
Zinc-Dependent Histone Deacetylases in Lung Endothelial Pathobiology

 , , ,
, , ,  , , and
, , and 
Abstract
1. Introduction
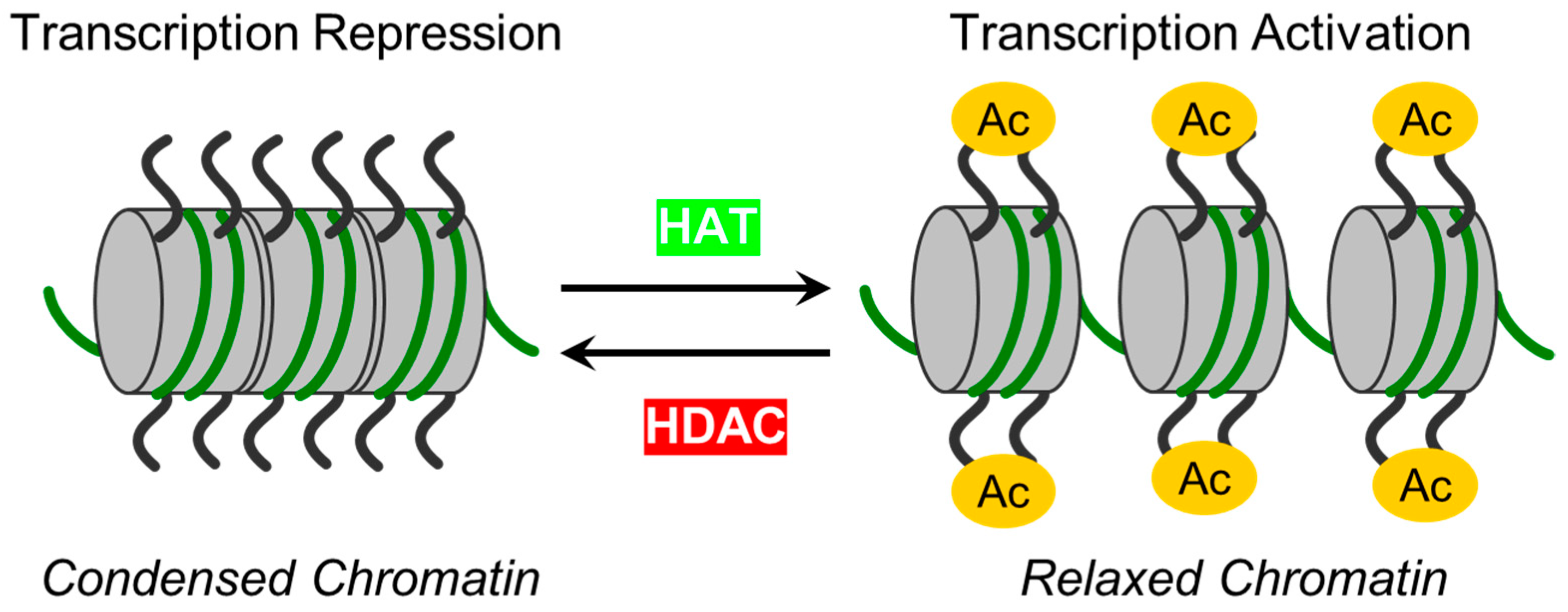
2. Zinc-Dependent HDACs: Classification, Functions, Regulations and Modulations
3. Zinc-Dependent HDACs in Endothelial Function
3.1. Class I HDACs: HDAC1, HDAC2, HDAC3, and HDAC8
3.2. Class IIa HDACs: HDAC4, HDAC5, HDAC7, and HDAC9
3.3. Class IIb HDACs: HDAC6 and HDAC10
3.4. Class IV HDAC: HDAC11
4. EC-Mediated Central Signaling Cascades and Their Regulation by Zinc-Dependent HDACs
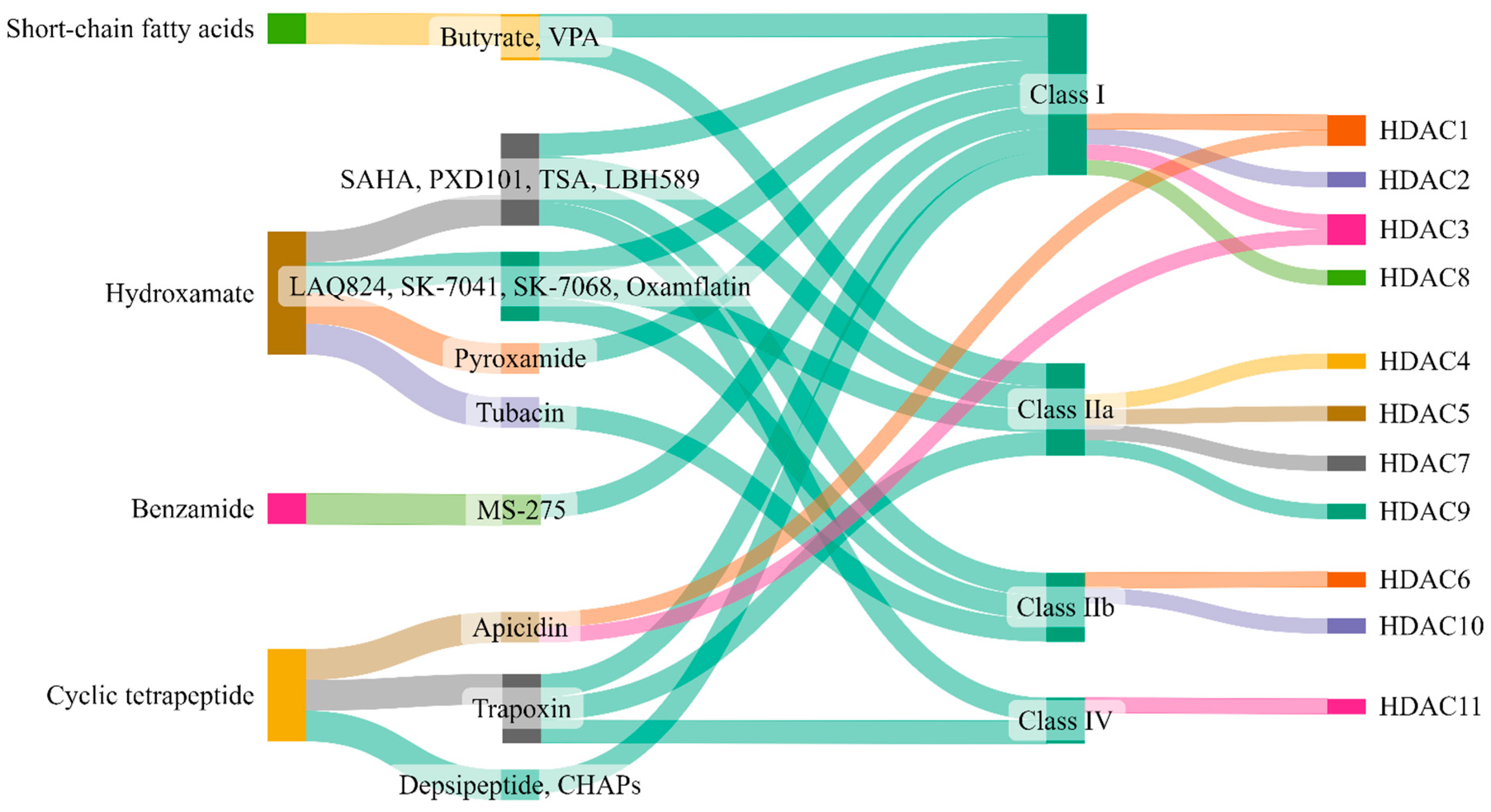
5. Therapeutic Targeting of Zinc-Dependent HDACs in Lung Injury
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mokrá, D. Acute lung injury—From pathophysiology to treatment. Physiol. Res. 2020, 69, S353–S366. [Google Scholar] [CrossRef] [PubMed]
- Long, M.E.; Mallampalli, R.K.; Horowitz, J.C. Pathogenesis of pneumonia and acute lung injury. Clin. Sci. 2022, 136, 747–769. [Google Scholar] [CrossRef] [PubMed]
- Rubenfeld, G.D.; Caldwell, E.; Peabody, E.; Weaver, J.; Martin, D.P.; Neff, M.; Stern, E.J.; Hudson, L.D. Incidence and outcomes of acute lung injury. N. Engl. J. Med. 2005, 353, 1685–1693. [Google Scholar] [CrossRef]
- Cusack, R.; Bos, L.D.; Povoa, P.; Martin-Loeches, I. Endothelial dysfunction triggers acute respiratory distress syndrome in patients with sepsis: A narrative review. Front. Med. 2023, 10, 1203827. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. ARDS Definition Task Force. Acute respiratory distress syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar]
- Steinberg, K.P.; Hudson, L.D.; Goodman, R.B.; Hough, C.L.; Lanken, P.N.; Hyzy, R.; Thompson, B.T.; Ancukiewicz, M.; National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N. Engl. J. Med. 2006, 354, 1671–1684. [Google Scholar]
- Wiedemann, H.P.; Wheeler, A.P.; Bernard, G.R.; Thompson, B.T.; Hayden, D.; deBoisblanc, B.; Connors, A.F., Jr.; Hite, R.D.; Harabin, A.L.; National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Comparison of two fluid-management strategies in acute lung injury. N. Engl. J. Med. 2006, 354, 2564–2575. [Google Scholar] [CrossRef]
- Arcaro, G.; Zenere, B.M.; Travia, D.; Zenti, M.G.; Covi, G.; Lechi, A.; Muggeo, M. Non-invasive detection of early endothelial dysfunction in hypercholesterolaemic subjects. Atherosclerosis 1995, 114, 247–254. [Google Scholar] [CrossRef]
- Maniatis, N.A.; Kotanidou, A.; Catravas, J.D.; Orfanos, S.E. Endothelial pathomechanisms in acute lung injury. Vasc. Pharmacol. 2008, 49, 119–133. [Google Scholar] [CrossRef]
- Vassiliou, A.G.; Kotanidou, A.; Dimopoulou, I.; Orfanos, S.E. Endothelial damage in acute respiratory distress syndrome. Int. J. Mol. Sci. 2020, 21, 8793. [Google Scholar] [CrossRef]
- Romero, M.J.; Yue, Q.; Singla, B.; Hamacher, J.; Sridhar, S.; Moseley, A.S.; Song, C.; Mraheil, M.A.; Fischer, B.; Zeitlinger, M.; et al. Direct endothelial ENaC activation mitigates vasculopathy induced by SARS-CoV2 spike protein. Front. Immunol. 2023, 14, 1241448. [Google Scholar] [CrossRef]
- Xu, S.W.; Ilyas, I.; Weng, J.P. Endothelial dysfunction in COVID-19: An overview of evidence, biomarkers, mechanisms and potential therapies. Acta Pharmacol. Sin. 2023, 44, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Huertas, A.; Guignabert, C.; Barberà, J.A.; Bärtsch, P.; Bhattacharya, J.; Bhattacharya, S.; Bonsignore, M.R.; Dewachter, L.; Dinh-Xuan, A.T.; Dorfmüller, P.; et al. Pulmonary vascular endothelium: The orchestra conductor in respiratory diseases: Highlights from basic research to therapy. Eur. Respir. J. 2018, 51, 1700745. [Google Scholar] [CrossRef] [PubMed]
- Simmons, S.; Erfinanda, L.; Bartz, C.; Kuebler, W.M. Novel mechanisms regulating endothelial barrier function in the pulmonary microcirculation. J. Physiol. 2019, 597, 997–1021. [Google Scholar] [CrossRef]
- Borek, I.; Birnhuber, A.; Voelkel, N.F.; Marsh, L.M.; Kwapiszewska, G. The vascular perspective on acute and chronic lung disease. J. Clin. Investig. 2023, 133, e170502. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef]
- Dudek, S.M.; Garcia, J.G. Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. (1985) 2001, 91, 1487–1500. [Google Scholar] [CrossRef]
- Lampugnani, M.G.; Corada, M.; Caveda, L.; Breviario, F.; Ayalon, O.; Geiger, B.; Dejana, E. The molecular organization of endothelial cell to cell junctions: Differential association of plakoglobin, beta-catenin, and alpha-catenin with vascular endothelial cadherin (VE-cadherin). J. Cell Biol. 1995, 129, 203–217. [Google Scholar] [CrossRef]
- Dejana, E.; Orsenigo, F.; Lampugnani, M.G. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 2008, 121, 2115–2122. [Google Scholar] [CrossRef]
- Aslam, M.; Gündüz, D.; Troidl, C.; Heger, J.; Hamm, C.W.; Schulz, R. Purinergic Regulation of Endothelial Barrier Function. Int. J. Mol. Sci. 2021, 22, 1207. [Google Scholar] [CrossRef]
- Fang, Z.; Wang, X.; Sun, X.; Hu, W.; Miao, Q.R. The role of histone protein acetylation in regulating endothelial function. Front. Cell Dev. Biol. 2021, 9, 672447. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E.; Wang, N.; Stamenovic, D. Tensegrity, cellular biophysics, and the mechanics of living systems. Rep. Prog. Phys. 2014, 77, 046603. [Google Scholar] [CrossRef] [PubMed]
- Widdicombe, J. Functional morphology and physiology of pulmonary rapidly adapting receptors (RARs). Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2003, 270, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.F.; Nelson, C.M.; Tan, J.L.; Chen, C.S. Cadherins, RhoA, and Rac1 are differentially required for stretch-mediated proliferation in endothelial versus smooth muscle cells. Circ. Res. 2007, 101, e44–e52. [Google Scholar] [CrossRef] [PubMed]
- Tzima, E.; Irani-Tehrani, M.; Kiosses, W.B.; Dejana, E.; Schultz, D.A.; Engelhardt, B.; Cao, G.; DeLisser, H.; Schwartz, M.A. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 2005, 437, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Wu, D.; Birukov, K.G. Mechanosensing and mechanoregulation of endothelial cell functions. Compr. Physiol. 2019, 9, 873–904. [Google Scholar]
- Kovacs, L.; Kovacs-Kasa, A.; Verin, A.D.; Fulton, D.; Lucas, R.; Su, Y. Histone deacetylases in vascular permeability and remodeling associated with acute lung injury. Vessel. Plus. 2018, 2, 15. [Google Scholar] [CrossRef]
- Lee, D.Y.; Chiu, J.J. Atherosclerosis and flow: Roles of epigenetic modulation in vascular endothelium. J. Biomed. Sci. 2019, 26, 56. [Google Scholar] [CrossRef]
- Luo, G.; Liu, B.; Fu, T.; Liu, Y.; Li, B.; Li, N.; Geng, Q. The role of histone deacetylases in acute lung injury-friend or foe. Int. J. Mol. Sci. 2023, 24, 7876. [Google Scholar] [CrossRef]
- Shen, Z.; Bei, Y.; Lin, H.; Wei, T.; Dai, Y.; Hu, Y.; Zhang, C.; Dai, H. The role of class IIa histone deacetylases in regulating endothelial function. Front. Physiol. 2023, 14, 1091794. [Google Scholar] [CrossRef]
- Zhang, H.N.; Dai, Y.; Zhang, C.H.; Omondi, A.M.; Ghosh, A.; Khanra, I.; Chakraborty, M.; Yu, X.B.; Liang, J. Sirtuins family as a target in endothelial cell dysfunction: Implications for vascular ageing. Biogerontology 2020, 21, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Bahl, S.; Seto, E. Regulation of histone deacetylase activities and functions by phosphorylation and its physiological relevance. Cell Mol. Life Sci. 2021, 78, 427–445. [Google Scholar] [CrossRef]
- Eom, G.H.; Kook, H. Posttranslational modifications of histone deacetylases: Implications for cardiovascular diseases. Pharmacol. Ther. 2014, 143, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Histone deacetylase 6 in health and disease. Epigenomics 2015, 7, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Iaconelli, J.; Xuan, L.; Karmacharya, R. HDAC6 modulates signaling pathways relevant to synaptic biology and neuronal differentiation in human stem-cell-derived neurons. Int. J. Mol. Sci. 2019, 20, 1605. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Kawaguchi, Y.; Lai, C.H.; Kovacs, J.J.; Higashimoto, Y.; Appella, E.; Yao, T.P. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002, 21, 6236–6245. [Google Scholar] [CrossRef]
- Banerjee, S.; Adhikari, N.; Amin, S.A.; Jha, T. Histone deacetylase 8 (HDAC8) and its inhibitors with selectivity to other isoforms: An overview. Eur. J. Med. Chem. 2019, 164, 214–240. [Google Scholar] [CrossRef]
- Kumar, S.; Attrish, D.; Srivastava, A.; Banerjee, J.; Tripathi, M.; Chandra, P.S.; Dixit, A.B. Non-histone substrates of histone deacetylases as potential therapeutic targets in epilepsy. Expert. Opin. Ther. Targets 2021, 25, 75–85. [Google Scholar] [CrossRef]
- Zheng, W. The Zinc-dependent HDACs: Non-histone substrates and catalytic deacylation beyond deacetylation. Mini Rev. Med. Chem. 2022, 22, 2478–2485. [Google Scholar] [CrossRef]
- Dunaway, L.S.; Pollock, J.S. HDAC1: An environmental sensor regulating endothelial function. Cardiovasc. Res. 2022, 118, 1885–1903. [Google Scholar] [CrossRef]
- Yao, H.; Rahman, I. Role of histone deacetylase 2 in epigenetics and cellular senescence: Implications in lung inflammaging and COPD. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L557–L566. [Google Scholar] [CrossRef]
- Pandey, D.; Hori, D.; Kim, J.H.; Bergman, Y.; Berkowitz, D.E.; Romer, L.H. NEDDylation promotes endothelial dysfunction: A role for HDAC2. J. Mol. Cell Cardiol. 2015, 81, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Ziesché, E.; Kettner-Buhrow, D.; Weber, A.; Wittwer, T.; Jurida, L.; Soelch, J.; Müller, H.; Newel, D.; Kronich, P.; Schneider, H.; et al. The coactivator role of histone deacetylase 3 in IL-1-signaling involves deacetylation of p65 NF-κB. Nucleic Acids Res. 2013, 41, 90–109. [Google Scholar] [CrossRef] [PubMed]
- Zampetaki, A.; Zeng, L.; Margariti, A.; Xiao, Q.; Li, H.; Zhang, Z.; Pepe, A.E.; Wang, G.; Habi, O.; deFalco, E.; et al. Histone deacetylase 3 is critical in endothelial survival and atherosclerosis development in response to disturbed flow. Circulation 2010, 121, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Liu, Q.; Adrianto, I.; Wu, X.; Glassbrook, J.; Khalasawi, N.; Yin, C.; Yi, Q.; Dong, Z.; Geissmann, F.; et al. Histone deacetylase 3 controls lung alveolar macrophage development and homeostasis. Nat. Commun. 2020, 11, 3822. [Google Scholar] [CrossRef] [PubMed]
- Kee, H.J.; Ryu, Y.; Seok, Y.M.; Choi, S.Y.; Sun, S.; Kim, G.R.; Jeong, M.H. Selective inhibition of histone deacetylase 8 improves vascular hypertrophy, relaxation, and inflammation in angiotensin II hypertensive mice. Clin. Hypertens. 2019, 25, 13. [Google Scholar] [CrossRef] [PubMed]
- Hou, F.; Wei, W.; Qin, X.; Liang, J.; Han, S.; Han, A.; Kong, Q. The posttranslational modification of HDAC4 in cell biology: Mechanisms and potential targets. J. Cell Biochem. 2020, 121, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Shao, C.; Rose, P.; Zhu, Y.Z. Epigenetics and vascular senescence-potential new therapeutic targets? Front. Pharmacol. 2020, 11, 535395. [Google Scholar] [CrossRef]
- Litke, C.; Bading, H.; Mauceri, D. Histone deacetylase 4 shapes neuronal morphology via a mechanism involving regulation of expression of vascular endothelial growth factor D. J. Biol. Chem. 2018, 293, 8196–8207. [Google Scholar] [CrossRef]
- Turpaev, K.T. Transcription factor KLF2 and its role in the regulation of inflammatory processes. Biochemistry 2020, 85, 54–67. [Google Scholar] [CrossRef]
- Margariti, A.; Zampetaki, A.; Xiao, Q.; Zhou, B.; Karamariti, E.; Martin, D.; Yin, X.; Mayr, M.; Li, H.; Zhang, Z.; et al. Histone deacetylase 7 controls endothelial cell growth through modulation of beta-catenin. Circ. Res. 2010, 106, 1202–1211. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, X.; Parra, M.; Verdin, E.; Bassel-Duby, R.; Olson, E.N. Control of endothelial cell proliferation and migration by VEGF signaling to histone deacetylase 7. Proc. Natl. Acad. Sci. USA 2008, 105, 7738–7743. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Young, B.D.; Li, S.; Qi, X.; Richardson, J.A.; Olson, E.N. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell 2006, 126, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Ho, C.C.; Reineke, E.; Lam, M.; Cheng, X.; Stanya, K.J.; Liu, Y.; Chakraborty, S.; Shih, H.M.; Kao, H.Y. Histone deacetylase 7 promotes PML sumoylation and is essential for PML nuclear body formation. Mol. Cell. Biol. 2008, 28, 5658–5667. [Google Scholar] [CrossRef] [PubMed]
- Turtoi, A.; Mottet, D.; Matheus, N.; Dumont, B.; Peixoto, P.; Hennequière, V.; Deroanne, C.; Colige, A.; De Pauw, E.; Bellahcène, A.; et al. The angiogenesis suppressor gene AKAP12 is under the epigenetic control of HDAC7 in endothelial cells. Angiogenesis 2012, 15, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Kasotakis, G.; Kintsurashvili, E.; Galvan, M.D.; Graham, C.; Purves, J.T.; Agarwal, S.; Corcoran, D.L.; Sullenger, B.A.; Palmer, S.M.; Remick, D.G. Histone deacetylase 7 inhibition in a murine model of gram-negative pneumonia-induced acute lung injury. Shock 2020, 53, 344–351. [Google Scholar] [CrossRef]
- Schiano, C.; Benincasa, G.; Franzese, M.; Della Mura, N.; Pane, K.; Salvatore, M.; Napoli, C. Epigenetic-sensitive pathways in personalized therapy of major cardiovascular diseases. Pharmacol. Ther. 2020, 210, 107514. [Google Scholar] [CrossRef]
- Gal, J.; Chen, J.; Na, D.Y.; Tichacek, L.; Barnett, K.R.; Zhu, H. The acetylation of Lysine-376 of G3BP1 regulates RNA binding and stress granule dynamics. Mol. Cell. Biol. 2019, 39, e00052-19. [Google Scholar] [CrossRef]
- Leucker, T.M.; Nomura, Y.; Kim, J.H.; Bhatta, A.; Wang, V.; Wecker, A.; Jandu, S.; Santhanam, L.; Berkowitz, D.; Romer, L.; et al. Cystathionine γ-lyase protects vascular endothelium: A role for inhibition of histone deacetylase 6. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H711–H720. [Google Scholar] [CrossRef]
- Kaluza, D.; Kroll, J.; Gesierich, S.; Yao, T.P.; Boon, R.A.; Hergenreider, E.; Tjwa, M.; Rössig, L.; Seto, E.; Augustin, H.G.; et al. Class IIb HDAC6 regulates endothelial cell migration and angiogenesis by deacetylation of cortactin. EMBO J. 2011, 30, 4142–4156. [Google Scholar] [CrossRef]
- Nomura, Y.; Nakano, M.; Woo Sung, H.; Han, M.; Pandey, D. Inhibition of HDAC6 Activity Protects against endothelial dysfunction and atherogenesis in vivo: A role for HDAC6 neddylation. Front. Physiol. 2021, 12, 675724. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, S.H.; Choi, M.C.; Lee, J.; Oh, D.Y.; Im, S.A.; Bang, Y.J.; Kim, T.Y. Class II histone deacetylases play pivotal roles in heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors. Biochem. Biophys. Res. Commun. 2008, 368, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Duan, B.; Ye, D.; Zhu, S.; Jia, W.; Lu, C.; Wang, G.; Guo, X.; Yu, Y.; Wu, C.; Kang, J. HDAC10 promotes angiogenesis in endothelial cells through the PTPN22/ERK axis. Oncotarget 2017, 8, 61338–61349. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Tan, R.; Sun, M.; Yuan, L.; Ruiz, M.; Dupuis, J.; Hu, Q.; Zhu, L. MiR-1249 on endothelial extracellular vesicles mediates cigarette smoke-induced pulmonary hypertension by inhibiting HDAC10 (Histone Deacetylase 10)-NFκB (Nuclear Factor κB)-CaSR (Calcium-Sensing Receptor) cascade. Hypertension 2022, 79, 2721–2732. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ge, J. Proteinase-activated receptor-2 modulates Ve-Cadherin expression to affect human vascular endothelial barrier function. J. Cell. Biochem. 2017, 118, 4587–4593. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Jin, Z.; Lv, X.; Zheng, Z.; Gao, H.; Deng, Y.; Liu, Y.; Chen, L.; Wang, W.; He, J.; et al. Hydroxytyrosol acetate inhibits vascular endothelial cell pyroptosis via the HDAC11 signaling pathway in atherosclerosis. Front. Pharmacol. 2021, 12, 656272. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Jin, Z.; Zheng, Z.; Lv, X.; Ren, L.; Yang, J.; Chen, D.; Wang, B.; Yang, W.; Chen, L.; et al. HDAC11 promotes both NLRP3/caspase-1/GSDMD and caspase-3/GSDME pathways causing pyroptosis via ERG in vascular endothelial cells. Cell Death Discov. 2022, 8, 112. [Google Scholar] [CrossRef] [PubMed]
- Porter, N.J.; Christianson, D.W. Structure, mechanism, and inhibition of the zinc-dependent histone deacetylases. Curr. Opin. Struct. Biol. 2019, 59, 9–18. [Google Scholar] [CrossRef]
- Shvedunova, M.; Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef]
- Schator, D.; Gomez-Valero, L.; Buchrieser, C.; Rolando, M. Patho-epigenetics: Histone deacetylases as targets of pathogens and therapeutics. microLife 2021, 2, uqab013. [Google Scholar] [CrossRef]
- Yoshida, M.; Kudo, N.; Kosono, S.; Ito, A. Chemical and structural biology of protein lysine deacetylases. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 297–321. [Google Scholar] [CrossRef]
- Zhao, T.C.; Wang, Z.; Zhao, T.Y. The important role of histone deacetylases in modulating vascular physiology and arteriosclerosis. Atherosclerosis 2020, 303, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Ito, M.; Elliott, W.M.; Cosio, B.; Caramori, G.; Kon, O.M.; Barczyk, A.; Hayashi, S.; Adcock, I.M.; Hogg, J.C.; et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N. Engl. J. Med. 2005, 352, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Caramori, G.; Lim, S.; Oates, T.; Chung, K.F.; Barnes, P.J.; Adcock, I.M. Expression and activity of histone deacetylases in human asthmatic airways. Am. J. Respir. Crit. Care Med. 2002, 166, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Kee, H.J.; Eom, G.H.; Joung, H.; Shin, S.; Kim, J.R.; Cho, Y.K.; Choe, N.; Sim, B.W.; Jo, D.; Jeong, M.H.; et al. Activation of histone deacetylase 2 by inducible heat shock protein 70 in cardiac hypertrophy. Circ. Res. 2008, 103, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Kee, H.J.; Jin, L.; Ryu, Y.; Sun, S.; Kim, G.R.; Jeong, M.H. Inhibition of class IIa histone deacetylase activity by gallic acid, sulforaphane, TMP269, and panobinostat. Biomed. Pharmacother. 2018, 101, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Eom, G.H. HDAC and HDAC inhibitor: From cancer to cardiovascular diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A. Targeting the Zinc-dependent histone deacetylases (HDACs) for drug discovery. In Chemical Epigenetics; Springer: Cham, Switzerland, 2020; Volume 33, ISBN 978-3-030-42981-2. [Google Scholar]
- Neugebauer, R.C.; Sippl, W.; Jung, M. Inhibitors of NAD+ dependent histone deacetylases (sirtuins). Curr. Pharm. Des. 2008, 14, 562–573. [Google Scholar]
- Agarwal, R.; Pattarawat, P.; Duff, M.R.; Wang, H.R.; Baudry, J.; Smith, J.C. Structure-based discovery of selective histone deacetylase (HDAC) 3 and 4 inhibitors. bioRxiv 2022. [Google Scholar] [CrossRef]
- Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone deacetylases (HDACs): Evolution, specificity, role in transcriptional complexes, and pharmacological actionability. Genes 2020, 11, 556. [Google Scholar] [CrossRef]
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Roles of histone deacetylases in epigenetic regulation: Emerging paradigms from studies with inhibitors. Clin. Epigenetics 2012, 4, 5. [Google Scholar] [CrossRef]
- Emiliani, S.; Fischle, W.; Van Lint, C.; Al-Abed, Y.; Verdin, E. Characterization of a human RPD3 ortholog, HDAC3. Proc. Natl. Acad. Sci. USA 1998, 95, 2795–2800. [Google Scholar] [CrossRef] [PubMed]
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 1996, 272, 408–411. [Google Scholar] [CrossRef] [PubMed]
- de Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Bradley, E.W.; Carpio, L.R.; van Wijnen, A.J.; McGee-Lawrence, M.E.; Westendorf, J.J. Histone deacetylases in bone development and skeletal disorders. Physiol. Rev. 2015, 95, 1359–1381. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.A.; Hagelkruys, A.; Seiser, C. Transcription and beyond: The role of mammalian class I lysine deacetylases. Chromosoma 2014, 123, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, J.S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Bertos, N.R.; Wang, A.H.; Yang, X.J. Class II histone deacetylases: Structure, function, and regulation. Biochem. Cell Biol. 2001, 79, 243–252. [Google Scholar] [CrossRef]
- Verdin, E.; Dequiedt, F.; Kasler, H.G. Class II histone deacetylases: Versatile regulators. Trends Genet. 2003, 19, 286–293. [Google Scholar] [CrossRef]
- Grozinger, C.M.; Schreiber, S.L. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc. Natl. Acad. Sci. USA 2000, 97, 7835–7840. [Google Scholar] [CrossRef]
- Di Giorgio, E.; Brancolini, C. Regulation of class IIa HDAC activities: It is not only matter of subcellular localization. Epigenomics 2016, 8, 251–269. [Google Scholar] [CrossRef] [PubMed]
- Lahm, A.; Paolini, C.; Pallaoro, M.; Nardi, M.C.; Jones, P.; Neddermann, P.; Sambucini, S.; Bottomley, M.J.; Lo Surdo, P.; Carfí, A.; et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. USA 2007, 104, 17335–17340. [Google Scholar] [CrossRef] [PubMed]
- Parra, M. Class IIa HDACs—New insights into their functions in physiology and pathology. FEBS J. 2015, 282, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Das Gupta, K.; Shakespear, M.R.; Curson, J.E.B.; Murthy, A.M.V.; Iyer, A.; Hodson, M.P.; Ramnath, D.; Tillu, V.A.; von Pein, J.B.; Reid, R.C.; et al. Class IIa Histone deacetylases drive toll-like receptor-inducible glycolysis and macrophage inflammatory responses via pyruvate kinase M2. Cell Rep. 2020, 30, 2712–2728.e8. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.D.; Cai, R.; Bhatia, U.; Asselbergs, F.A.; Song, C.; Terry, R.; Trogani, N.; Widmer, R.; Atadja, P.; Cohen, D. Isolation and characterization of a novel class II histone deacetylase, HDAC10. J. Biol. Chem. 2002, 277, 6656–6666. [Google Scholar] [CrossRef] [PubMed]
- Guardiola, A.R.; Yao, T.P. Molecular cloning and characterization of a novel histone deacetylase HDAC10. J. Biol. Chem. 2002, 277, 3350–3356. [Google Scholar] [CrossRef]
- Tong, J.J.; Liu, J.; Bertos, N.R.; Yang, X.J. Identification of HDAC10, a novel class II human histone deacetylase containing a leucine-rich domain. Nucleic Acids Res. 2002, 30, 1114–1123. [Google Scholar] [CrossRef]
- Núñez-Álvarez, Y.; Suelves, M. HDAC11: A multifaceted histone deacetylase with proficient fatty deacylase activity and its roles in physiological processes. FEBS J. 2022, 289, 2771–2792. [Google Scholar] [CrossRef]
- Cao, J.; Sun, L.; Aramsangtienchai, P.; Spiegelman, N.A.; Zhang, X.; Huang, W.; Seto, E.; Lin, H. HDAC11 regulates type I interferon signaling through defatty-acylation of SHMT2. Proc. Natl. Acad. Sci. USA 2019, 116, 5487–5492. [Google Scholar] [CrossRef]
- Kee, H.J.; Kook, H. Roles and targets of class I and IIa histone deacetylases in cardiac hypertrophy. J. Biomed. Biotechnol. 2011, 2011, 928326. [Google Scholar] [CrossRef]
- Dzreyan, V.A.; Demyanenko, S.V. The role of post-translational protein acetylation and deacetylation in the apoptosis of neurons of the peripheral nervous system. Biochem. Mosc. Suppl. Ser. A 2023, 17, 249–263. [Google Scholar] [CrossRef]
- Ali, I.; Conrad, R.J.; Verdin, E.; Ott, M. Lysine acetylation goes global: From epigenetics to metabolism and therapeutics. Chem. Rev. 2018, 118, 1216–1252. [Google Scholar] [CrossRef] [PubMed]
- Millán-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone post-translational modifications—Cause and consequence of genome function. Nat. Rev. Genet. 2022, 23, 563–580. [Google Scholar] [CrossRef]
- Chen, H.P.; Zhao, Y.T.; Zhao, T.C. Histone deacetylases and mechanisms of regulation of gene expression. Crit. Rev. Oncog. 2015, 20, 35–47. [Google Scholar] [CrossRef] [PubMed]
- King, J.; Patel, M.; Chandrasekaran, S. Metabolism, HDACs, and HDAC inhibitors: A systems biology perspective. Metabolites 2021, 11, 792. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; He, Y.; Fu, W.; Sahebkar, A.; Tan, Y.; Xu, S.; Li, H. Histone deacetylases (HDACs) and atherosclerosis: A mechanistic and pharmacological review. Front. Cell Dev. Biol. 2020, 8, 581015. [Google Scholar] [CrossRef]
- Cross, D.; Drury, R.; Hill, J.; Pollard, A.J. Epigenetics in Sepsis: Understanding its role in endothelial dysfunction, immunosuppression, and potential therapeutics. Front. Immunol. 2019, 10, 1363. [Google Scholar] [CrossRef]
- Zemskov, E.A.; Gross, C.M.; Aggarwal, S.; Zemskova, M.A.; Wu, X.; Gu, C.; Wang, T.; Tang, H.; Black, S.M. NF-κB-dependent repression of Sox18 transcription factor requires the epigenetic regulators histone deacetylases 1 and 2 in acute lung injury. Front. Physiol. 2022, 13, 947537. [Google Scholar] [CrossRef]
- Kashio, T.; Shirakura, K.; Kinoshita, M.; Morita, M.; Ishiba, R.; Muraoka, K.; Kanbara, T.; Tanaka, M.; Funatsu, R.; Hino, N.; et al. HDAC inhibitor, MS-275, increases vascular permeability by suppressing Robo4 expression in endothelial cells. Tissue Barriers 2021, 9, 1911195. [Google Scholar] [CrossRef]
- Rolando, M.; Stefani, C.; Doye, A.; Acosta, M.I.; Visvikis, O.; Yevick, H.G.; Buchrieser, C.; Mettouchi, A.; Bassereau, P.; Lemichez, E. Contractile actin cables induced by Bacillus anthracis lethal toxin depend on the histone acetylation machinery. Cytoskeleton 2015, 72, 542–556. [Google Scholar] [CrossRef]
- Pandey, D.; Sikka, G.; Bergman, Y.; Kim, J.H.; Ryoo, S.; Romer, L.; Berkowitz, D. Transcriptional regulation of endothelial arginase 2 by histone deacetylase 2. Arter. Thromb. Vasc. Biol. 2014, 34, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Hori, D.; Nomura, Y.; Nakano, M.; Han, M.; Bhatta, A.; Chen, K.; Akiyoshi, K.; Pandey, D. Endothelial-specific overexpression of histone deacetylase 2 protects mice against endothelial dysfunction and atherosclerosis. Cell Physiol. Biochem. 2020, 54, 947–958. [Google Scholar]
- Hou, Q.; Hu, K.; Liu, X.; Quan, J.; Liu, Z. HADC regulates the diabetic vascular endothelial dysfunction by targetting MnSOD. Biosci. Rep. 2018, 38, BSR20181042. [Google Scholar] [CrossRef] [PubMed]
- Bedenbender, K.; Scheller, N.; Fischer, S.; Leiting, S.; Preissner, K.T.; Schmeck, B.T.; Vollmeister, E. Inflammation-mediated deacetylation of the ribonuclease 1 promoter via histone deacetylase 2 in endothelial cells. FASEB J. 2019, 33, 9017–9029. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.G.; Liu, F.; Verin, A.D.; Birukova, A.; Dechert, M.A.; Gerthoffer, W.T.; Bamberg, J.R.; English, D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Investig. 2001, 108, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.D.; Barabutis, N.; Birmpas, C.; Dimitropoulou, C.; Thangjam, G.; Cherian-Shaw, M.; Dennison, J.; Catravas, J.D. Histone deacetylase inhibitors prevent pulmonary endothelial hyperpermeability and acute lung injury by regulating heat shock protein 90 function. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L1410–L1419. [Google Scholar] [CrossRef]
- Zhao, Q.; Yu, Z.; Zhang, F.; Huang, L.; Xing, C.; Liu, N.; Xu, Y.; Wang, X. HDAC3 inhibition prevents oxygen glucose deprivation/reoxygenation-induced transendothelial permeability by elevating PPARγ activity in vitro. J. Neurochem. 2019, 149, 298–310. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, F.; Yu, Z.; Guo, S.; Liu, N.; Jiang, Y.; Lo, E.H.; Xu, Y.; Wang, X. HDAC3 inhibition prevents blood-brain barrier permeability through Nrf2 activation in type 2 diabetes male mice. J. Neuroinflammation 2019, 16, 103. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Huang, S.; Chen, G.; Sun, J.; Chen, Y.; Wang, N.; Dong, Y.; Shen, E.; Hu, Z.; Gong, W.; Jin, L.; et al. Histone deacetylase 3 inhibition alleviates type 2 diabetes mellitus-induced endothelial dysfunction via Nrf2. Cell Commun. Signal. 2021, 19, 35. [Google Scholar] [CrossRef] [PubMed]
- Celermajer, D.S.; Sorensen, K.E.; Gooch, V.M.; Spiegelhalter, D.J.; Miller, O.I.; Sullivan, I.D.; Lloyd, J.K.; Deanfield, J.E. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet 1992, 340, 1111–1115. [Google Scholar] [CrossRef]
- Chen, L.; Shang, C.; Wang, B.; Wang, G.; Jin, Z.; Yao, F.; Yue, Z.; Bai, L.; Wang, R.; Zhao, S.; et al. HDAC3 inhibitor suppresses endothelial-to-mesenchymal transition via modulating inflammatory response in atherosclerosis. Biochem. Pharmacol. 2021, 192, 114716. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Lee, C.I.; Lin, T.E.; Lim, S.H.; Zhou, J.; Tseng, Y.C.; Chien, S.; Chiu, J.J. Role of histone deacetylases in transcription factor regulation and cell cycle modulation in endothelial cells in response to disturbed flow. Proc. Natl. Acad. Sci. USA 2012, 109, 1967–1972. [Google Scholar] [CrossRef] [PubMed]
- Waltregny, D.; Glénisson, W.; Tran, S.L.; North, B.J.; Verdin, E.; Colige, A.; Castronovo, V. Histone deacetylase HDAC8 associates with smooth muscle alpha-actin and is essential for smooth muscle cell contractility. FASEB J. 2005, 19, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, S.; Cleary, R.A.; Wang, R.; Gannon, O.J.; Seto, E.; Tang, D.D. Histone deacetylase 8 regulates cortactin deacetylation and contraction in smooth muscle tissues. Am. J. Physiol. Cell Physiol. 2014, 307, C288–C295. [Google Scholar] [CrossRef] [PubMed]
- Bandela, M.; Belvitch, P.; Garcia, J.G.N.; Dudek, S.M. Cortactin in lung cell function and disease. Int. J. Mol. Sci. 2022, 23, 4606. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Kettmann, R.; Dequiedt, F. Class IIa histone deacetylases: Regulating the regulators. Oncogene 2007, 26, 5450–5467. [Google Scholar] [CrossRef]
- Martin, M.; Kettmann, R.; Dequiedt, F. Class IIa histone deacetylases: Conducting development and differentiation. Int. J. Dev. Biol. 2009, 53, 291–301. [Google Scholar] [CrossRef]
- Kovacs-Kasa, A.; Kovacs, L.; Cherian-Shaw, M.; Patel, V.; Meadows, M.L.; Fulton, D.J.; Su, Y.; Verin, A.D. Inhibition of Class IIa HDACs improves endothelial barrier function in endotoxin-induced acute lung injury. J. Cell Physiol. 2021, 236, 2893–2905. [Google Scholar] [CrossRef]
- Lobera, M.; Madauss, K.P.; Pohlhaus, D.T.; Wright, Q.G.; Trocha, M.; Schmidt, D.R.; Baloglu, E.; Trump, R.P.; Head, M.S.; Hofmann, G.A.; et al. Selective class IIa histone deacetylase inhibition via a non-chelating zinc-binding group. Nat. Chem. Biol. 2013, 9, 319–325. [Google Scholar] [CrossRef]
- Li, M.; Zheng, Y.; Yuan, H.; Liu, Y.; Wen, X. Effects of dynamic changes in histone acetylation and deacetylase activity on pulmonary fibrosis. Int. Immunopharmacol. 2017, 52, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Xiao, C.; Long, F.; Su, Z.; Jia, W.; Qin, M.; Huang, M.; Wu, W.; Suguro, R.; Liu, X.; et al. HDAC4 regulates vascular inflammation via activation of autophagy. Cardiovasc. Res. 2018, 114, 1016–1028. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Cheng, H.; Chen, X.; Gu, J.; Su, W.; Cai, G.; Yan, Y.; Wang, C.; Xia, X.; Zhang, K.; et al. The activation of histone deacetylases 4 prevented endothelial dysfunction: A crucial mechanism of HuangqiGuizhiWuwu Decoction in improving microcirculation dysfunction in diabetes. J. Ethnopharmacol. 2023, 307, 116240. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.M.; Wang, X.; Li, C.X.; Liu, X.Y.; Guo, X.J.; Li, Y.; Chen, Y.L.; Ye, H.X.; Chen, H.S. SP1 promotes HDAC4 expression and inhibits HMGB1 expression to reduce intestinal barrier dysfunction, oxidative stress, and inflammatory response after sepsis. J. Innate Immun. 2022, 14, 366–379. [Google Scholar] [CrossRef]
- Hu, K.; Huang, M.J.; Ling, S.; Li, Y.X.; Cao, X.Y.; Chen, Y.F.; Lei, J.M.; Fu, W.Z.; Tan, B.F. LncRNA CASC11 upregulation promotes HDAC4 to alleviate oxidized low-density lipoprotein-induced injury of cardiac microvascular endothelial cells. Kaohsiung J. Med. Sci. 2023, 39, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, X.; Li, Q.; Zhou, S.M.; Hu, B.; Hu, G.W.; Niu, X.; Guo, S.C.; Wang, Y.; Deng, Z.F. Role of phosphorylated HDAC4 in stroke-induced angiogenesis. Biomed. Res. Int. 2017, 2017, 2957538. [Google Scholar] [CrossRef]
- Schader, T.; Löwe, O.; Reschke, C.; Malacarne, P.; Hahner, F.; Müller, N.; Gajos-Draus, A.; Backs, J.; Schröder, K. Oxidation of HDAC4 by Nox4-derived H2O2 maintains tube formation by endothelial cells. Redox Biol. 2020, 36, 101669. [Google Scholar] [CrossRef]
- Hrgovic, I.; Doll, M.; Pinter, A.; Kaufmann, R.; Kippenberger, S.; Meissner, M. Histone deacetylase inhibitors interfere with angiogenesis by decreasing endothelial VEGFR-2 protein half-life in part via a VE-cadherin-dependent mechanism. Exp. Dermatol. 2017, 26, 194–201. [Google Scholar] [CrossRef]
- Zecchin, A.; Pattarini, L.; Gutierrez, M.I.; Mano, M.; Mai, A.; Valente, S.; Myers, M.P.; Pantano, S.; Giacca, M. Reversible acetylation regulates vascular endothelial growth factor receptor-2 activity. J. Mol. Cell Biol. 2014, 6, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Urbich, C.; Rössig, L.; Kaluza, D.; Potente, M.; Boeckel, J.N.; Knau, A.; Diehl, F.; Geng, J.G.; Hofmann, W.K.; Zeiher, A.M.; et al. HDAC5 is a repressor of angiogenesis and determines the angiogenic gene expression pattern of endothelial cells. Blood 2009, 113, 5669–5679. [Google Scholar] [CrossRef]
- Tsou, P.S.; Wren, J.D.; Amin, M.A.; Schiopu, E.; Fox, D.A.; Khanna, D.; Sawalha, A.H. Histone deacetylase 5 is overexpressed in scleroderma endothelial cells and impairs Angiogenesis via repression of proangiogenic factors. Arthritis Rheumatol. 2016, 68, 2975–2985. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Tan, J.; Yuan, Y.; Shen, J.; Chen, Y. Semaglutide ameliorates lipopolysaccharide-induced acute lung injury through inhibiting HDAC5-mediated activation of NF-κB signaling pathway. Hum. Exp. Toxicol. 2022, 41, 9603271221125931. [Google Scholar] [CrossRef]
- Wang, Y.; Abrol, R.; Mak, J.Y.W.; Das Gupta, K.; Ramnath, D.; Karunakaran, D.; Fairlie, D.P.; Sweet, M.J. Histone deacetylase 7: A signalling hub controlling development, inflammation, metabolism and disease. FEBS J. 2023, 290, 2805–2832. [Google Scholar] [CrossRef]
- Hsu, A.; Duan, Q.; McMahon, S.; Huang, Y.; Wood, S.A.; Gray, N.S.; Wang, B.; Bruneau, B.G.; Haldar, S.M. Salt-inducible kinase 1 maintains HDAC7 stability to promote pathologic cardiac remodeling. J. Clin. Investig. 2020, 130, 2966–2977. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.T.; Gao, C.; Liu, Y.; Guo, S.; Wang, A.; Wang, B.; Erdjument-Bromage, H.; Miyagi, M.; Tempst, P.; Kao, H.Y. Monoubiquitination of filamin B regulates vascular endothelial growth factor-mediated trafficking of histone deacetylase 7. Mol. Cell Biol. 2013, 33, 1546–1560. [Google Scholar] [CrossRef]
- Yang, J.; Moraga, A.; Xu, J.; Zhao, Y.; Luo, P.; Lao, K.H.; Margariti, A.; Zhao, Q.; Ding, W.; Wang, G.; et al. A histone deacetylase 7-derived peptide promotes vascular regeneration via facilitating 14-3-3γ phosphorylation. Stem Cells. 2020, 38, 556–573. [Google Scholar] [CrossRef]
- Martin, M.; Geudens, I.; Bruyr, J.; Potente, M.; Bleuart, A.; Lebrun, M.; Simonis, N.; Deroanne, C.; Twizere, J.C.; Soubeyran, P.; et al. PP2A regulatory subunit Bα controls endothelial contractility and vessel lumen integrity via regulation of HDAC7. EMBO J. 2013, 32, 2491–2503. [Google Scholar] [CrossRef]
- Parra, M.; Mahmoudi, T.; Verdin, E. Myosin phosphatase dephosphorylates HDAC7, controls its nucleocytoplasmic shuttling, and inhibits apoptosis in thymocytes. Genes Dev. 2007, 21, 638–643. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kovacs-Kasa, A.; Gorshkov, B.A.; Kim, K.M.; Kumar, S.; Black, S.M.; Fulton, D.J.; Dimitropoulou, C.; Catravas, J.D.; Verin, A.D. The protective role of MLCP-mediated ERM dephosphorylation in endotoxin-induced lung injury in vitro and in vivo. Sci. Rep. 2016, 6, 39018. [Google Scholar] [CrossRef]
- Kim, K.M.; Csortos, C.; Czikora, I.; Fulton, D.; Umapathy, N.S.; Olah, G.; Verin, A.D. Molecular characterization of myosin phosphatase in endothelium. J. Cell Physiol. 2012, 227, 1701–1708. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Ma, Z.; Zhang, Y.; Han, L.; Shao, C.; Feng, Y.; Gao, F.; Di, S.; Zhang, Z.; Zhang, J.; et al. HDAC7 promotes NSCLC proliferation and metastasis via stabilization by deubiquitinase USP10 and activation of β-catenin-FGF18 pathway. J. Exp. Clin. Cancer Res. 2022, 41, 91. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Wei, X.; Wang, Z.; Han, H.; Fu, Y.; Liu, J.; Zhang, Y.; Guo, J.; Dong, C.; Zhou, D.; et al. HDAC9 exacerbates endothelial injury in cerebral ischaemia/reperfusion injury. J. Cell Mol. Med. 2016, 20, 1139–1149. [Google Scholar] [CrossRef]
- Lu, S.; Li, H.; Li, K.; Fan, X.D. HDAC9 promotes brain ischemic injury by provoking IκBα/NF-κB and MAPKs signaling pathways. Biochem. Biophys. Res. Commun. 2018, 503, 1322–1329. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, L.; Cheng, Q.; Wu, Y. Aberrant expression and regulatory role of histone deacetylase 9 in vascular endothelial cell injury in intracranial aneurysm. Biomol. Biomed. 2023, 24, 61–72. [Google Scholar] [CrossRef]
- Brancolini, C.; Di Giorgio, E.; Formisano, L.; Gagliano, T. Quis custodiet ipsos custodes (who controls the controllers)? Two decades of studies on HDAC9. Life 2021, 11, 90. [Google Scholar] [CrossRef]
- Joo, E.E.; Yamada, K.M. Post-polymerization crosstalk between the actin cytoskeleton and microtubule network. Bioarchitecture 2016, 6, 53–59. [Google Scholar] [CrossRef]
- Valenzuela-Fernández, A.; Cabrero, J.R.; Serrador, J.M.; Sánchez-Madrid, F. HDAC6: A key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008, 18, 291–297. [Google Scholar] [CrossRef]
- Karki, P.; Birukova, A.A. Microtubules as major regulators of endothelial function: Implication for lung injury. Front. Physiol. 2021, 12, 758313. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Ma, M.; Ma, Z.; Fu, J. HDAC6 inhibition prevents TNF-α-induced caspase 3 activation in lung endothelial cell and maintains cell-cell junctions. Oncotarget 2016, 7, 54714–54722. [Google Scholar] [CrossRef] [PubMed]
- Smith, Q.; Macklin, B.; Chan, X.Y.; Jones, H.; Trempel, M.; Yoder, M.C.; Gerecht, S. Differential HDAC6 activity modulates ciliogenesis and subsequent mechanosensing of endothelial cells derived from pluripotent stem cells. Cell Rep. 2018, 24, 895–908.e6. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.S.; Hubbert, C.C.; Lu, J.; Lee, Y.S.; Lee, J.Y.; Yao, T.P. Histone deacetylase 6 regulates growth factor-induced actin remodeling and endocytosis. Mol. Cell Biol. 2007, 27, 8637–8647. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yuan, Z.; Zhang, Y.; Yong, S.; Salas-Burgos, A.; Koomen, J.; Olashaw, N.; Koomen, J.; Olashaw, N.; Parsons, J.T.; et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol. Cell. 2007, 27, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168–1179. [Google Scholar] [CrossRef]
- Yu, J.; Ma, Z.; Shetty, S.; Ma, M.; Fu, J. Selective HDAC6 inhibition prevents TNF-α-induced lung endothelial cell barrier disruption and endotoxin-induced pulmonary edema. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L39–L47. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, Y.; Ballestrem, C.; Carramusa, L.; Mazitschek, R.; Khochbin, S.; Bershadsky, A. Regulation of microtubule dynamics by inhibition of the tubulin deacetylase HDAC6. J. Cell Sci. 2009, 122, 3531–3541. [Google Scholar] [CrossRef]
- Li, D.; Xie, S.; Ren, Y.; Huo, L.; Gao, J.; Cui, D.; Liu, M.; Zhou, J. Microtubule-associated deacetylase HDAC6 promotes angiogenesis by regulating cell migration in an EB1-dependent manner. Protein Cell. 2011, 2, 150–160. [Google Scholar] [CrossRef]
- Zhang, Q.Q.; Zhang, W.J.; Chang, S. HDAC6 inhibition: A significant potential regulator and therapeutic option to translate into clinical practice in renal transplantation. Front. Immunol. 2023, 14, 1168848. [Google Scholar] [CrossRef]
- Saito, S.; Lasky, J.A.; Guo, W.; Nguyen, H.; Mai, A.; Danchuk, S.; Sullivan, D.E.; Shan, B. Pharmacological inhibition of HDAC6 attenuates endothelial barrier dysfunction induced by thrombin. Biochem. Biophys. Res. Commun. 2011, 408, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Gorshkov, B.A.; Zemskova, M.A.; Verin, A.D.; Bogatcheva, N.V. Taxol alleviates 2-methoxyestradiol-induced endothelial permeability. Vasc. Pharmacol. 2012, 56, 56–63. [Google Scholar] [CrossRef]
- Borgas, D.; Chambers, E.; Newton, J.; Ko, J.; Rivera, S.; Rounds, S.; Lu, Q. Cigarette smoke disrupted lung endothelial barrier integrity and increased susceptibility to acute lung injury via histone deacetylase 6. Am. J. Respir. Cell Mol. Biol. 2016, 54, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Karki, P.; Ke, Y.; Tian, Y.; Ohmura, T.; Sitikov, A.; Sarich, N.; Montgomery, C.P.; Birukova, A.A. Staphylococcus aureus-induced endothelial permeability and inflammation are mediated by microtubule destabilization. J. Biol. Chem. 2019, 294, 3369–3384. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, N.; Wroblowski, S.; Knyphausen, P.; de Boor, S.; Brenig, J.; Zienert, A.Y.; Meyer-Teschendorf, K.; Praefcke, G.J.K.; Nolte, H.; Krüger, M.; et al. Structural and mechanistic insights into the regulation of the fundamental rho regulator RhoGDIα by lysine acetylation. J. Biol. Chem. 2016, 291, 5484–5499. [Google Scholar] [CrossRef]
- Menden, H.; Xia, S.; Mabry, S.M.; Noel-MacDonnell, J.; Rajasingh, J.; Ye, S.Q.; Sampath, V. Histone deacetylase 6 regulates endothelial MyD88-dependent canonical TLR signaling, lung inflammation, and alveolar remodeling in the developing lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 317, L332–L346. [Google Scholar] [CrossRef]
- Zhang, H.P.; Wang, L.; Fu, J.J.; Fan, T.; Wang, Z.L.; Wang, G. Association between histone hyperacetylation status in memory T lymphocytes and allergen-induced eosinophilic airway inflammation. Respirology 2016, 21, 850–857. [Google Scholar] [CrossRef]
- Liao, W.; Sun, J.; Liu, W.; Li, W.; Jia, J.; Ou, F.; Su, K.; Zheng, Y.; Zhang, Z.; Sun, Y. HDAC10 upregulation contributes to interleukin 1β-mediated inflammatory activation of synovium-derived mesenchymal stem cells in temporomandibular joint. J. Cell Physiol. 2019, 234, 12646–12662. [Google Scholar] [CrossRef]
- Wang, L.; Zheng, S.; Zhang, L.; Xiao, H.; Gan, H.; Chen, H.; Zhai, X.; Liang, P.; Zhao, J.; Li, Y. Histone deacetylation 10 alleviates inflammation after intracerebral hemorrhage via the PTPN22/NLRP3 pathway in rats. Neuroscience 2020, 432, 247–259. [Google Scholar] [CrossRef]
- Tian, Y.; Shi, H.; Zhang, D.; Wang, C.; Zhao, F.; Li, L.; Xu, Z.; Jiang, J.; Li, J. Nebulized inhalation of LPAE-HDAC10 inhibits acetylation-mediated ROS/NF-κB pathway for silicosis treatment. J. Control. Release 2023, 364, 618–631. [Google Scholar] [CrossRef]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Ma, Z.; Huang, D.; Zhang, J.; Li, J.; Zhi, P.; Zhang, L.; Feng, Y.; Ge, X.; Zhai, J.; et al. Clinicopathological characteristics and prognostic significance of HDAC11 protein expression in non-small cell lung cancer: A retrospective study. Transl. Lung Cancer Res. 2022, 11, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Xie, C.; Chen, Q.; Zhuang, S. HDAC11, an emerging therapeutic target for metabolic disorders. Front. Endocrinol. 2022, 13, 989305. [Google Scholar] [CrossRef] [PubMed]
- Bala, S.; Csak, T.; Kodys, K.; Catalano, D.; Ambade, A.; Furi, I.; Lowe, P.; Cho, Y.; Iracheta-Vellve, A.; Szabo, G. Alcohol-induced miR-155 and HDAC11 inhibit negative regulators of the TLR4 pathway and lead to increased LPS responsiveness of Kupffer cells in alcoholic liver disease. J. Leukoc. Biol. 2017, 102, 487–498. [Google Scholar] [CrossRef]
- Zheng, C.; Zhong, M.; Qi, Z.; Shen, F.; Zhao, Q.; Wu, L.; Huang, Y.; Tsang, S.Y.; Yao, X. Histone deacetylase inhibitors relax mouse aorta partly through their inhibitory action on L-type Ca2+ channels. J. Pharmacol. Exp. Ther. 2017, 363, 211–220. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Liu, B.; Geng, B.; Li, N.; Geng, Q. The role of HDAC3 and its inhibitors in regulation of oxidative stress and chronic diseases. Cell Death Discov. 2023, 9, 131. [Google Scholar] [CrossRef] [PubMed]
- Reichert, N.; Choukrallah, M.A.; Matthias, P. Multiple roles of class I HDACs in proliferation, differentiation, and development. Cell Mol. Life Sci. 2012, 69, 2173–2187. [Google Scholar] [CrossRef] [PubMed]
- Telles, E.; Seto, E. Modulation of cell cycle regulators by HDACs. Front. Biosci. (Sch. Ed.) 2012, 4, 831–839. [Google Scholar]
- Ramaiah, M.J.; Tangutur, A.D.; Manyam, R.R. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci. 2021, 277, 119504. [Google Scholar] [CrossRef]
- Peng, Z.; Shu, B.; Zhang, Y.; Wang, M. Endothelial response to pathophysiological stress. Arter. Thromb. Vasc. Biol. 2019, 39, e233–e243. [Google Scholar] [CrossRef]
- Dzobo, K.E.; Hanford, K.M.L.; Kroon, J. Vascular metabolism as driver of Atherosclerosis: Linking endothelial metabolism to inflammation. Immunometabolism 2021, 3, e210020. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.T.; Hung, W.C. Inhibition of proliferation, sprouting, tube formation and Tie2 signaling of lymphatic endothelial cells by the histone deacetylase inhibitor SAHA. Oncol. Rep. 2013, 30, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Sun, M.; Ramchandran, R.; Raj, J.U. IGF-1 signaling in neonatal hypoxia-induced pulmonary hypertension: Role of epigenetic regulation. Vasc. Pharmacol. 2015, 73, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhou, Y.; Loberg, A.; Tahara, S.M.; Malik, P.; Kalra, V.K. Activated transcription factor 3 in association with histone deacetylase 6 negatively regulates microRNA 199a2 transcription by chromatin remodeling and reduces endothelin-1 expression. Mol. Cell Biol. 2016, 36, 2838–2854. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Li, G.; Su, F.; Cai, Y.; Shi, L.; Meng, Y.; Liu, Z.; Sun, J.; Wang, M.; Qian, M.; et al. HDAC8 cooperates with SMAD3/4 complex to suppress SIRT7 and promote cell survival and migration. Nucleic Acids Res. 2020, 48, 2912–2923. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.F.; Lu, J.Y.; Zhang, Y.J.; Zhang, L.X.; Lu, G.D.; Xie, Z.J.; Cheng, M.B.; Shen, Y.F.; Zhang, Y. C/EBPα negatively regulates SIRT7 expression via recruiting HDAC3 to the upstream-promoter of hepatocellular carcinoma cells. Biochim. Biophys. Acta 2016, 1859, 348–354. [Google Scholar] [CrossRef]
- Singh, T.; Kaur, P.; Singh, P.; Singh, S.; Munshi, A. Differential molecular mechanistic behavior of HDACs in cancer progression. Med. Oncol. 2022, 39, 171. [Google Scholar] [CrossRef]
- Lee, Y.; Gustafsson, A.B. Role of apoptosis in cardiovascular disease. Apoptosis 2009, 14, 536–548. [Google Scholar] [CrossRef]
- Tricot, O.; Mallat, Z.; Heymes, C.; Belmin, J.; Lesèche, G.; Tedgui, A. Relation between endothelial cell apoptosis and blood flow direction in human atherosclerotic plaques. Circulation 2000, 101, 2450–2453. [Google Scholar] [CrossRef]
- Bombeli, T.; Schwartz, B.R.; Harlan, J.M. Endothelial cells undergoing apoptosis become proadhesive for nonactivated platelets. Blood 1999, 93, 3831–3838. [Google Scholar] [CrossRef]
- Cancel, L.M.; Tarbell, J.M. The role of apoptosis in LDL transport through cultured endothelial cell monolayers. Atherosclerosis 2010, 208, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Tajadura, V.; Hansen, M.H.; Smith, J.; Charles, H.; Rickman, M.; Farrell-Dillon, K.; Claro, V.; Warboys, C.; Ferro, A. β-catenin promotes endothelial survival by regulating eNOS activity and flow-dependent anti-apoptotic gene expression. Cell Death Dis. 2020, 11, 493. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Peng, K.; Wu, Q.; Wang, Y.; Fan, X.; Zhang, D.M.; Passerini, A.G.; Sun, C. HDAC1 and 2 regulate endothelial VCAM-1 expression and atherogenesis by suppressing methylation of the GATA6 promoter. Theranostics 2021, 11, 5605–5619. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Cheng, N.; Du, J.; Zhang, H.; Zhang, C. MicroRNA-200b-3p promotes endothelial cell apoptosis by targeting HDAC4 in atherosclerosis. BMC Cardiovasc. Disord. 2021, 21, 172. [Google Scholar] [CrossRef] [PubMed]
- Shakespear, M.R.; Halili, M.A.; Irvine, K.M.; Fairlie, D.P.; Sweet, M.J. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011, 32, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Wei, T.; Shen, Z.; Bei, Y.; Lin, H.; Dai, H. Classical HDACs in the regulation of neuroinflammation. Neurochem. Int. 2021, 150, 105182. [Google Scholar] [CrossRef] [PubMed]
- Rajendrasozhan, S.; Yang, S.R.; Edirisinghe, I.; Yao, H.; Adenuga, D.; Rahman, I. Deacetylases and NF-kappaB in redox regulation of cigarette smoke-induced lung inflammation: Epigenetics in pathogenesis of COPD. Antioxid. Redox Signal. 2008, 10, 799–811. [Google Scholar] [CrossRef]
- Hu, Y.; Suliman, B.A. Roles of HDACs in the responses of innate immune cells and as targets in inflammatory diseases. Adv. Exp. Med. Biol. 2017, 1024, 91–110. [Google Scholar]
- Gatla, H.R.; Muniraj, N.; Thevkar, P.; Yavvari, S.; Sukhavasi, S.; Makena, M.R. Regulation of chemokines and cytokines by histone deacetylases and an update on histone decetylase inhibitors in human diseases. Int. J. Mol. Sci. 2019, 20, 1110. [Google Scholar] [CrossRef]
- Luan, Y.; Liu, H.; Luan, Y.; Yang, Y.; Yang, J.; Ren, K.D. New insight in HDACs: Potential therapeutic targets for the treatment of Atherosclerosis. Front. Pharmacol. 2022, 13, 863677. [Google Scholar] [CrossRef]
- Kulthinee, S.; Yano, N.; Zhuang, S.; Wang, L.; Zhao, T.C. Critical functions of histone deacetylases (HDACs) in modulating inflammation associated with cardiovascular diseases. Pathophysiology 2022, 29, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.; Verin, A.D.; Black, S.M.; Catravas, J.D. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem. Pharmacol. 2009, 77, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Zhang, B.; Wei, X.; Wang, Z.; Fan, B.; Du, P.; Zhang, Y.; Jian, W.; Chen, L.; Wang, L.; et al. HDAC4/5-HMGB1 signalling mediated by NADPH oxidase activity contributes to cerebral ischaemia/reperfusion injury. J. Cell Mol. Med. 2013, 17, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, Z.; Liu, J. Role of HDACs in normal and malignant hematopoiesis. Mol. Cancer 2020, 19, 5, Erratum in Mol. Cancer 2020, 19, 55. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.; Bertrand, P. Inside HDACs with more selective HDAC inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483. [Google Scholar] [CrossRef] [PubMed]
- Bieliauskas, A.V.; Pflum, M.K. Isoform-selective histone deacetylase inhibitors. Chem. Soc. Rev. 2008, 37, 1402–1413. [Google Scholar] [CrossRef]
- Li, Y.; Wang, F.; Chen, X.; Wang, J.; Zhao, Y.; Li, Y.; He, B. Zinc-dependent deacetylase (HDAC) inhibitors with different zinc binding groups. Curr. Top. Med. Chem. 2019, 19, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lin, S.; Gu, Z.; Chen, L.; He, B. Zinc-dependent deacetylases (HDACs) as potential targets for treating Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2022, 76, 129015. [Google Scholar] [CrossRef]
- Jin, G.; Wang, K.; Zhao, Y.; Yuan, S.; He, Z.; Zhang, J. Targeting histone deacetylases for heart diseases. Bioorg. Chem. 2023, 138, 106601. [Google Scholar] [CrossRef]
- von Knethen, A.; Brüne, B. Histone deacetylation inhibitors as therapy concept in sepsis. Int. J. Mol. Sci. 2019, 20, 346. [Google Scholar] [CrossRef]
- Shanmugam, G.; Rakshit, S.; Sarkar, K. HDAC inhibitors: Targets for tumor therapy, immune modulation and lung diseases. Transl. Oncol. 2022, 16, 101312. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Tang, S.E.; Ko, F.C.; Wu, G.C.; Huang, K.L.; Chu, S.J. Valproic acid attenuates acute lung injury induced by ischemia-reperfusion in rats. Anesthesiology 2015, 122, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Kasotakis, G.; Galvan, M.D.; Osathanugrah, P.; Dharia, N.; Bufe, L.; Breed, Z.; Mizgerd, J.P.; Remick, D.G. Timing of valproic acid in acute lung injury: Prevention is the best therapy? J. Surg. Res. 2017, 220, 206–212. [Google Scholar] [CrossRef]
- Kasotakis, G.; Galvan, M.; King, E.; Sarkar, B.; Stucchi, A.; Mizgerd, J.P.; Burke, P.A.; Remick, D. Valproic acid mitigates the inflammatory response and prevents acute respiratory distress syndrome in a murine model of Escherichia coli pneumonia at the expense of bacterial clearance. J. Trauma Acute Care Surg. 2017, 82, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Cheng, B.; Yang, L.; Li, G.; Yuan, Y.; Luo, G.; Shu, Z.; Jiang, H. LncRNA ZEB1-AS1 knockdown alleviates oxidative low-density lipoprotein-induced endothelial cell injury via the miR-590-5p/HDAC9 axis. Cent. Eur. J. Immunol. 2021, 46, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Larsson, P.; Ulfhammer, E.; Magnusson, M.; Bergh, N.; Lunke, S.; El-Osta, A.; Medcalf, R.L.; Svensson, P.A.; Karlsson, L.; Jern, S. Role of histone acetylation in the stimulatory effect of valproic acid on vascular endothelial tissue-type plasminogen activator expression. PLoS ONE. 2012, 7, e31573. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Okada, M.; Mizuno, W.; Oda, M.; Ide, N.; Morita, T.; Hara, Y.; Yamawaki, H. HDAC4 mediates development of hypertension via vascular inflammation in spontaneous hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1894–H1904. [Google Scholar] [CrossRef]
- Granger, A.; Abdullah, I.; Huebner, F.; Stout, A.; Wang, T.; Huebner, T.; Epstein, J.A.; Gruber, P.J. Histone deacetylase inhibition reduces myocardial ischemia-reperfusion injury in mice. FASEB J. 2008, 22, 3549–3560. [Google Scholar] [CrossRef]
- Thangavel, J.; Malik, A.B.; Elias, H.K.; Rajasingh, S.; Simpson, A.D.; Sundivakkam, P.K.; Vogel, S.M.; Xuan, Y.T.; Dawn, B.; Rajasingh, J. Combinatorial therapy with acetylation and methylation modifiers attenuates lung vascular hyperpermeability in endotoxemia-induced mouse inflammatory lung injury. Am. J. Pathol. 2014, 184, 2237–2249. [Google Scholar] [CrossRef]
- Thangavel, J.; Samanta, S.; Rajasingh, S.; Barani, B.; Xuan, Y.T.; Dawn, B.; Rajasingh, J. Epigenetic modifiers reduce inflammation and modulate macrophage phenotype during endotoxemia-induced acute lung injury. J. Cell Sci. 2015, 128, 3094–3105. [Google Scholar] [CrossRef]
- Luo, Z.F.; Jiang, X.H.; Liu, H.; He, L.Y.; Luo, X.; Chen, F.C.; Tan, Y.L. miR-23b attenuates LPS-induced inflammatory responses in acute lung injury via inhibition of HDAC2. Biochem. Genet. 2021, 59, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Asare, Y.; Campbell-James, T.A.; Bokov, Y.; Yu, L.L.; Prestel, M.; El Bounkari, O.; Roth, S.; El Bounkari, O.; Roth, S.; Megens, R.T.A.; et al. Histone deacetylase 9 activates IKK to regulate atherosclerotic plaque vulnerability. Circ. Res. 2020, 127, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Illi, B.; Dello Russo, C.; Colussi, C.; Rosati, J.; Pallaoro, M.; Spallotta, F.; Rotili, D.; Spallotta, F.; Rotili, D.; Valente, S.; et al. Nitric oxide modulates chromatin folding in human endothelial cells via protein phosphatase 2A activation and class II histone deacetylases nuclear shuttling. Circ. Res. 2008, 102, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.T.; Antony, L.; Dalrymple, S.L.; Brennen, W.N.; Gerber, S.; Hammers, H.; Wissing, M.; Kachhap, S.; Luo, J.; Xing, L.; et al. Tasquinimod is an allosteric modulator of HDAC4 survival signaling within the compromised cancer microenvironment. Cancer Res. 2013, 73, 1386–1399. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Hou, X.; Li, T.; Yu, J.; Chen, H.; Liu, N.; Qiu, A.; Zhuang, S. Pharmacological and genetic inhibition of HDAC4 alleviates renal injury and fibrosis in mice. Front. Pharmacol. 2022, 13, 929334. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, X.; Shetty, S.; Hou, G.; Wang, Q.; Fu, J. HDAC6 inhibition blocks inflammatory signaling and caspase-1 activation in LPS-induced acute lung injury. Toxicol. Appl. Pharmacol. 2019, 370, 178–183. [Google Scholar] [CrossRef]
- Chi, Z.; Byeon, H.E.; Seo, E.; Nguyen, Q.T.; Lee, W.; Jeong, Y.; Choi, J.; Pandey, D.; Berkowitz, D.E.; Kim, J.H.; et al. Histone deacetylase 6 inhibitor tubastatin A attenuates angiotensin II-induced hypertension by preventing cystathionine γ-lyase protein degradation. Pharmacol. Res. 2019, 146, 104281. [Google Scholar] [CrossRef]
- Zhou, B.; Zeng, S.; Li, N.; Yu, L.; Yang, G.; Yang, Y.; Zhang, X.; Fang, M.; Xia, J.; Xu, Y. Angiogenic factor with G patch and FHA domains 1 is a novel regulator of vascular injury. Arter. Thromb. Vasc. Biol. 2017, 37, 675–684. [Google Scholar] [CrossRef]
- dos Santos, C.C.; Han, B.; Andrade, C.F.; Bai, X.; Uhlig, S.; Hubmayr, R.; Tsang, M.; Lodyga, M.; Keshavjee, S.; Slutsky, A.S.; et al. DNA microarray analysis of gene expression in alveolar epithelial cells in response to TNF-alpha, LPS, and cyclic stretch. Physiol. Genom. 2004, 19, 331–342. [Google Scholar] [CrossRef]
- dos Santos, C.C.; Okutani, D.; Hu, P.; Han, B.; Crimi, E.; He, X.; Keshavjee, S.; Greenwood, C.; Slutsky, A.S.; Zhang, H.; et al. Differential gene profiling in acute lung injury identifies injury-specific gene expression. Crit. Care Med. 2008, 36, 855–865. [Google Scholar] [CrossRef]
- Lynn, H.; Sun, X.; Casanova, N.; Gonzales-Garay, M.; Bime, C.; Garcia, J.G.N. Genomic and genetic approaches to deciphering acute respiratory distress syndrome risk and mortality. Antioxid. Redox Signal. 2019, 31, 1027–1052. [Google Scholar] [CrossRef] [PubMed]
- Göttlicher, M.; Minucci, S.; Zhu, P.; Krämer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef] [PubMed]
- Sztajnkrycer, M.D. Valproic acid toxicity: Overview and management. J. Toxicol. Clin. Toxicol. 2002, 40, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Verin, A.D. Letter to the Editor: “Histone deacetylase 7 inhibition in a murine model of Gram-negative Pneumonia-induced acute lung injury”. Shock 2020, 53, 375. [Google Scholar] [CrossRef]
- Li, L.F.; Lee, C.S.; Lin, C.W.; Chen, N.H.; Chuang, L.P.; Hung, C.Y.; Liu, Y.Y. Trichostatin A attenuates ventilation-augmented epithelial-mesenchymal transition in mice with bleomycin-induced acute lung injury by suppressing the Akt pathway. PLoS ONE 2017, 12, e0172571. [Google Scholar] [CrossRef]
- Chen, H.Y.; Li, L.; Fu, Z.J. Histone deacetylase inhibitors trichostatin A and suberoylanilide hydroxamic acid attenuate ventilator-induced lung injury. Pharmazie 2014, 69, 55–59. [Google Scholar]
- Ni, Y.F.; Wang, J.; Yan, X.L.; Tian, F.; Zhao, J.B.; Wang, Y.J.; Jiang, T. Histone deacetylase inhibitor, butyrate, attenuates lipopolysaccharide-induced acute lung injury in mice. Respir. Res. 2010, 11, 33. [Google Scholar] [CrossRef]
- Zhang, L.; Jin, S.; Wang, C.; Jiang, R.; Wan, J. Histone deacetylase inhibitors attenuate acute lung injury during cecal ligation and puncture-induced polymicrobial sepsis. World J. Surg. 2010, 34, 1676–1683. [Google Scholar] [CrossRef]
- Pooladanda, V.; Thatikonda, S.; Bale, S.; Pattnaik, B.; Sigalapalli, D.K.; Bathini, N.B.; Singh, S.B.; Godugu, C. Nimbolide protects against endotoxin-induced acute respiratory distress syndrome by inhibiting TNF-α mediated NF-κB and HDAC-3 nuclear translocation. Cell Death Dis. 2019, 10, 81. [Google Scholar] [CrossRef]
- Leus, N.G.; van der Wouden, P.E.; van den Bosch, T.; Hooghiemstra, W.T.R.; Ourailidou, M.E.; Kistemaker, L.E.; Bischoff, R.; Gosens, R.; Haisma, H.J.; Dekker, F.J. HDAC 3-selective inhibitor RGFP966 demonstrates anti-inflammatory properties in RAW 264.7 macrophages and mouse precision-cut lung slices by attenuating NF-κB p65 transcriptional activity. Biochem. Pharmacol. 2016, 108, 58–74. [Google Scholar] [CrossRef]
- Nguyen, H.C.B.; Adlanmerini, M.; Hauck, A.K.; Lazar, M.A. Dichotomous engagement of HDAC3 activity governs inflammatory responses. Nature 2020, 584, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Ning, L.; Rui, X.; Bo, W.; Qing, G. The critical roles of histone deacetylase 3 in the pathogenesis of solid organ injury. Cell Death Dis. 2021, 12, 734. [Google Scholar] [CrossRef] [PubMed]
- Lauffer, B.E.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 2013, 288, 26926–26943. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Jung, S.M.; Park, K.S.; Kim, K.J. Integrative analysis of lung molecular signatures reveals key drivers of idiopathic pulmonary fibrosis. BMC Pulm. Med. 2021, 21, 404. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.M.; Park, K.S.; Kim, K.J. Integrative analysis of lung molecular signatures reveals key drivers of systemic sclerosis-associated interstitial lung disease. Ann. Rheum. Dis. 2022, 81, 108–116. [Google Scholar] [CrossRef]
- Bergougnoux, A.; Petit, A.; Knabe, L.; Bribes, E.; Chiron, R.; De Sario, A.; Claustres, M.; Molinari, N.; Vachier, I.; Taulan-Cadars, M.; et al. The HDAC inhibitor SAHA does not rescue CFTR membrane expression in Cystic Fibrosis. Int. J. Biochem. Cell Biol. 2017, 88, 124–132. [Google Scholar] [CrossRef]
- Barnes, P.J. Histone deacetylase-2 and airway disease. Ther. Adv. Respir. Dis. 2009, 3, 235–243. [Google Scholar] [CrossRef]
- Barnes, P.J. Role of HDAC2 in the pathophysiology of COPD. Annu. Rev. Physiol. 2009, 71, 451–464. [Google Scholar] [CrossRef]
- Kee, H.J.; Kim, I.; Jeong, M.H. Zinc-dependent histone deacetylases: Potential therapeutic targets for arterial hypertension. Biochem. Pharmacol. 2022, 202, 115111. [Google Scholar] [CrossRef]
- Su, Y.; Han, W.; Kovacs-Kasa, A.; Verin, A.D.; Kovacs, L. HDAC6 Activates ERK in airway and pulmonary vascular remodeling of chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2021, 65, 603–614. [Google Scholar] [CrossRef]
- Wang, Y.; Wallach, J.; Duane, S.; Wang, Y.; Wu, J.; Wang, J.; Adejare, A.; Ma, H. Developing selective histone deacetylases (HDACs) inhibitors through ebselen and analogs. Drug Des. Dev. Ther. 2017, 11, 1369–1382. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zheng, R.; Sun, S. Drug Repurposing: Escitalopram attenuates acute lung injury by inhibiting the SIK2/ HDAC4/ NF-κB signaling cascade. Biochem. Biophys. Res. Commun. 2022, 599, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, Y.; Tu, T.; Schmull, S.; Han, Y.; Wang, W.; Li, H. Dual inhibition of HDAC and tyrosine kinase signaling pathways with CUDC-907 attenuates TGFβ1 induced lung and tumor fibrosis. Cell Death Dis. 2020, 11, 765. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Isoform | Subcellular Distribution | Preferential Expression | A.A. Length | Non-Histone Substrates | Activities/Functions in Vasculature |
|---|---|---|---|---|---|---|
| Class I | HDAC1 | Nucleus | Ubiquitous | 482 | p53, SHP, MyoD, STAT3, E2F1, AMPK, NF-kB, RB1, CtIP, ATF4, SRF [36,37,38,39] | Facilitates the impact of external and environmental stimuli on ECs [40] |
| HDAC2 | Nucleus | Ubiquitous | 488 | YY1, BCL6, GCCR, STAT3 [37] | Protect against DNA damage response and the onset of cellular senescence [41], critical for vascular homeostasis and endothelial health [42] | |
| HDAC3 | Nucleus/ Cytoplasm | Ubiquitous | 428 | YY1, SHP, p65, GATA1, MEF2D, STAT3, ATF4, SUMO-LXR [37,38,39,43] | Preserves endothelial integrity [44]; Controls lung alveolar macrophage development and homeostasis [45] | |
| HDAC8 | Nucleus/ Cytoplasm | Ubiquitous | 377 | Actin, SMC3 [37]; KMT2D, NCOA3, TUBA1A [39] | Culprit in hypertension [46] | |
| Class IIa | HDAC4 | Nucleus/ Cytoplasm | Heart, SM, Brain | 1084 | HP1, GATA1 [37]; SRF, ATF4, SUMO-LXR [38]; human transcription factor HIF-1α [39] | Regulates cellular senescence, apoptosis and autophagy, acts as inflammatory mediator [47,48] and a regulator of vascular endothelial growth factor D [49] |
| HDAC5 | Nucleus/ Cytoplasm | Heart, SM, Brain | 1122 | HP1, SMAD7 [37]; p53 [39] | Controls activity of KLF2, KLF2 activation in ECs; induces eNOS expression resulting in vasodilation [50] | |
| HDAC7 | Nucleus/ Cytoplasm | Heart, Placenta, Pancreas, SM | 952 | PLAG1, PLAG2 [37]; HIF-1α [38] | Suppresses EC proliferation [51], controls EC proliferation and migration [52], maintains vascular integrity in embryogenesis [53], promotes promyelocytic leukemia protein sumoylation [54], Promotes angiogenesis [55]; involves in E. coli-induced ALI [56] | |
| HDAC9 | Nucleus/ Cytoplasm | SM, Brain | 1011 | NA | Inflammatory mediator [57] | |
| Class IIb | HDAC6 | Cytoplasm | Heart, Liver, Kidney, Pancreas | 1215 | HSP90, SHP, SMAD, α-tubulin [37], G3BP1 [58]; Survivin, AKT, β-catenin, Peroxiredoxin, MMP-9 [38]; p53, ERK1, human cortactin [39] | Crucial in EC function [59], Regulates EC migration and angiogenesis [60], Important in atherosclerosis [61] and HSP90-mediated VEGFR regulation [62] |
| HDAC10 | Cytoplasm | Liver, Spleen, Kidney | 669 | AKT, β-catenin, MMP-9 [38]; N-acetylputrescine, N8-acetylspermidine [39] | Accelerates angiogenesis in EC via PTPN22/ERK axis [63], Pulmonary hypertension [64], Regulates HSP90-mediated VEGFR [62] | |
| Class IV | HDAC11 | Nucleus | Brain, Heart, SM, Kidney, & Testis | 347 | MyoD [38]; SHMT2 [39] | Compromises the vascular endothelial barrier function [65], Key player in atherosclerosis [66], Triggers caspase-mediated pathways (NLRP3/caspase-1/GSDMD; caspase-3/GSDME) causing pyroptosis [67] |
| Class | Inhibitor | Mode of Action | Reference/s |
|---|---|---|---|
| Class I | Valproic acid | Attenuate parameters of lung injury like oxidative stress, apoptosis, and inflammation, enhance HO-1 activity (ALI) | [223] |
| Reduces levels of IL-6 and tumor necrosis factor (ALI) | [224] | ||
| Reduces neutrophil influx into lungs and local tissue destruction via decreasing myeloperoxidase activity. Ameliorate pulmonary as well as systemic inflammatory response (ARDS) | [225] | ||
| Antagonizes the inflammatory damage of vascular tissues | [226] | ||
| Inhibits VEGFR-2 protein expression in angiogenesis | [142] | ||
| Increases histone acetylation in thrombopoiesis | [227] | ||
| Sodium butyrate | Inhibits VEGFR-2 protein expression in angiogenesis | [142] | |
| Trichostatin A | Alleviates HDAC4-mediated vascular inflammatory responses in hypertension | [228] | |
| Prevents I/R injury-induced activation of gene programs that include cell death and vascular permeability | [229] | ||
| Inhibits VEGFR-2 protein expression in angiogenesis | [142] | ||
| Trichostatin A + 5-Aza 2-deoxycytidine | Inhibits the eNOS-Cav1-MLC2 signaling pathway, enhances acetylation of histone markers and improves EC permeability (ALI) | [230] | |
| Reduce inflammation and promote an anti-inflammatory M2 macrophage by inhibiting MAPK-HuR-TNF and activating STAT3-Bcl2 pathways (ALI) | [231] | ||
| miR-23b (HDAC2) | Reduces levels of IL-1β, IL-6, and TNF-α and inhibit HDAC2 (ALI) | [232] | |
| PCI34051 (HDAC8) | Reduces blood pressure via attenuating a component of the RAS or modulating nitric oxide signaling pathways (Hypertension) | [46] | |
| Class IIa | Valproic acid | Same as Class I | - |
| Sodium butyrate | Same as Class I | - | |
| Trichostatin A | Same as Class I | - | |
| TMP 195 | Limits proinflammatory responses in Atherosclerosis | [233] | |
| MC1568 | Abolishes NO-induced formation of macromolecular complexes and regulates downstream gene expression | [234] | |
| Tasquinimod (HDAC4) | Allosterically binds to HDAC4 and prevents HDAC4/nuclear receptor corepressor (N-CoR); HDAC3 complex formation, which inhibits HDAC4-regulated histone deacetylation and transcription, thus reduces inflammation in Angiogenesis | [235,236] | |
| Class IIb | Sodium butyrate | Same as Class I | - |
| Trichostatin A | Same as Class I | - | |
| Class IIb (HDAC6) | CAY10603 | Prevent ɑ-tubulin deacetylation, protects against inflammation, blocks IĸB phosphorylation, and reduces caspase-1 activation, particularly in epithelial cells (ALI) | [237] |
| Tubastatin A | Inhibit angiotensin II-induced hypertension via protecting cystathionine γ-lyase protein degradation | [238] | |
| Class IV | Trichostatin A | Same as Class I | - |
| Hydroxytyrosol acetate | Inhibits pyroptosis by possible targeting of HDAC11 in TNF-α-induced HUVECs (Atherosclerosis) | [66] | |
| Quisinostat | Aggf1 regulates the pathophysiology of vascular endothelium; therefore, HDAC11 inhibitors restore the expression of Aggf1 in vascular injury | [239] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, R.S.; Maloney, M.E.; Lucas, R.; Fulton, D.J.R.; Patel, V.; Bagi, Z.; Kovacs-Kasa, A.; Kovacs, L.; Su, Y.; Verin, A.D. Zinc-Dependent Histone Deacetylases in Lung Endothelial Pathobiology. Biomolecules 2024, 14, 140. https://doi.org/10.3390/biom14020140
Patil RS, Maloney ME, Lucas R, Fulton DJR, Patel V, Bagi Z, Kovacs-Kasa A, Kovacs L, Su Y, Verin AD. Zinc-Dependent Histone Deacetylases in Lung Endothelial Pathobiology. Biomolecules. 2024; 14(2):140. https://doi.org/10.3390/biom14020140
Chicago/Turabian StylePatil, Rahul S., McKenzie E. Maloney, Rudolf Lucas, David J. R. Fulton, Vijay Patel, Zsolt Bagi, Anita Kovacs-Kasa, Laszlo Kovacs, Yunchao Su, and Alexander D. Verin. 2024. "Zinc-Dependent Histone Deacetylases in Lung Endothelial Pathobiology" Biomolecules 14, no. 2: 140. https://doi.org/10.3390/biom14020140
APA StylePatil, R. S., Maloney, M. E., Lucas, R., Fulton, D. J. R., Patel, V., Bagi, Z., Kovacs-Kasa, A., Kovacs, L., Su, Y., & Verin, A. D. (2024). Zinc-Dependent Histone Deacetylases in Lung Endothelial Pathobiology. Biomolecules, 14(2), 140. https://doi.org/10.3390/biom14020140