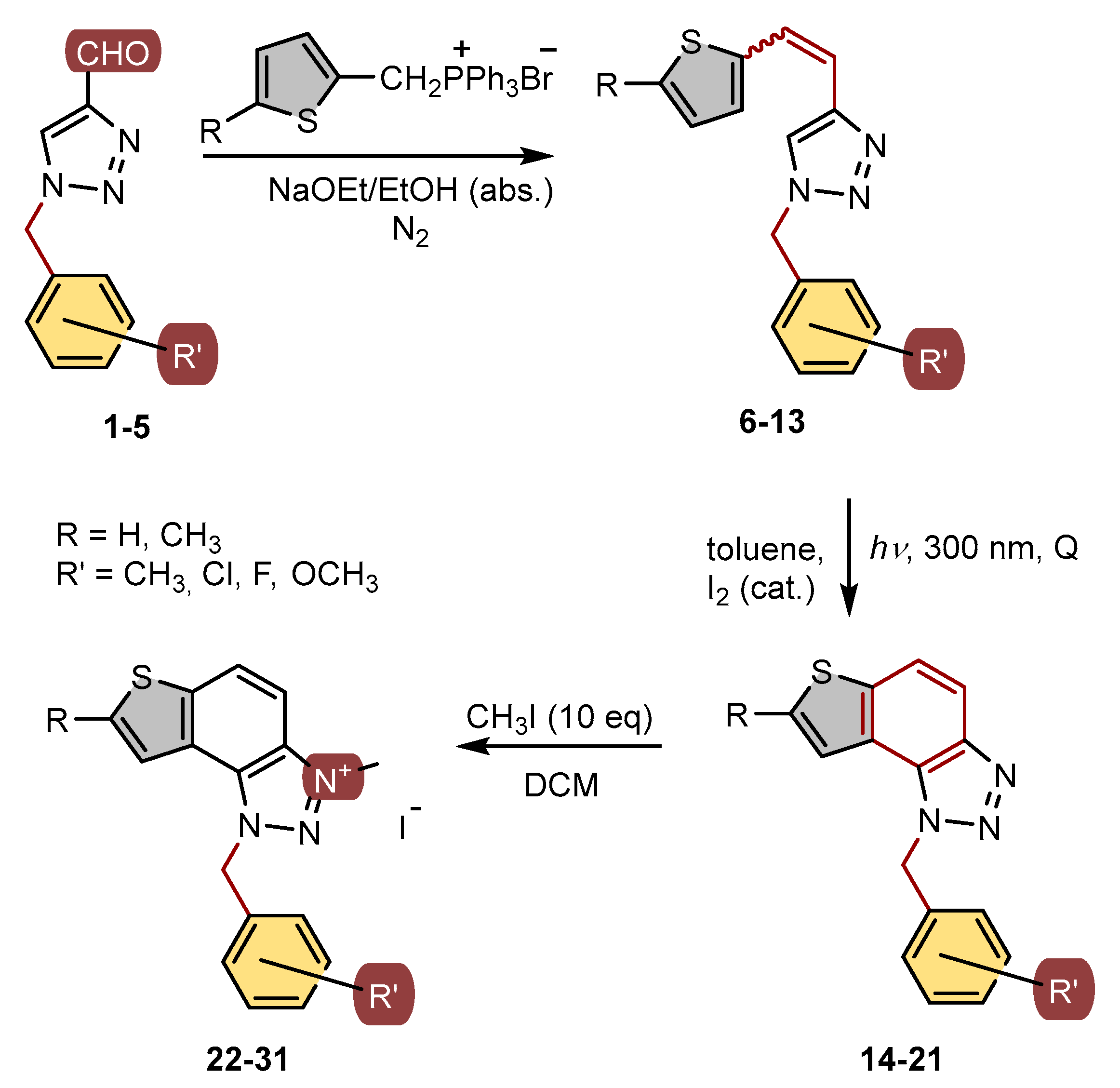
2. Materials and Methods
2.1. General Procedure
1H NMR and
13C NMR (CDCl
3) techniques were used to confirm the structure of the synthesized compounds, and the spectra were recorded on a Bruker Avance instrument at 300 and 600 MHz for
1H NMR and at 75 and 150 MHz for
13C NMR. Chemical shifts (
δ) are expressed in parts per million values (ppm) and coupling constants (
J) in Hz. As a standard, tetramethylsilane (TMS) was used. The following signals in the
1H NMR spectra were specific and did not correspond to compounds: the signal for water in chloroform at about 1.50 ppm; the signal for chloroform at 7.24 ppm; the signal for dichloromethane in chloroform at 5.26 ppm; and the signal for acetone in chloroform at 2.17 ppm. Each
13C NMR spectrum contained one specific signal (group of three peak lines) at 77 ppm corresponding to the solvent-deuterated chloroform used. Triazolium salts
23,
25,
26,
30 and
31 were also recorded in CD
3OD to solve the problem of relatively low solubility. High-resolution mass spectrometry (HRMS) analyses were performed on a MALDI TOF/TOF analyzer mass spectrometer fitted with an Nd:YAG laser at 355 nm (fitting rate of 200 Hz). Photochemical reactions were carried out in a 50.0 mL solution in quartz cuvettes that transmitted light. For this purpose, a Rayonet photochemical reactor equipped with UV lamps (10) with a wavelength of 313 nm was used. All solvents used in this work were purified by distillation and were commercially available. The phosphonium salts were synthesized in our laboratory, and 1-(4-nitrophenyl)-1
H-1,2,3-triazole-4-carbaldehyde used was previously synthesized in our laboratory [
21]. After each Wittig reaction, an extraction was performed three times, separating the organic and aqueous layers.
The organic layer was dried over anhydrous magnesium sulfate, MgSO4. Thin-layer chromatography was performed on plates coated with silica gel (0.2 mm, 60/Kieselguhr F254) immersed in 10 mL of the dissolution system. Column chromatography was performed in glass columns of different diameters. The columns were filled with silica gel (60 Å, technical grade) of different heights. Abbreviations used in the experimental part of the work are the following: CAN—acetonitrile; DCM—dichloromethane; E—diethyl ether; EtOAc—ethyl acetate; PE—petroleum ether; NMR—nuclear magnetic resonance; UV—ultraviolet spectroscopy; NaOEt—sodium ethoxide; s—singlet; d—doublet; t—triplet; m—multiplet; dd—doublet of doublets; and q—quartet. UV/Vis spectra were recorded on a Varian Cary 100 Bio spectrometer and fluorescence spectra on a Varian Cary Eclipse fluorimeter in appropriate quartz cuvettes (path length = 1 cm).
2.2. Synthesis of Phosphonium Salts 1′ and 2′
In a 250 mL three-necked flask, 7.9 g (0.063 mol) of 2-thiophene methanol was dissolved in 72.3 mL of dry solvent diethyl ether and added to the flask. Then, 5.71 g (0.022 mol) of phosphorus tribromide (PBr3) was dissolved in 7.23 mL of dry diethyl ether and added dropwise to the reaction flask over 40 min. The reaction mixture was stirred at room temperature on a magnetic stirrer for 1 h. Subsequently, 15 mL of methanol and 100 mL of distilled water were added, and the mixture was extracted with a total of 300 mL of diethyl ether. The ether layer was dried overnight with anhydrous magnesium sulfate (MgSO4) filtered, and evaporated to dryness using a rotary evaporator, leaving 2-thiophene bromide as a yellow oil in the flask. This oil was then dissolved in 20 mL of toluene, while 16.52 g (0.037 mol) of triphenylphosphine (PPh3) was dissolved in 50 mL of toluene, and both solutions were added to the reaction flask. The mixture was stirred on a magnetic stirrer for three days and then filtered through a Büchner funnel under reduced pressure. The resulting light-yellow salt was dried in a desiccator for 12 h. The dry thiophene-phosphonium salt 1′ obtained was used in all subsequent experiments for synthesizing compounds 6–8.
![Biomolecules 14 01391 i018]()
In a 250 mL round flask, 4.925 g (0.0440 mol) of dimethylthiophene was dissolved in 133 mL of carbon tetrachloride (CCl4). Then, 7.83 g (1 equivalent) of N-bromosuccinimide (NBS) was added to the mixture. The chemicals were stirred in an oil bath at a reflux temperature (150 °C). Once reflux started, a small amount of α,α-azobisisobutyronitrile (AIBN) catalyst was added. The oil bath temperature was then reduced to approximately 100 °C due to excessive reflux. With reflux established, the reaction mixture was illuminated with a lamp to initiate the reaction. After 1.5 h, bromide formation began, accompanied by the appearance of white, fluffy succinimide crystals on the surface. After another hour, a second dose of AIBN was added, and the mixture was refluxed for an additional 3 h. The succinimide was filtered out using pleated filter paper, and the clear orange liquid was evaporated using a rotary evaporator, leaving a brown, oily precipitate of thiophene bromide in the flask. In the second step, the thiophene bromide was dissolved in 10 mL of benzene with added PPh3. The mixture was heated to reflux (80 °C, with the mixer set to 110 °C) and stirred at that temperature for 1 h. The heat was then turned off, and the mixture was stirred at room temperature for the next 24 h. The resulting salt was filtered using a Büchner funnel, and the crystallizer was weighed before and after adding the salt. The 2-methylthiophene salt 2′ was dried in a desiccator under vacuum for 4–5 h and then used for synthesizing compounds 9–13.
2.3. Synthesis of Triazole Aldehydes 1–5
Triazole aldehydes
1–
5 were synthesized in small glass vials from 200 mg of 1-(4-nitrophenyl)-1
H-1,2,3-triazole-4-carbaldehyde [
21] dissolved in 2 mL of dry 1,4-dioxane. The 1.2 eq of appropriate benzylamine was then added to the reaction mixture, and the mixture was briefly purged with argon. Depending on the applied benzylamine, the reaction took place for a certain period of time (24–48 h). The course of the reaction was monitored by thin-layer chromatography. At the end of the reaction, the solvent was removed by evaporation on a rotary vacuum evaporator. The solid product was purified by column chromatography using an appropriate solvent system, mostly DCM/EtOAc.
![Biomolecules 14 01391 i019]()
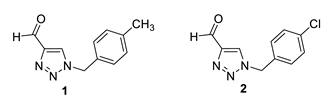
1-(4-methylbenzyl)-1H-1,2,3-triazole-4-carbaldehyde (1): 46 mg (isolated 23%), yellow oil; Rf (DCM) = 0.15; 1H NMR (CDCl3, 600 MHz) δ/ppm: 10.11 (s, 1H), 7.95 (s, 1H), 7.22 (d, J = 8.7 Hz, 2H), 7.20 (d, J = 8.7 Hz, 2H), 5.54 (s, 2H), 2.37 (s, 3H).
1-(4-chlorobenzyl)-1H-1,2,3-triazole-4-carbaldehyde (2): 131 mg (isolated 65%), yellow oil; Rf (DCM) = 0.40; 1H NMR (CDCl3, 300 MHz) δ/ppm: 10.13 (s, 1H), 8.01 (s, 1H), 7.39 (d, J = 7.4 Hz, 2H), 7.25 (d, J = 8.9 Hz, 2H), 5.57 (s, 2H).
![Biomolecules 14 01391 i020]()
1-(3-methylbenzyl)-1H-1,2,3-triazole-4-carbaldehyde (3): 130 mg (isolated 65%), yellow oil; Rf (DCM) = 0.22; 1H NMR (CDCl3, 600 MHz) δ/ppm: 10.12 (s, 1H), 7.98 (s, 1H), 7.29 (t, J = 7.3 Hz, 1H), 7.21 (d, J = 8.1 Hz, 1H), 7.10 (d, J = 6.5 Hz, 2H), 5.54 (s, 2H), 2.36 (s, 3H).
1-(3-methoxybenzyl)-1H-1,2,3-triazole-4-carbaldehyde (4): 121 mg (isolated 61%), yellow oil; Rf (DCM) = 0.30; 1H NMR (CDCl3, 300 MHz) δ/ppm: 10.13 (s, 1H), 7.98 (s, 1H), 7.32 (t, J = 7.8 Hz, 1H), 6.93 (dd, J = 8.7, 2.6 Hz, 1H), 6.88 (d, J = 7.8 Hz, 1H), 6.82 (s, 1H), 5.55 (s, 2H), 3.80 (s, 3H).
![Biomolecules 14 01391 i021]()
1-(3-fluorobenzyl)-1H-1,2,3-triazole-4-carbaldehyde (5): 163 mg (isolated 82%), yellow oil; Rf (DCM) = 0.42; 1H NMR (CDCl3, 600 MHz) δ/ppm: 10.14 (s, 1H), 8.03 (s, 1H), 7.41–7.37 (m, 1H), 7.11 (dd, J = 8.0, 2.4 Hz, 1H), 7.08 (d, J = 8.0 Hz, 1H), 7.00 (d, J = 8.8 Hz, 1H), 5.59 (s, 2H).
2.4. Synthesis of Triazolostilbenes 6–13 by Wittig Reaction
The apparatus, in a three-necked flask, a dropping funnel, a chlorine-calcium tube, and a balloon filled with nitrogen, was blown with argon for 5 min. A magnet was placed in the flask, the addition funnel was closed, and 30 mL of absolute ethanol was poured in (depending on the amount of starting reactants). A portion of absolute ethanol (10 mL) was poured into the flask, and the required amount of triphenylphosphonium salt was added. Sodium previously weighed in PE on an analytical balance with a precision of 0.0001 g was added to the remaining amount of absolute ethanol. After all the sodium had reacted in the ethanol with the evolution of hydrogen, a little portion of NaOEt was added to the flask. The aldehyde was dissolved in ethanol and transferred to a flask, and then the rest of the NaOEt from the funnel was added dropwise. The flask was closed with a glass stopper, and the reaction mixture was left to stir in a magnetic stirrer at room temperature (6—108 h, 7—72 h, 8—48 h, 9—96 h, 10—96 h, 11—96 h, 12—168 h, and 13—120 h). Compounds 6–13 were synthesized as mixtures of isomers and their pure isomers were not isolated due to the next reaction step.
![Biomolecules 14 01391 i022]()
1-(4-methylbenzyl)-4-(2-(thiophen-2-yl)vinyl)-1H-1,2,3-triazole (6): 37 mg (mixture of isomers, cis: trans = 1: 1, 80%), white oil; Rf (PE/E = 80%) = 0.48.
1-(4-chlorobenzyl)-4-(2-(thiophen-2-yl)vinyl)-1H-1,2,3-triazole (7): 145 mg (mixture of isomers, cis: trans = 1: 0.6, 90%), white oil; Rf (PE/E = 60%) = 0.25. MS (ESI) m/z (%, fragment): 302 (100); HRMS (m/z) for C15H12ClN3S: [M + H]+calcd = 302.0441, and [M + H]+measured = 302.0440.
![Biomolecules 14 01391 i023]()
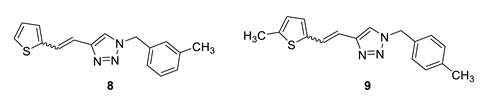
1-(3-methylbenzyl)-4-(2-(thiophen-2-yl)vinyl)-1H-1,2,3-triazole (8): 108 mg (mixture of isomers, cis: trans = 1: 0.8, 91%), white oil; Rf (PE/E = 80%) = 0.67.
1-(4-methylbenzyl)-4-(2-(5-methylthiophen-2-yl)vinyl)-1H-1,2,3-triazole (9): 13 mg (mixture of isomers, cis: trans = 1: 0.9, 13%), white oil; Rf (PE/E = 50%) = 0.60. MS (ESI) m/z (%, fragment): 296 (100); HRMS (m/z) for C17H17N3S: [M + H]+calcd = 296.1143, and [M + H]+measured = 296.1147.
![Biomolecules 14 01391 i024]()
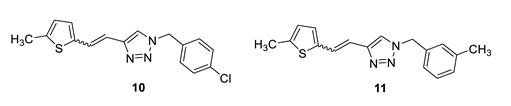
1-(4-chlorobenzyl)-4-(2-(5-methylthiophen-2-yl)vinyl)-1H-1,2,3-triazole (10): 50 mg (mixture of isomers, cis: trans = 1: 0.8, 32%), white oil; Rf (PE/E = 50%) = 0.50. MS (ESI) m/z (%, fragment): 316 (100); HRMS (m/z) for C16H14ClN3S: [M + H]+calcd = 316.0597, and [M + H]+measured = 316.0599.
1-(3-methylbenzyl)-4-(2-(5-methylthiophen-2-yl)vinyl)-1H-1,2,3-triazole (11): 12 mg (mixture of isomers, cis: trans = 1: 0.7, 10%), white oil; Rf (PE/E = 90%) = 0.58. MS (ESI) m/z (%, fragment): 296 (100); HRMS (m/z) for C17H17N3S: [M + H]+calcd = 296.1143, and [M + H]+measured = 296.1139.
![Biomolecules 14 01391 i025]()
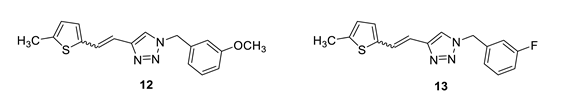
1-(3-methoxybenzyl)-4-(2-(5-methylthiophen-2-yl)vinyl)-1H-1,2,3-triazole (12): 35 mg (mixture of isomers, cis: trans = 1: 0.7, 24%), white oil; Rf (PE/E = 50%) = 0.50. MS (ESI) m/z (%, fragment): 312 (100); HRMS (m/z) for C17H17N3OS: [M + H]+calcd = 312.1092, and [M + H]+measured = 312.1098.
1-(3-fluorobenzyl)-4-(2-(5-methylthiophen-2-yl)vinyl)-1H-1,2,3-triazole (13): 16 mg (mixture of isomers, cis: trans = 1: 0.7, 23%), white oil; Rf (PE/E = 50%) = 0.54.
2.5. Synthesis of Photoproducts 14–21
The corresponding 1,2,3-triazolostilbenes 6–13, as mixtures of isomers, were dissolved in 50 mL of toluene. These mixtures were then transferred to quartz tubes, which allowed light to pass through. A small amount of iodine was added as an oxidizing agent. The solutions, with a concentration of approximately 10−3 mol L−1, were exposed to light for 1–4 h using ten lamps emitting at a wavelength of about 313 nm in a Rayonet photoreactor. The resulting products, 14–21, were purified through column chromatography. Spectroscopic data for photoproducts 14–21 are provided below.
![Biomolecules 14 01391 i026]()
1-(4-methylbenzyl)-1H-thieno [3’,2’:3,4]benzo [1,2-d][1,2,3]triazole (14): 27 mg (isolated 73%), yellow oil; Rf (PE/E (40%)) = 0.34; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.99 (d, J = 9.0 Hz, 1H), 7.80 (d, J = 9.0 Hz, 1H), 7.55 (d, J = 6.0 Hz, 1H), 7.51 (d, J = 5.6 Hz, 1H), 7.12 (d, J = 8.2 Hz, 2H), 7.09 (d, J = 8.2 Hz, 2H), 6.08 (s, 2H), 2.29 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 144.7, 140.0, 138.1, 132.1, 129.8, 128.7, 127.7, 126.5, 122.7, 120.3, 119.2, 116.1, 52.9, 21.1; MS (ESI) m/z (%, fragment): 280 (100); HRMS (m/z) for C16H13N3S: [M + H]+calcd = 280.0830, and [M + H]+measured = 280.0832.
1-(4-chlorobenzyl)-1H-thieno [3’,2’:3,4]benzo [1,2-d][1,2,3]triazole (15): 36 mg (isolated 25%), yellow oil; Rf (PE/E (80%)) = 0.25; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.00 (d, J = 8.6 Hz, 1H), 7.83 (d, J = 8.1 Hz, 1H), 7.58 (d, J = 5.4 Hz, 1H), 7.46 (d, J = 5.4 Hz, 1H), 7.29 (d, J = 8.6 Hz, 2H), 7.13 (d, J = 9.2 Hz, 2H), 6.09 (s, 2H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 144.7, 140.2, 134.3, 133.6, 129.4, 128.6, 128.0, 127.9, 122.5, 119.9, 119.4, 116.1, 52.4; MS (ESI) m/z (%, fragment): 300 (100); HRMS (m/z) for C15H10ClN3S: [M + H]+calcd = 300.0284, and [M + H]+measured = 300.0283.
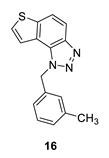
1-(3-methylbenzyl)-1H-thieno [3’,2’:3,4]benzo [1,2-d][1,2,3]triazole (16): 26 mg (isolated 24%), yellow oil; Rf (PE/E (40%)) = 0.35; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.00 (d, J = 8.9 Hz, 1H), 7.81 (d, J = 9.1 Hz, 1H), 7.55 (d, J = 5.9 Hz, 1H), 7.51 (d, J = 5.7 Hz, 1H), 7.20 (t, J = 8.0 Hz, 1H), 7.09 (d, J = 8.0 Hz, 1H), 6.99 (d, J = 8.4 Hz, 2H), 6.09 (s, 2H), 2.27 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 144.7, 140.0, 139.0, 135.1, 129.1, 128.9, 127.7, 127.2, 123.6, 122.8, 120.3, 119.2, 116.1, 53.1, 21.4; MS (ESI) m/z (%, fragment): 280 (100); HRMS (m/z) for C16H13N3S: [M + H]+calcd = 280.0830, and [M + H]+measured = 280.0828.
![Biomolecules 14 01391 i027]()
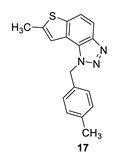
7-methyl-1-(4-methylbenzyl)-1H-thieno [3’,2’:3,4]benzo [1,2-d][1,2,3]triazole (17): 12 mg (isolated 15%), yellow oil; Rf (PE/E (30%)) = 0.46; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.91 (d, J = 8.6 Hz, 1H), 7.68 (d, J = 8.7 Hz, 1H), 7.18 (s, 1H), 7.12 (d, J = 8.3 Hz, 2H), 7.06 (d, J = 8.3 Hz, 2H), 6.04 (s, 2H), 2.60 (s, 3H), 2.30 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 142.6, 138.0, 132.3, 129.7, 126.6, 123.0, 118.9, 118.1, 115.0, 52.8, 21.1, 16.1; MS (ESI) m/z (%, fragment): 294 (100); HRMS (m/z) for C17H15N3S: [M + H]+calcd = 294.0987, and [M + H]+measured = 294.0988.
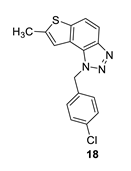
1-(4-chlorobenzyl)-7-methyl-1H-thieno [3’,2’:3,4]benzo [1,2-d][1,2,3]triazole (18): 14 mg (isolated 28%), colourless oil; Rf (PE/E = 50%) = 0.51; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.91 (d, J = 9.0 Hz, 1H), 7.70 (d, J = 9.0 Hz, 1H), 7.30 (d, J = 8.4 Hz, 2H), 7.15–7.09 (m, 3H), 6.05 (s, 2H), 2.61 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 144.7, 143.0, 139.5, 134.3, 133.9, 129.3, 128.0, 122.8, 119.1, 117.7, 115.1, 52.3, 16.2; MS (ESI) m/z (%, fragment): 314 (100); HRMS (m/z) for C16H12ClN3S: [M + H]+calcd = 314.0440, and [M + H]+measured = 314.0441.
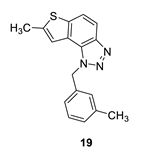
7-methyl-1-(3-methylbenzyl)-1H-thieno [3’,2’:3,4]benzo [1,2-d][1,2,3]triazole (19): 13 mg (isolated 29%), yellow oil; Rf (PE/E (40%)) = 0.46; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.92 (d, J = 8.9 Hz, 1H), 7.65 (d, J = 8.8 Hz, 1H), 7.23–7.18 (m, 2H), 7.09 (d, J = 7.5 Hz, 1H), 7.03–6.96 (m, 2H), 6.04 (s, 2H), 2.60 (s, 3H), 2.28 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 142.6, 139.3, 138.9, 135.3, 129.1, 128.9, 127.3, 123.6, 123.0, 118.9, 118.1, 115.1, 52.9, 21.4, 16.2; MS (ESI) m/z (%, fragment): 294 (100); HRMS (m/z) for C17H15N3S: [M + H]+calcd = 294.0987, and [M + H]+measured = 294.0981.
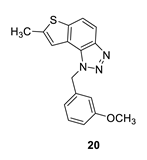
1-(3-methoxybenzyl)-7-methyl-1H-thieno [3’,2’:3,4]benzo [1,2-d][1,2,3]triazole (20): 14 mg (isolated 35%), yellow oil; Rf (PE/E (40%)) = 0.38; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.91 (d, J = 8.9 Hz, 1H), 7.68 (d, J = 8.9 Hz, 1H), 7.22 (d, J = 7.9 Hz, 1H), 7.17 (s, 1H), 6.85–6.75 (m, 2H), 6.70 (s, 1H), 6.05 (s, 2H), 3.71 (s, 3H), 2.60 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 160.1, 144.7, 142.6, 139.3, 136.9, 130.2, 128.3, 123.0, 118.9, 118.8, 118.1, 115.0, 113.6, 112.3, 55.2, 52.8, 16.2; MS (ESI) m/z (%, fragment): 310 (100); HRMS (m/z) for C17H15N3OS: [M + H]+calcd = 310.0936, and [M + H]+measured = 310.0939.
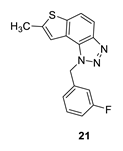
1-(3-fluorobenzyl)-7-methyl-1H-thieno [3’,2’:3,4]benzo [1,2-d][1,2,3]triazole (21): 8 mg (isolated 16%), colourless oil; Rf (PE/E (40%)) = 0.44; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.85 (d, J = 8.9 Hz, 1H), 7.63 (d, J = 8.9 Hz, 1H), 7.27–7.19 (m, 1H), 7.05 (s, 1H), 6.96–6.87 (m, 2H), 6.80 (d, J = 9.5 Hz, 1H), 6.01 (s, 2H), 2.51 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 161.9 (d, JCF = 247.8 Hz), 143.7, 141.9, 138.5, 136.8 (d, JCF = 7.4 Hz), 129.8 (d, JCF = 8.5 Hz), 127.1, 121.8, 121.1 (d, JCF = 3.1 Hz), 118.1, 116.7, 114.3 (d, JCF = 20.9 Hz), 114.1, 112.7 (d, JCF = 22.4 Hz), 51.4, 15.3; MS (ESI) m/z (%, fragment): 298 (100); HRMS (m/z) for C16H12FN3S: [M + H]+calcd = 298.0736, and [M + H]+measured = 298.0734.
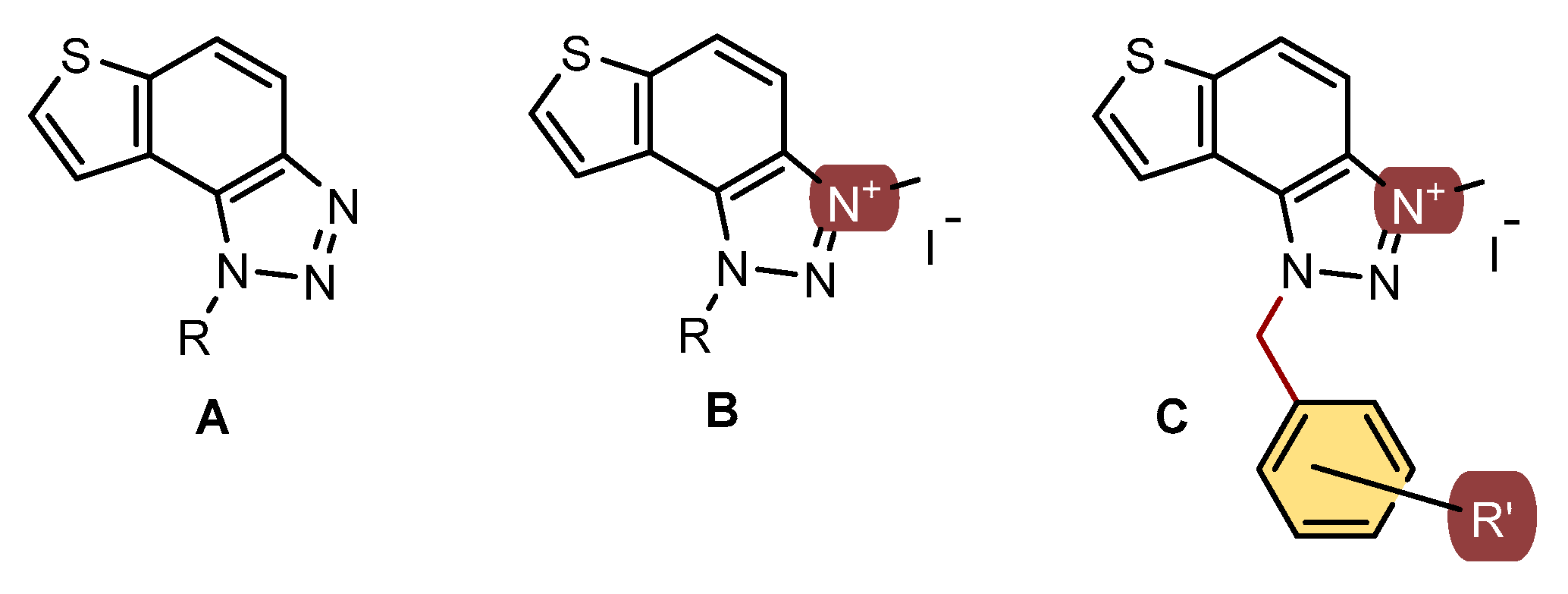
2.6. Synthesis of Triazolium Salts 22–31
In a glass reaction tube with a magnetic stirring bar, the corresponding thienobenzo-triazoles 14–21 (0.2 mmol, 1 equivalent), dry dichloromethane (0.4 mL), and iodomethane (2 mmol, 10 equivalents) were added. The reaction mixtures were purged with argon and then stirred at 60 °C for 24 h. After this period, the mixtures were cooled to 0 °C and diluted with 5 mL of diethyl ether. Following precipitation, the tubes were centrifuged three times for 10 min at 3000 rpm. The solvent was decanted, and the precipitate was washed three to five times with diethyl ether. The crude powders were then dried under high vacuum, yielding the pure triazolium salts 22–29. The solubility of triazolium salts 22–29 is low in CDCl3, which is reflected in the signals, especially in the 13C NMR spectra.
![Biomolecules 14 01391 i028]()
3-methyl-1-(4-methylbenzyl)-1H-thieno [3’,2’:3,4]benzo [1,2-d][1,2,3]triazol-3-ium (22): 8 mg (isolated 30%), yellow powder; Rf (DCM (100%)) = 0.19; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.99 (d, J = 9.1 Hz, 1H), 7.80 (dd, J = 9.1, 0.7 Hz, 1H), 7.55 (d, J = 5.5 Hz, 1H), 7.51 (dd, J = 5.5, 0.7 Hz, 1H), 7.10 (q, J = 17.3, 8.7 Hz, 4H), 2.29 (s, 3H), 1.59 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 143.1, 139.3, 135.1, 132.8, 130.9, 130.4, 127.8, 127.5, 127.2, 126.5, 120.6, 109.0, 57.2, 40.2, 21.2; MS (ESI) m/z (%, fragment): 294 (100); HRMS (m/z) for C17H16N3S+: [M + H]+calcd = 294.0987, and [M + H]+measured = 294.0985.
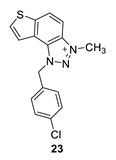
1-(4-chlorobenzyl)-3-methyl-1H-thieno [3’,2’:3,4]benzo [1,2-d][1,2,3]triazol-3-ium (23): 15 mg (isolated 42%), yellow powder; Rf (DCM (100%)) = 0.15; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.34 (d, J = 8.6 Hz, 1H), 8.10 (d, J = 8.8 Hz, 1H), 7.98 (d, J = 4.9 Hz, 1H), 7.85 (d, J = 5.7 Hz, 1H), 7.43–7.40 (m, 4H), 6.45 (s, 2H), 4.86 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 150.9, 130.9, 130.1, 129.5, 129.4, 128.9, 122.6, 121.3, 57.3, 34.6 (signals for 4 quaternary carbons are missing); MS (ESI) m/z (%, fragment): 314 (100); HRMS (m/z) for C16H13ClN3S+: [M + H]+calcd = 314.0440, and [M + H]+measured = 314.0436.
3-methyl-1-(3-methylbenzyl)-1H-thieno [3’,2’:3,4]benzo [1,2-d][1,2,3]triazol-3-ium (24): 4 mg (isolated 15%), yellow powder; Rf (DCM (100%)) = 0.20; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.34 (d, J = 11.1 Hz, 1H), 8.25 (d, J = 11.1 Hz, 1H), 7.94 (d, J = 6.3 Hz, 1H), 7.78 (d, J = 6.3 Hz, 1H), 7.31 (t, J = 7.4 Hz, 1H), 7.22 (d, J = 7.4 Hz, 1H), 7.18–7.16 (m, 2H), 6.34 (s, 2H), 4.92 (s, 3H), 2.35 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 142.7, 132.8, 130.7, 127.6, 127.1, 124.5, 122.7, 120.5, 116.0, 53.4, 30.9, 21.5 (signals for 5 quaternary carbons are missing); MS (ESI) m/z (%, fragment): 294 (100); HRMS (m/z) for C17H16N3S+: [M + H]+calcd = 294.0987, and [M + H]+measured = 294.0991.
![Biomolecules 14 01391 i029]()
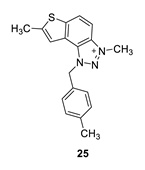
3,7-dimethyl-1-(4-methylbenzyl)-1H-thieno [3’,2’:3,4]benzo [1,2-d][1,2,3]triazol-3-ium (25): 6 mg (isolated 27%), white powder; Rf (DCM (100%)) = 0.22; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.21 (d, J = 6.6 Hz, 1H), 8.07–8.04 (m, 1H), 7.52 (s, 1H), 7.30–7.28 (m, 1H), 7.28–7.27 (m, 1H), 7.24–7.23 (m, 1H), 7.23–7.21 (m, 1H), 6.31 (s, 2H), 4.84 (s, 3H), 2.74 (s, 3H), 2.36 (s, 3H); MS (ESI) m/z (%, fragment): 308 (100); HRMS (m/z) for C18H18N3S+: [M + H]+calcd = 308.1216, and [M + H]+measured = 308.1218.
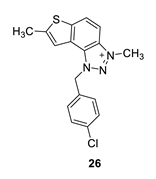
1-(4-chlorobenzyl)-3,7-dimethyl-1H-thieno [3’,2’:3,4]benzo [1,2-d][1,2,3]triazol-3-ium (26): 5 mg, (isolated 22%), white powder; Ryf (DCM (100%)) = 0.21; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.20 (d, J = 6.2 Hz, 1H), 7.98–7.90 (m, 1H), 7.58 (s, 1H), 7.44–7.38 (m, 4H), 6.41 (s, 2H), 4.79 (s, 3H), 2.75 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 156.2, 149.1, 136.0, 129.9, 129.4, 127.3, 126.9, 123.4, 118.4, 65.9, 16.6, 15.3 (signals for 3 quaternary carbons are missing); MS (ESI) m/z (%, fragment): 328 (100); HRMS (m/z) for C17H15ClN3S+: [M + H]+calcd = 328.0122, and [M + H]+measured = 328.0120.
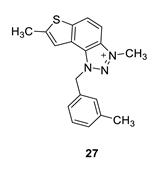
3,7-dimethyl-1-(3-methylbenzyl)-1H-thieno [3’,2’:3,4]benzo [1,2-d][1,2,3]triazol-3-ium (27): 2 mg (isolated 15%), white powder; Rf (DCM (100%)) = 0.24; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.21 (d, J = 9.3 Hz), 8.11–8.09 (m, 1H), 7.49 (s, 1H), 7.31 (t, J = 7.6 Hz, 1H), 7.23 (d, J = 7.1 Hz, 1H), 7.19–7.16 (m, 2H), 6.31 (s, 2H), 4.86 (s, 3H), 2.73 (s, 3H), 2.36 (s, 3H); MS (ESI) m/z (%, fragment): MS (ESI) m/z (%, fragment): 308 (100); HRMS (m/z) for C18H18N3S+: [M + H]+calcd = 308.1216, and [M + H]+measured = 308.1218.
![Biomolecules 14 01391 i030]()
1-(3-methoxybenzyl)-3,7-dimethyl-1H-thieno [3’,2’:3,4]benzo [1,2-d][1,2,3]triazol-3-ium (28): 4 mg, (isolated 26%), yellow powder; Rf (DCM (100%)) = 0.18; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.19 (br s, 1H), 8.00 (br s, 1H), 7.53 (br s, 1H), 7.30 (d, J = 8.4 Hz, 1H), 7.13–7.10 (m, 1H), 6.95–6.91 (m, 2H), 6.35 (br s, 2H), 4.81 (br s, 3H), 3.81 (s, 3H), 2.73 (s, 3H); MS (ESI) m/z (%, fragment): 324 (100).
1-(3-fluorobenzyl)-3,7-dimethyl-1H-thieno [3’,2’:3,4]benzo [1,2-d][1,2,3]triazol-3-ium (29): obtained only in traces according to NMR analysis, not isolated and evaluated. Its synthesis should be optimized in the following investigations.
Two additional triazolium salts,
30 and
31, were also prepared from the previously obtained and characterized neutral thienobenzo-triazole [
20] for comparison of the non-charged and charged analogues.
![Biomolecules 14 01391 i031]()
1-(3-chlorobenzyl)-3-methyl-1H-thieno [3’,2’:3,4]benzo [1,2-d][1,2,3]triazol-3-ium (30): 10 mg (isolated 43%), yellow powder; Rf (DCM (100%)) = 0.20; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.39–8.31 (m, 2H), 8.13–8.07 (m, 2H), 8.02–7.96 (m, 2H), 7.86–7.82 (m, 2H), 6.47 (s, 2H), 4.87 (s, 3H); MS (ESI) m/z (%, fragment): 314 (100); HRMS (m/z) for C16H13ClN3S+: [M + H]+calcd = 314.0440, and [M + H]+measured = 314.0436.
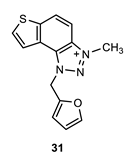
1-(furan-2-ylmethyl)-3-methyl-1H-thieno [3’,2’:3,4]benzo [1,2-d][1,2,3]triazol-3-ium (31): 6 mg (isolated 29%), yellow powder; Rf (DCM (100%)) = 0.17; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.34 (d, J = 9.0 Hz, 1H), 8.11 (d, J = 9.0 Hz, 1H), 7.99 (d, J = 5.8 Hz, 1H), 7.86 (d, J = 5.8 Hz, 1H), 7.50–7.47 (m, 2H), 7.13 (d, J = 3.8 Hz, 2H), 6.43 (s, 2H), 4.82 (s, 3H); MS (ESI) m/z (%, fragment): 270 (100); HRMS (m/z) for C14H12N3OS+: [M + H]+calcd = 270.0623, and [M + H]+measured = 270.0627.
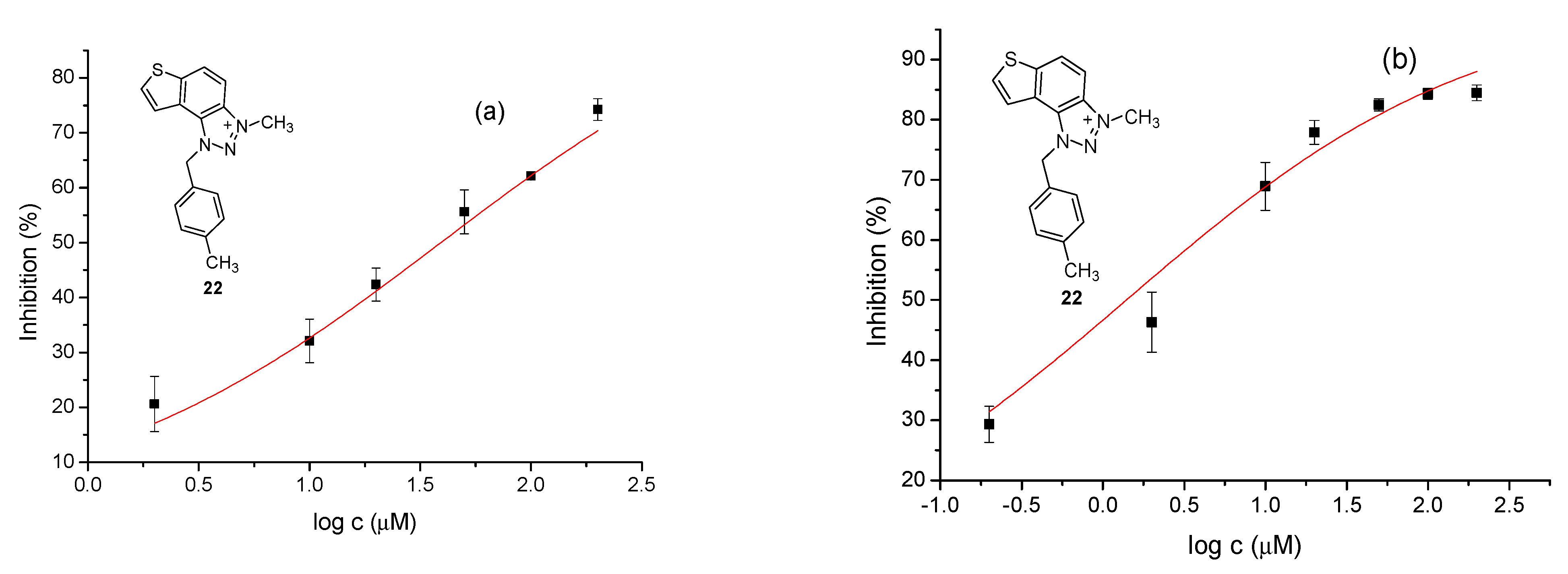
2.7. Inhibition Activity
The modified Ellman’s method was used to determine the synthesized triazole compounds’ cholinesterase inhibitory potential [
22]. Acetylcholinesterase from electric eel (eeAChE, type VI-S), butyrylcholinesterase from equine serum (eqBChE, E.C. 3.1.1.8), acetylthiocholine iodide (ATChI),
S-butyrylthiocholine iodide (BTChI), Trisma base, and reference standard galantamine were purchased from Sigma Aldrich (ST. Louis, MO). Ellman’s reagent 5,50-dithiobis-(2-nitrobenzoic acid) (DTNB) was purchased from Zwijndrecht (Belgium). All tested samples were dissolved in ethanol in concentrations ranging from 0.1 to 500 μM (final concentration in reaction mixture). Reaction mixtures were prepared as follows: 10 μL of tested solution of different concentrations, 10 μL of enzyme (AChE and BChE final concentrations 0.03 U/mL, prepared in 20 mM of Tris buffer, pH 7.5), and 10 μL of DTNB (final concentration 0.3 mM, prepared in 50 mM of Tris buffer, pH 8) were added to 180 μL of Tris buffer (50 mM, pH 8.0). The reaction was started by adding 10 μL of substrate ATChI/BTChI (final concentration 0.5 mM, prepared in Tris buffer, pH 8.0). The tested sample was replaced by 10 μL of buffer solution in the control measurement. A blank test for each sample contained the same solutions only without enzymes; its volume was replaced by adding 10 μL of buffer. The absorbance of the reaction mixture was measured at 405 nm over 6 min at room temperature using a 96-well microplate reader (Bio Tek 800TSUV Absorbance Reader, Agilent). Experiments were run in triplicate. The percentage of enzyme inhibition was calculated according to the equation: Inhibition (%) = [(A
C − A
T)/A
C] × 100 where A
C is the control enzyme activity, without the test sample, and A
T is the enzyme activity with the test sample, and represented as mean value ± standard deviation. Data of mean inhibition values were used for the IC
50 value calculation by nonlinear curve fitting of compound concentrations (log) vs. response. The inhibition contribution from ethanol was measured and subtracted from the calculation.
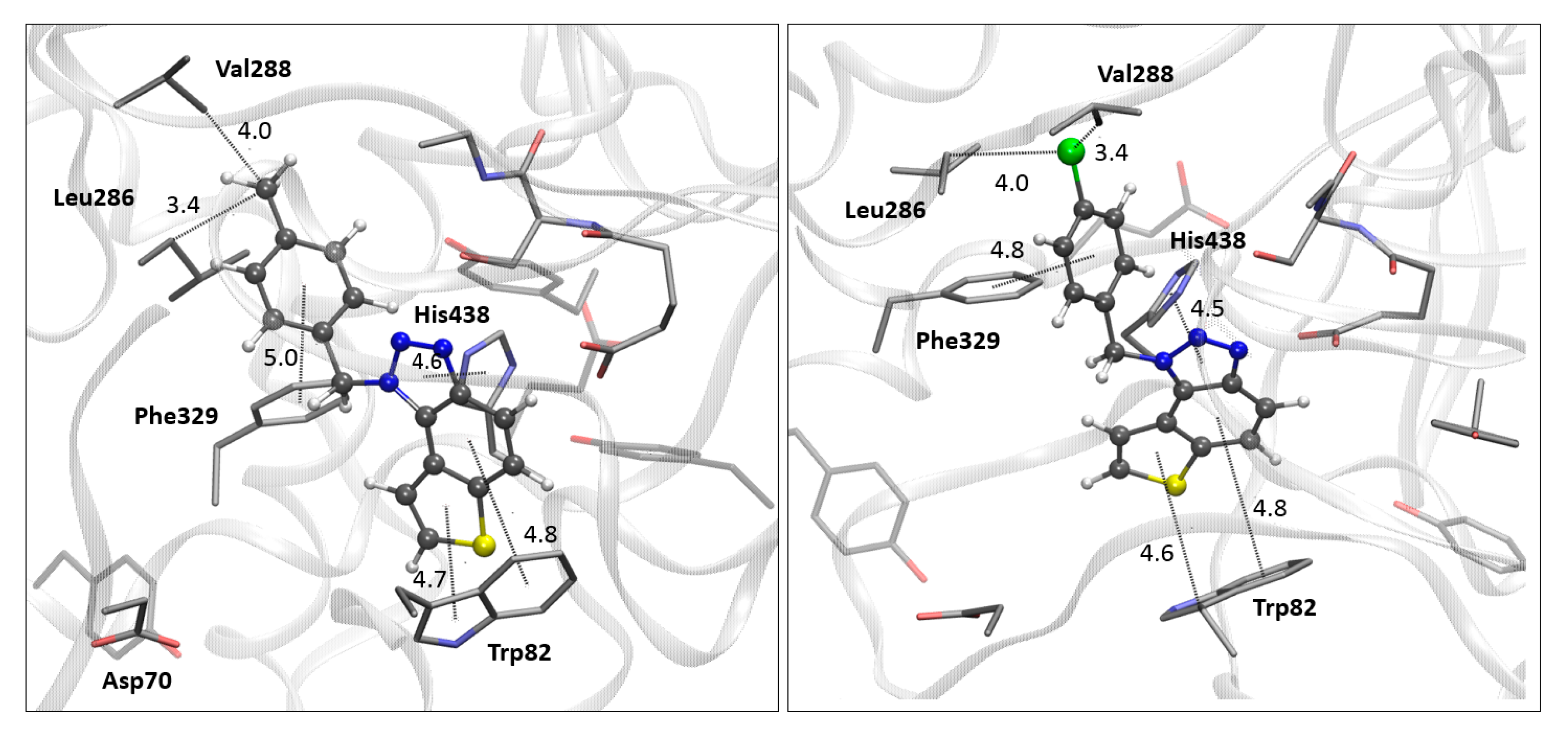
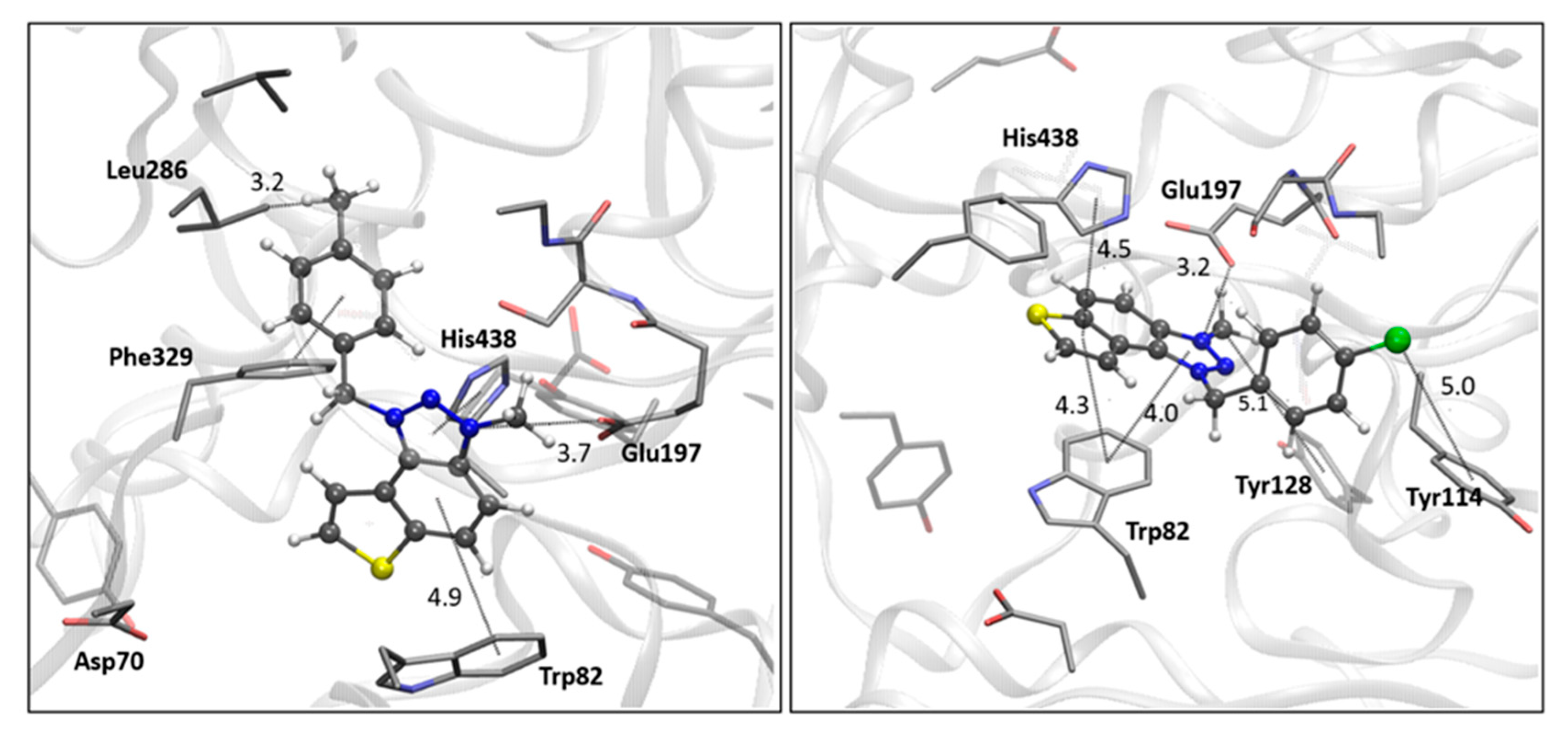
2.8. Computational Study
The structures of the ligands used for the docking study were obtained by geometry optimization at the B3LYP/6-31G(d) level of theory using the Gaussian16 program package [
23]. A vibrational analysis of the optimized geometries confirmed the absence of imaginary frequencies (NImag = 0), indicating that the structures correspond to minima on the potential energy surface. Optimized ligand structures were further prepared for docking by adding Gasteiger charges, detecting rotatable bonds, identifying aromatic carbons, and setting torsional degrees of freedom. Crystallographic data for AChE (PDB ID: 4EY7) and BChE (PDB ID: 7AIY) were obtained from the Protein Data Bank [
24,
25] and prepared for docking by removing non-amino acid residues, adding polar hydrogens, and assigning Kollman charges. Molecular docking was performed using Autodock4.0 [
26], employing the Lamarckian Genetic Algorithm, which generated 25 docking poses for each ligand. The enzyme residues were kept rigid throughout the process. The search space was defined by setting a GridBox around the center of the active site for each ChE (details given in
Supplementary Materials), with box dimensions of 60 × 60 × 60 points and a grid spacing of 0.375 Å.


 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}