC1a, to extracellular protons, leading to increased neuronal excitability and potentially contributing to CN-induced neurotoxicity. Specifically, we propose that CN enhances ASIC1a currents by binding to a high-affinity zinc site on the channel. This mechanism, involving zinc binding, is known to modulate ASIC channel activity [
28]. The potentiation of ASIC currents by CN is likely mediated through this high-affinity zinc-binding site in the extracellular domain of ASIC1a.
To test this hypothesis, we will examine the effects of CN on ASIC currents in cultured mouse cortical neurons, utilizing a combination of pharmacological agents, genetic knockout (KO) models, and electrophysiological techniques. By investigating the mechanisms underlying the CN-induced potentiation of ASIC currents, this study aims to provide novel insights into how CN modulates ion channel function and assess the potential contribution of ASICs to CN-induced neuronal injury.
2. Materials and Methods
2.1. Primary Cortical Neuronal Cultures
The care and handling of animals in this study were conducted in accordance with the guidelines and protocols approved by the Institutional Animal Care and Use Committee at the University of Missouri–Kansas City. All procedures complied with the ethical standards set forth by the National Institutes of Health guidelines for the use of animals. Every effort was made to minimize animal use and alleviate any discomfort. Primary cultures of mouse cortical neurons were prepared using established methods [
28,
29]. In brief, time-pregnant BL/6J mice (embryonic day 16), including ASIC1a and ASIC2 knockout (KO) mice, were anesthetized with halothane and euthanized by cervical dislocation. Fetuses were promptly removed and placed in cold Hanks’ solution that lacked calcium and magnesium. The cortex was carefully dissected, incubated with 0.05% trypsin-EDTA for 10 min at 37 °C, and then dissociated by triturating with fire-polished glass pipettes. The resulting cell suspension was plated onto poly-L-ornithine-coated 35 mm culture dishes. Neurons were cultured in Neurobasal medium supplemented with B27 and maintained at 37 °C in a humidified incubator with 5% CO
2. Cultures were fed twice a week and used for electrophysiological recordings between 10 and 15 days after plating. The ASIC1a and ASIC2 KO mice used in this study were bred at our institution and generously provided by Drs. Wemmie and Welsh from the University of Iowa.
2.2. Transient Expression of Functional ASICs in CHO Cells
The tissue culture methods for Chinese Hamster Ovary (CHO) cells (American Type Culture Collection, Manassas, VA, USA) and the protocol for transfecting CHO cells with various ASIC subunits have been previously described in detail [
28,
29,
30]. Briefly, CHO cells were cultured in standard F12 medium (American Type Culture Collection, Manassas, VA, USA) supplemented with 10% fetal bovine serum and incubated at 37 °C in a CO
2 incubator. Cells were trypsinized with trypsin–EDTA, plated onto 35 mm culture dishes at 10–15% confluence, and allowed to recover for 24 h at 37 °C. Once the cells reached approximately 50–70% confluence, they were transiently transfected with expression vectors containing cDNA for rat ASIC subunits—ASIC1a, ASIC1b, ASIC2a, ASIC2b, and ASIC3—along with enhanced green fluorescent protein at a 1:0.25 molar ratio (Invitrogen, San Diego, CA, USA) using X-tremeGENE HP transfection reagent (Roche Diagnostics, Indianapolis, IN, USA). For co-expression experiments involving ASIC1a and either ASIC2a or ASIC2b, a 2:1 molar ratio of ASIC1a (2) to ASIC2a (1) or ASIC2b (1) was used. Electrophysiological recordings were carried out 48 to 72 h post-transfection. The cDNA clones for rat ASIC1a, ASIC1b, ASIC2a, ASIC2b, and ASIC3 were generously provided by Dr. M. Lazdunski (Institut de Pharmacologie Moléculaire et Cellulaire, Centre National de la Recherche Scientifique, Valbonne, France).
2.3. Electrophysiology
Whole-cell voltage-clamp recordings were conducted as previously described [
28,
29,
30]. Patch electrodes, with resistances ranging from 3 to 6 MΩ when filled with intracellular solution, were made from thin-walled borosilicate glass (1.5 mm diameter, WPI, Sarasota, FL, USA) using a two-stage puller (PC-10, Narishige, Tokyo, Japan). Whole-cell ASIC currents were induced by lowering the pH from 7.4 to various levels (e.g., pH 6.5) at a holding potential of −60 mV and recordings were made using Axopatch 200B amplifiers (Axon CNS, Molecular Devices, Foster City, CA, USA). Data were filtered at 2 kHz and digitized at 5 Hz using Digidata 1440 DAC units (Axon CNS, Molecular Devices, Foster City, CA, USA). Data acquisition was performed using pCLAMP software (Version 10.2, Axon CNS, Molecular Devices, Foster City, CA, USA).
Typically, ASIC channels were activated by reducing the pH from 7.4 to the target levels (e.g., pH 6.5) for 4 to 7 s, with recordings taken every 2 min to allow for full recovery from desensitization. A voltage step of −10 mV from the holding potential (−60 mV, unless otherwise noted) was applied periodically during each experiment to monitor cell capacitance and access resistance. Recordings in which the access resistance or capacitance changed by more than 10% during the experiment were excluded from the analysis.
2.4. Point Mutagenesis
Site-directed mutagenesis was performed as previously described [
28,
29,
30]. Briefly, the point mutation in ASIC1a was introduced using the Quick-Change Site-Directed Mutagenesis system (Stratagene, La Jolla, CA, USA), according to the manufacturer’s instructions. The primers for mutagenesis were obtained from Sigma-Genosys (The Woodlands, TX, USA). The mutation at lysine 133 in ASIC1a was confirmed through restriction enzyme digestion and DNA sequencing. To ensure precision, the entire rat ASIC1a cDNA was sequenced to verify that no unintended mutations were introduced.
2.5. Solutions and Compounds
Standard extracellular fluid (ECF) contained (mM) 140 NaCl, 5.4 KCl, 2.0 CaCl
2, 1.0 MgCl
2, 20 HEPES, and 10 glucose (pH 7.4; 320~330 mOsm). For solutions with a pH of 6.0 or lower, MES was used instead of HEPES for more reliable pH buffering [
28,
29,
30]. The pipette solution contained (mM) 140 K-Gluconate, 10 HEPES, 11 EGTA, 2 TEA, 1 CaCl
2, 2 MgCl
2, and 4 K
2ATP (pH 7.2~7.3; 290~300 mOsm). All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA). A multi-barrel perfusion system (SF-77, Hamden, CT, USA) was employed to achieve a rapid exchange of extracellular solutions. For the pretreatment with cyanide protocol, cyanide was present in the ECF of both pH 7.4 and lower pH (e.g., 6.5). This method was described in our previous publications [
28,
29,
30].
2.6. Data Analysis
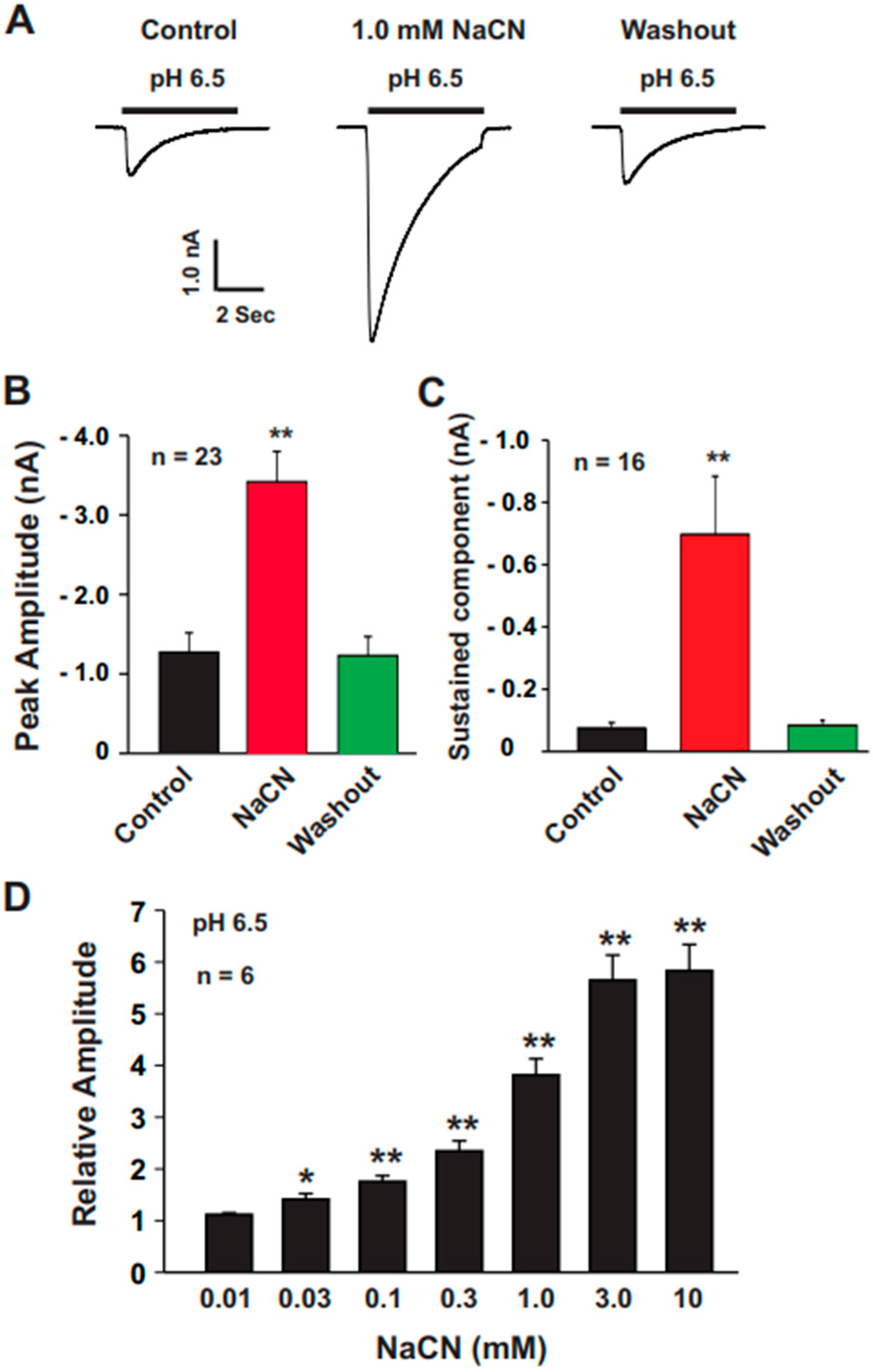
All data were analyzed using Clampfit 10.2 software (Axon CNS, Molecular Devices, Foster City, CA, USA). For experiments involving varying concentrations of CN (0.01, 0.03, 0.1, 0.3, 1.0, 3.0, and 10.0 mM), ASIC currents were normalized to control values obtained in the absence of CN treatment.
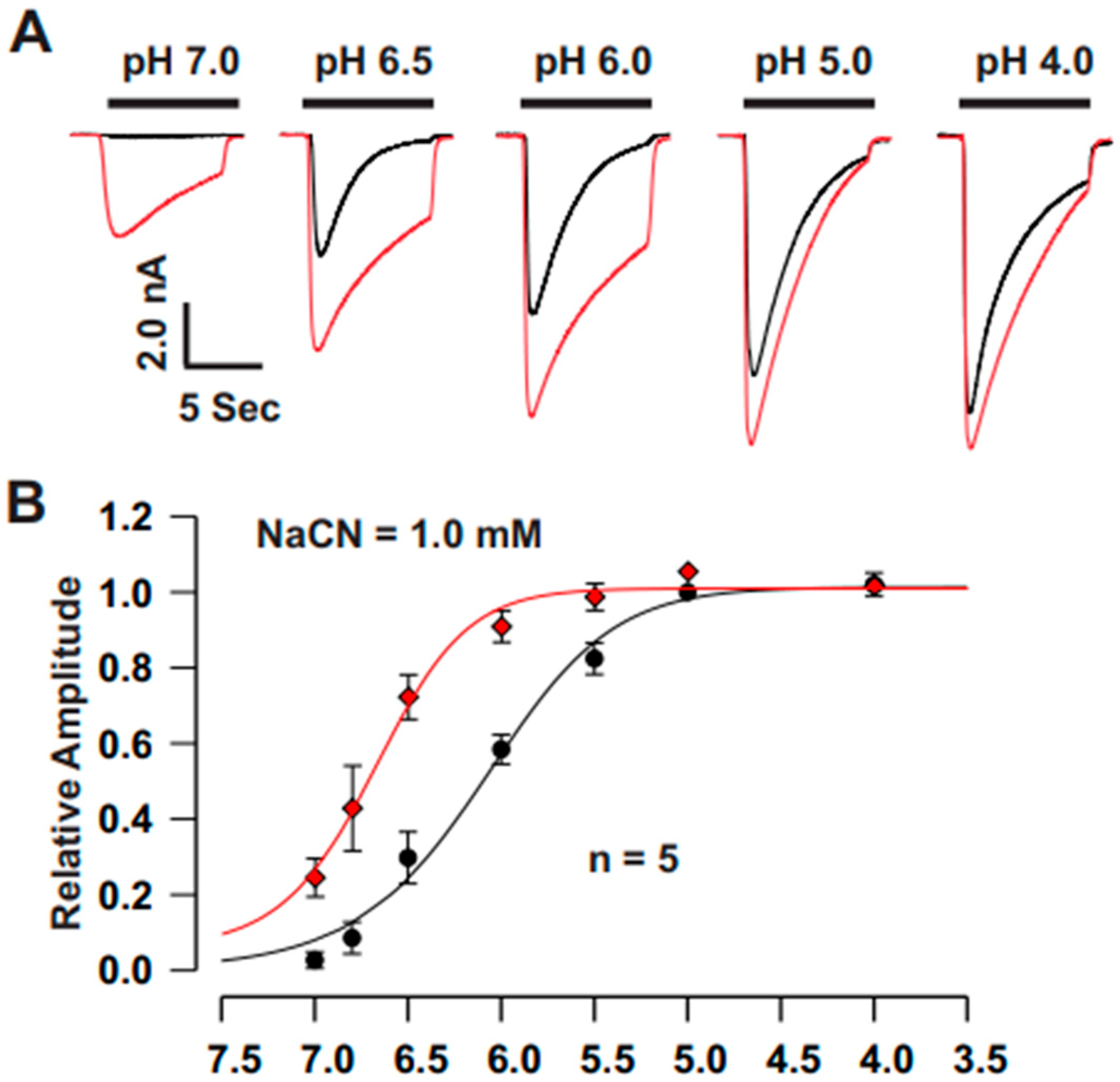
For pH activation curves, the ECF from one barrel of the perfusion system was maintained at pH 7.4, while the ECF from the second barrel was sequentially switched to pH values of 7.0, 6.8, 6.5, 6.0, 5.0, and 4.0 using the SF-77B fast perfusion system (Warner Instrument Co., CT, USA). The acid-induced currents at each pH were normalized to the peak current measured at pH 4.0. These normalized values were then fitted to the Hill equation using SigmaPlot 10 software to determine the pH50 values (the pH at which the current reached half of its maximum). To evaluate the sustained component of the ASIC currents, measurements were taken 6 s after the pH drop.
2.7. Statistics
Statistical analyses were conducted using SigmaPlot software. Data were presented as the mean ± standard error of the mean (SEM) for each experimental subgroup. Significant differences between mean values of the experimental groups were evaluated using a Student’s t-test for two-group comparisons and a one-way analysis of variance (ANOVA) for multiple pairwise comparisons, with post-hoc testing performed using the Bonferroni method. Differences were considered statistically significant when p < 0.05.
4. Discussion
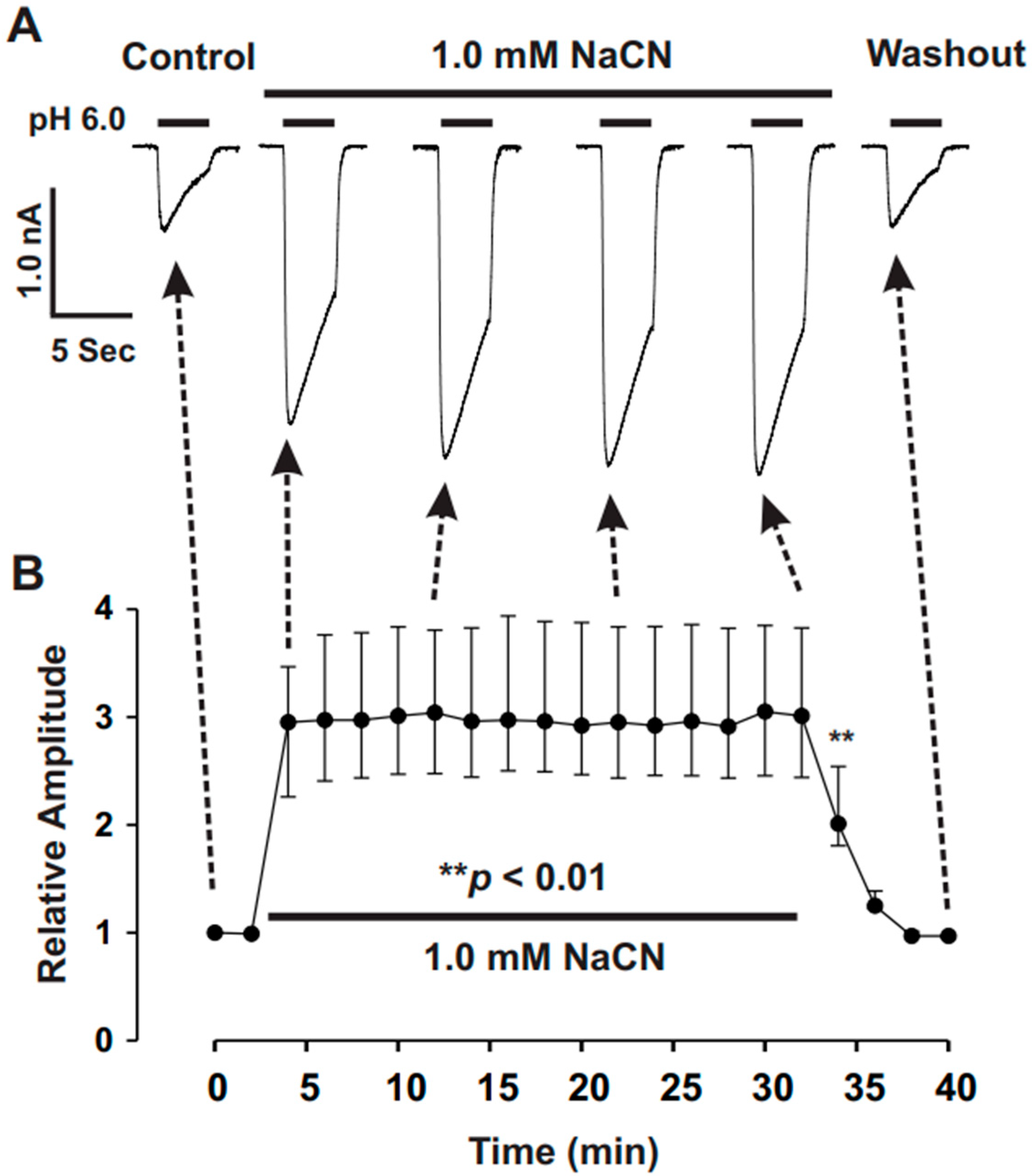
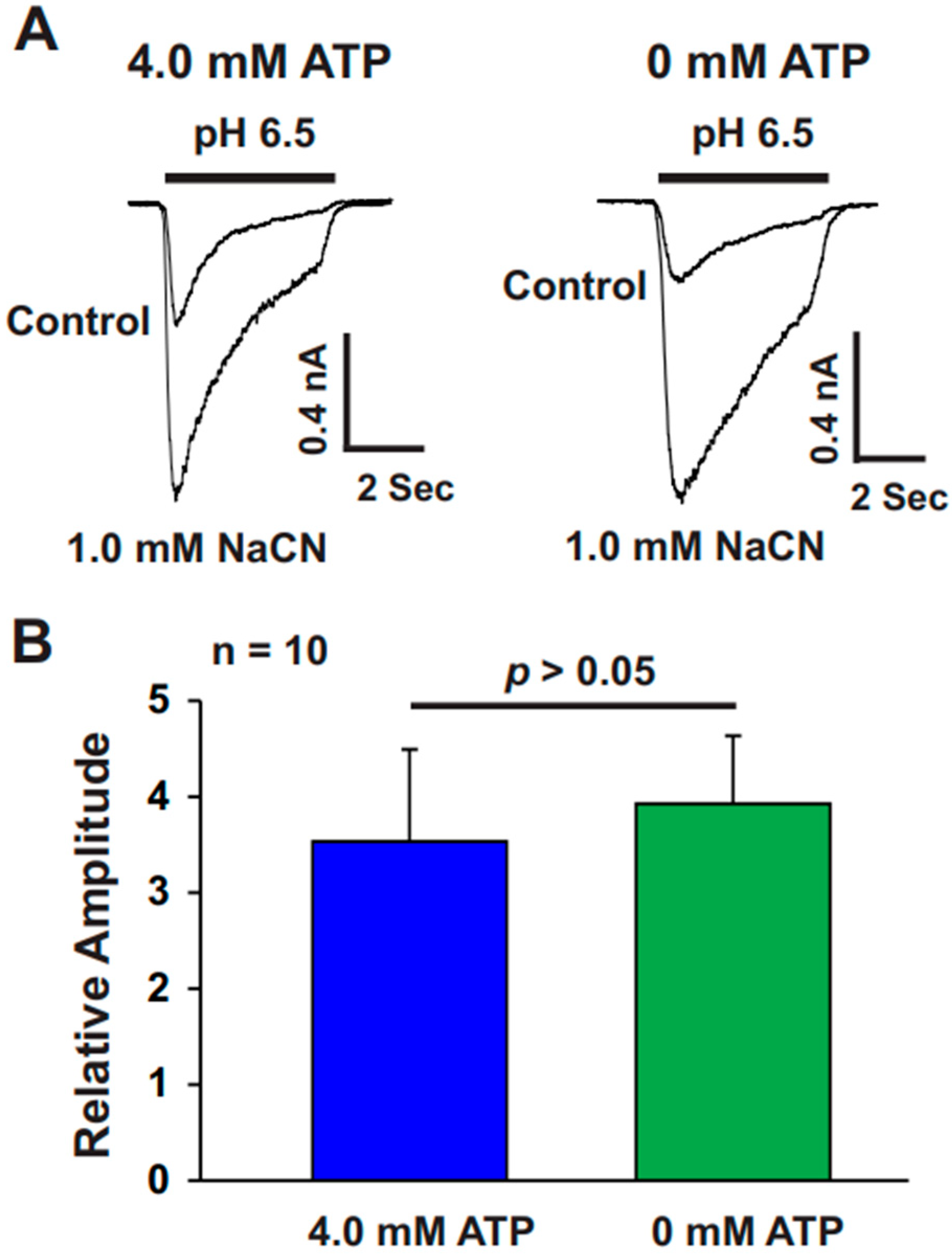
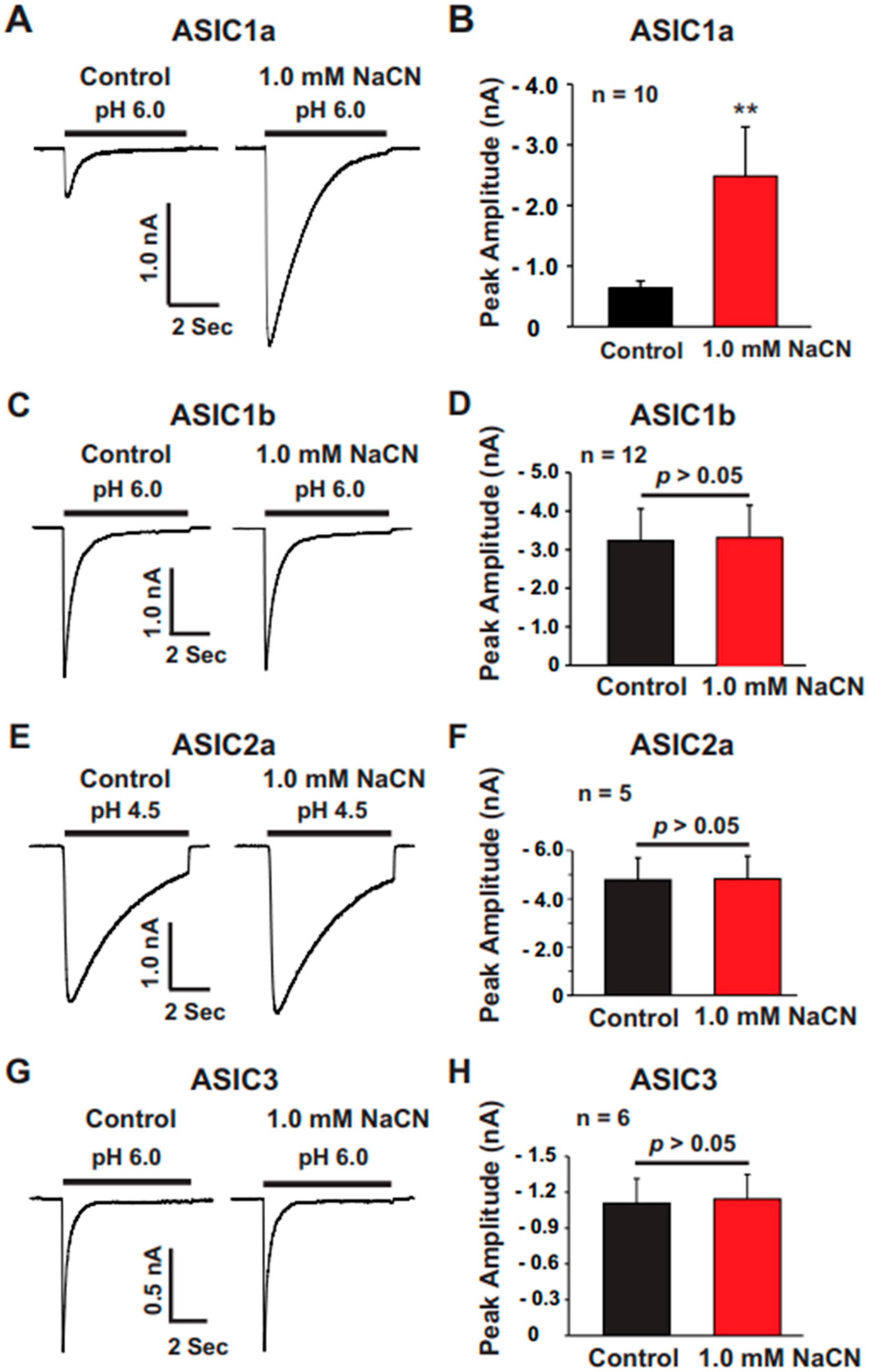
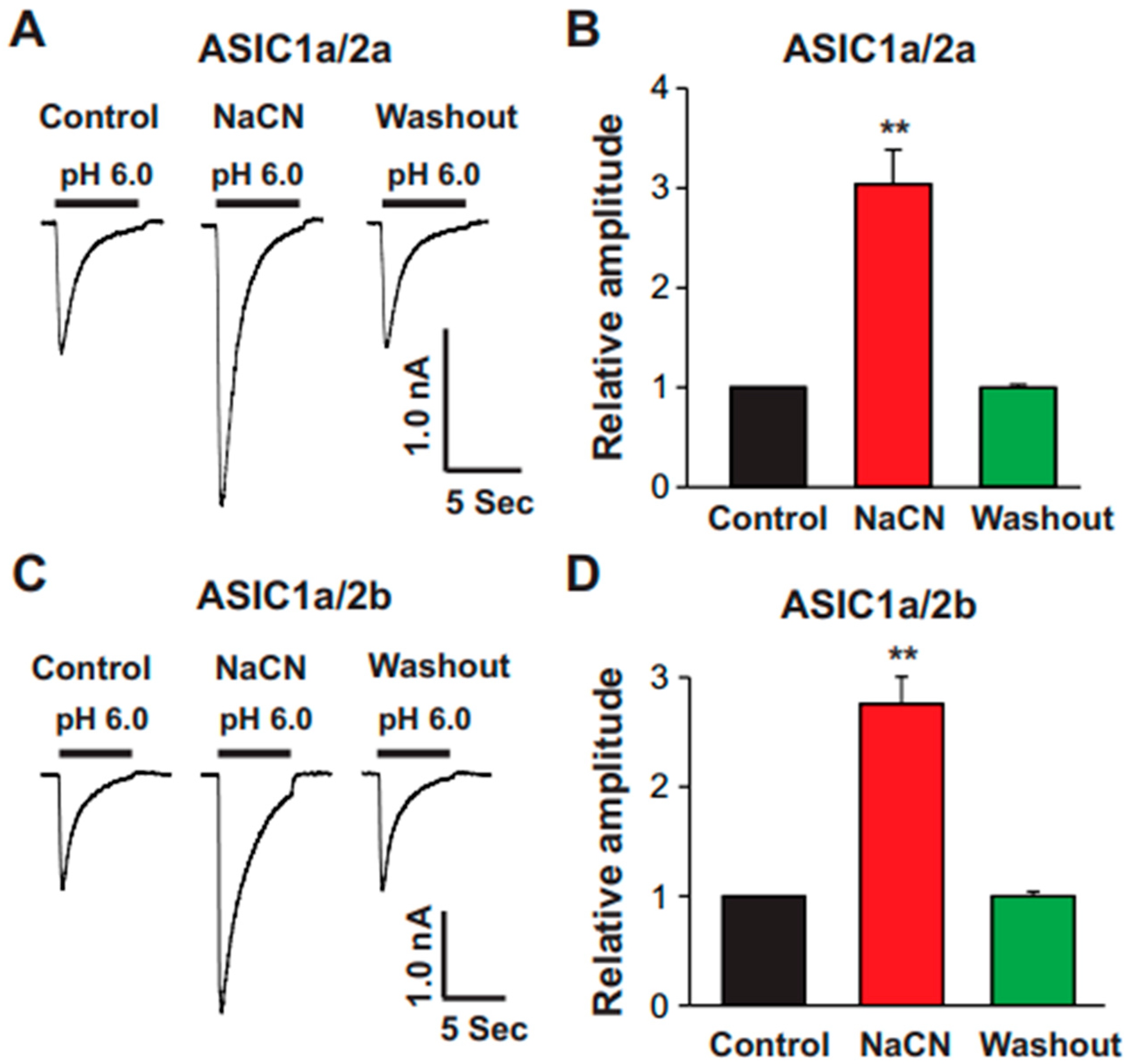
The present study provides significant insights into the effect of CN on ASICs in cultured mouse cortical neurons, with a particular focus on the potentiation of ASIC currents by CN. The key findings of this study are as follows: (1). We found that CN rapidly potentiated the ASIC currents in cultured mouse cortical neurons in a dose-dependent manner, shifting the pH dose–response curve to the left. This suggests that CN enhances the sensitivity of ASICs to changes in extracellular pH. (2). The potentiation of ASIC currents by CN was observed regardless of whether ATP was present in the recording pipette or not. The results indicate that the perturbation of intracellular ATP levels was not involved in the CN-mediated potentiation of ASIC currents, although the main target of CN is mitochondrial respiration. (3). We further revealed that the potentiation effect of CN on ASIC currents was tightly linked to the interaction with zinc. Specifically, CN was shown to bind with high affinity to zinc, and this binding was essential for the potentiation of ASIC currents. The use of a high-affinity zinc chelator, TPEN, blocked the potentiation of ASIC currents by CN, while treatment with low-affinity zinc had no such effect, supporting the critical role of zinc in this process. (4). We also explored the specificity of CN’s effect on different ASIC subtypes. CN potentiated ASIC currents in neurons from ASIC2 KO mice but not from ASIC1a KO mice. Additionally, CN had no effect on ASIC1b, 2a, or 3 channels but potentiated currents from homomeric ASIC1a and heteromeric ASIC1a/2 channels. This indicates that CN’s potentiating effect is specific to certain ASIC subtypes, particularly ASIC1a. (5). We identified lysine 133 (K133) in the extracellular domain of the ASIC1a subunit as a key residue responsible for the potentiation effect of CN. The mutation of K133 to arginine (K133R) abolished the CN-induced potentiation of ASIC1a currents, further reinforcing the importance of this residue in CN’s mechanism of action.
The findings of this study highlight the unique interaction between CN and ASIC channels, particularly ASIC1a, and provide a deeper understanding of how CN can modulate ion channel activity in the brain. The potentiation of ASIC currents by CN is explained primarily through its high-affinity interaction with zinc, a metal ion known to modulate the activity of ion channels. CN is a potent, rapid-acting toxin, and its effect on ASIC currents is mediated by its interaction with zinc. In the present study, we found that TPEN (10 nM), a high-affinity zinc chelator, potentiated ASIC currents, and CN at a concentration of 1.0 mM also potentiated ASIC currents in the same neuron to a similar extent. However, when TPEN and CN were applied together, the potentiation was similar to that observed with either TPEN or CN alone; thus, our data suggest that TPEN and CN likely act through similar mechanisms to potentiate ASIC currents. Zinc is known to act as a potent modulator of ASICs, particularly in the extracellular domain of ASIC1a, which is rich in histidine and cysteine residues that can interact with metal ions [
25,
26]. The presence of high-affinity zinc-binding sites on ASICs, particularly at the K133 residue in ASIC1a, is crucial for this modulation [
28]. The observation that CN potentiates ASIC currents in neurons from ASIC2 KO mice but not from ASIC1a KO mice suggests that ASIC1a is the primary target for CN-induced potentiation. The results from CHO cells expressing homomeric and heteromeric ASIC subtypes also support this conclusion, showing that CN potentiates currents specifically in ASIC1a and ASIC1a/2 channels and not in ASIC1b, 2a, or 3 channels. This specificity likely arises from structural differences in the extracellular domains of these channels, which may impact their ability to interact with zinc and other modulators such as CN. The mutation of K133 to arginine in the extracellular domain of ASIC1a provides crucial insight into the mechanism of CN’s action. K133 is located in a region that is known to interact with zinc, and its mutation disrupts the high-affinity binding of zinc to the ASIC1a subunit, thereby preventing the potentiation of ASIC1a currents by CN. This suggests that the K133 residue is a key structural feature that mediates CN’s effect on ASIC1a and likely contributes to the specificity of CN’s action on this channel subtype. While lysine and zinc would normally repel each other due to their positive charges, other factors such as the structural context of the ASIC1a subunit, coordination with other amino acid residues, or the divalent nature of zinc might allow for their interaction. The mutation of lysine 133 likely disrupts this delicate interaction, which is why it blocks the high-affinity zinc inhibition of the ASIC1a current. Further studies are needed to clarify the exact mechanism of interaction between zinc and lysine at the extracellular site of the ASIC1a channel.
The findings of this study hold significant implications for our understanding of how CN affects neuronal ion channels and contribute to the broader field of neurotoxicity and neurodegeneration. CN is a well-known toxicant, and its rapid action on the nervous system is often linked to its ability to disrupt cellular respiration and energy metabolism. However, this study adds another layer of complexity by showing that CN can also directly modulate ion channel activity, particularly ASICs. By potentiating ASIC currents, CN may exacerbate neuronal excitability and contribute to neurotoxic effects. This mechanism may be particularly relevant in the context of acute CN poisoning, where rapid neuronal depolarization could lead to excitotoxicity and neuronal damage. ASICs are involved in a range of neurological and psychological disorders, including ischemia [
6,
7], epilepsy [
10,
11], and chronic pain [
12,
13]. By modulating ASIC activity, CN may influence the pathophysiology of these conditions. Understanding how CN interacts with ASICs could open new avenues for therapeutic interventions. For example, targeting the interaction between CN and zinc could provide strategies for mitigating the neurotoxic effects of CN exposure. The identification of the K133 residue as a critical site for the CN-induced potentiation of ASIC1a currents could inform the development of drugs that specifically modulate ASIC1a activity. This could be particularly valuable in conditions where ASICs play a pathological role, such as in ischemic stroke or neurodegenerative diseases. The ability to selectively block or enhance ASIC1a activity could have therapeutic benefits for a variety of neurological disorders.
The current study lays the foundation for several important directions in future research. While this study focused on the interaction between CN and ASICs, the role of zinc in neurotoxicity and neurodegeneration warrants further exploration. Zinc dysregulation is implicated in a variety of neurological conditions [
35,
36], including Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Future studies could investigate how other environmental toxins or physiological conditions that alter zinc homeostasis might affect ASIC function and contribute to disease progression. This study focused on cortical neurons, but CN’s effects on other neuronal subtypes and brain regions remain to be explored. Investigating whether CN has similar effects on ASIC channels in different parts of the brain, such as the hippocampus or basal ganglia, could provide valuable insights into how CN affects brain function more broadly. The identification of K133 as a critical residue in CN-induced potentiation opens the door for the design of small molecules or peptide inhibitors that could block the interaction between CN and ASIC1a. These compounds could be tested for their ability to mitigate CN toxicity and could serve as potential therapeutics for poisoning or other conditions involving excessive ASIC1a activity. While this study focused on acute exposure to CN, it would be interesting to examine the long-term effects of chronic or sub-lethal CN exposure on ASIC function and neuronal health. The chronic modulation of ASIC activity could have lasting consequences on neuronal excitability, synaptic plasticity, and brain function, which should be explored in future studies. Finally, further studies in animal models of CN toxicity would be valuable to confirm the findings observed in cultured neurons. In vivo studies could help elucidate the physiological and behavioral consequences of the CN-induced potentiation of ASIC currents, providing a more comprehensive understanding of CN’s neurotoxic effects.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}