Abstract
Heme is the reactive center of several metal-based proteins that are involved in multiple biological processes. However, free heme, defined as the labile heme pool, has toxic properties that are derived from its hydrophobic nature and the Fe-atom. Therefore, the heme concentration must be tightly controlled to maintain cellular homeostasis and to avoid pathological conditions. Therefore, different systems have been developed to scavenge either Hb (i.e., haptoglobin (Hp)) or the free heme (i.e., high-density lipoproteins (HDL), low-density lipoproteins (LDL), hemopexin (Hx), and human serum albumin (HSA)). In the first seconds after heme appearance in the plasma, more than 80% of the heme binds to HDL and LDL, and only the remaining 20% binds to Hx and HSA. Then, HSA slowly removes most of the heme from HDL and LDL, and finally, heme transits to Hx, which releases it into hepatic parenchymal cells. The Hx:heme or HSA:heme complexes are internalized via endocytosis mediated by the CD91 and CD71 receptors, respectively. As heme constitutes a major iron source for pathogens, bacteria have evolved hemophores that can extract and uptake heme from host proteins, including HSA:heme. Here, the molecular mechanisms underlying heme scavenging and delivery from HSA are reviewed. Moreover, the relevance of HSA in disease states associated with increased heme plasma concentrations are discussed.
Keywords:
catabolism; CD71 receptor; heme; heme export; heme import; heme scavenging; hemoglobin; hemopexin; hemophore; human serum albumin 1. The Double Face of the Heme
Heme is an iron-containing porphyrin that constitutes the prosthetic moiety of heme proteins [1]. Heme is synthesized through a series of reactions that take place in the mitochondrion and in the cytoplasm of all eukaryotic cells [2]. Daily, human erythroid cells synthesize 75% of the total body heme (~300 mg of heme/day), which is incorporated in hemoglobin (Hb), whereas hepatocytes produce ~50 mg of heme/day, which represents the central site of catalases, cytochrome P450, cytochrome B5, myoglobin (Mb), cytochrome c, and other mitochondrial cytochromes [1,2]. Heme is the catalytic center of hemoproteins exerting several crucial biological functions such as oxygen sensing, cell respiration and metabolism, growth, self-renewal, and differentiation. Indeed, hemoproteins include: (i) Hb and Mb that allow oxygen transport and storage; (ii) cytochromes, which are involved in cell respiration in mitochondria (i.e., cytochrome c, cytochrome c oxidase (COX), and cytochrome reductase); (iii) drug-metabolizing cytochromes P450; (iv) enzymes (e.g., catalases, peroxidases, guanyl cyclases, nitric oxide synthases, histidine kinases, cyclic nucleotide phosphodiesterases); and (v) heme-responsive transcription factors with basic helix–loop–helix (bHLH) DNA-binding domain motif. Of note, heme is also part of cyanocobalamin (also named vitamin B12) [1,3,4,5].
In contrast to the important heme-based biological functions of hemoproteins, the free heme, defined as labile heme pool, has toxic properties that originate from its hydrophobic nature and from the presence of the Fe-atom [2]. The labile heme pool is derived either from newly synthesized heme that has not yet been incorporated into hemoproteins or from heme that has been released from hemoproteins under oxidative conditions. The labile heme pool acts as an “alarmin” [6] as it is sensed by pattern recognition receptors such as the Toll like receptor [7], and NACHT, LRR, and PYD domains-containing protein 3 (NALP3) [8]. The labile heme pool may increase after extracellular heme overload, increased heme synthesis, accelerated hemoprotein breakdown, impaired incorporation into apo-proteins, and impaired heme-oxygenase (HO) activity [1,9,10].
Free heme is an abundant source of ferrous iron (Fe(II)) that can participate in the Fenton reaction, a process during which reactive oxygen species (ROS) are produced. Free heme is extremely toxic for cells. Indeed, heme (i) intercalates biologic membranes altering lipid bilayers; (ii) is strongly pro-inflammatory, inducing the recruitment of leukocytes, platelets, and red blood cells (RBCs) to the vascular endothelium; (iii) oxidizes low-density lipoproteins (LDLs); and (iv) inactivates nitric oxide, thus impairing vascular functions [1]. Furthermore, ROS produced by the heme-driven Fenton reaction can (i) damage lipid membranes, proteins, and nucleic acids; (ii) activate cell signaling pathways and oxidant-sensitive pro-inflammatory transcription factors; (iii) alter protein expression; and (iv) perturb membrane channels [1,2,5]. All these events contribute to promoting cell death [2,5].
Here, the molecular mechanisms underlying the role of human serum albumin (HSA) in heme scavenging and delivery are discussed, highlighting the competition between mammalian cells and pathogens in heme up-taking.
2. Regulation of Heme Levels
Hb is the main blood hemoprotein responsible for O2 delivery into the circulatory system, also playing a key role in ROS and reactive nitrogen species (RNS) detoxification [11,12,13]. Although Hb is normally confined to RBCs, low levels of extra-erythrocytic Hb and free heme in the plasma may be due to physiological phenomena associated with intravascular hemolysis, which occur during the destruction of senescent erythrocytes and the enucleation of erythroblasts [11,14,15]. Because of the potential extracellular toxicity of free heme, its concentration is tightly controlled to maintain cellular homeostasis and to avoid pathological conditions. To this purpose, mammals have developed different systems able to scavenge either Hb (i.e., haptoglobin (Hp)) or free heme (i.e., high-density lipoproteins (HDL), low-density lipoproteins (LDL), hemopexin (Hx), and human serum albumin (HSA)) [1,2,16,17,18,19] (Figure 1). During the physiological turnover of RBCs, the small fraction of free extracellular Hb released into the plasma (~10 %) [20] is captured by Hp and transported to reticulo-endothelial macrophages located in the liver and in the spleen, which represent the main sites for the clearance of aged and damaged RBCs. Then, the Hp:Hb complex is captured by the Hp scavenger receptor (i.e., CD163) and is internalized [1,17] (Figure 1). Following RBCs phagocytosis or Hp-mediated Hb internalization, Hb is degraded and the heme is either recycled for de novo erythropoiesis or catabolized.
2.1. Heme Scavenging in Plasma
Heme scavenging by HDL, LDL, HSA, and Hx provides protection against free heme oxidative damage, limits access by pathogens to heme, and contributes to iron homeostasis by recycling the heme iron. In the first seconds after heme appearance, more than 80% of this macrocycle binds to HDL and LDL, and only the remaining 20% binds to Hx and HSA. In particular, HSA slowly removes most of the heme from HDL and LDL and transfers it to Hx, which finally releases the macrocycle into hepatic parenchymal cells via endocytosis mediated by the CD91 receptor [1,2,11,16,17,18,21] (Figure 1). Of note, free heme binds to HSA (Kd ~ 1.0 × 10−9 M) with a lower affinity compared with Hx (Kd < 10−9 M) [11].
Although HDL and LDL are the most oxidatively intolerant plasma components, they bind heme with a high affinity (Kd ranging between 10−11 M and 10−10 M) and at a faster rate than HSA and Hx. Of note, the kinetics of heme release from HDL and LDL is faster than the kinetics of heme-induced lipoprotein oxidation [18,22,23].
RBCs clearance and extracellular Hb scavenging are relatively modest events under steady-state conditions, but are drastically enhanced in hemolytic disorders (e.g., anemia, vasculopathy, endothelial dysfunction, and infections), when high levels of extracellular Hb and heme ultimately lead to the saturation and depletion of Hp and Hx scavenging systems [24], causing heme-mediated oxidative damage to tissues [1,2,25]. When the buffering capacity of plasma Hp is exceeded, hypoxia induces the quick oxidation of Hb to methemoglobin (metHb) [26]. Cell-free Hb that becomes oxidized or denatured prior to clearance is prone to release free heme [11,15,27]. As the Hx plasma concentration (~1.5 × 10−5 M) is about two orders of magnitude lower than that of HSA (~7.5 × 10−4 M) [11], the release of a massive quantity of heme reduces the bioavailability of Hx, and consequently HSA acts as the main heme scavenger [28,29]. Indeed, in patients with hemolytic disorders, the plasmatic level of the HSA:heme complex increases from ~1.0 × 10−6 M in physiological conditions, to ~4.0 × 10−5 M [23,24,30].

Figure 1.
Overview of heme scavenging in blood vessels. The intravascular hemolysis of red blood cells (RBCs) induces the release of hemoglobin (Hb), which is scavenged by high-density lipoproteins (HDLs), low-density lipoproteins (LDLs), human serum albumin (HSA), and hemopexin (Hx). First, the free heme binds to HDL and LDL, then it moves to HSA, and lastly it is transferred to Hx. The HSA:heme complex [31] is internalized by the CD71 receptor, whereas the Hx:heme complex [32] binds to the CD91 receptor and is then moved into macrophages. The figure has been partially generated through the website Servier Medical Art licensed under a Creative Commons Attribution 3.0 imported license. The three-dimensional structures of Hb (PDB ID: 1JY7) [33], Hp:Hb (PDB ID: 4F4O) [34,35], HSA:heme (PDB ID: 1N5U) [31], and Hx:heme (PDB ID: 1QJS) [32] complexes have been drawn using UCSF-Chimera [36].
Figure 1.
Overview of heme scavenging in blood vessels. The intravascular hemolysis of red blood cells (RBCs) induces the release of hemoglobin (Hb), which is scavenged by high-density lipoproteins (HDLs), low-density lipoproteins (LDLs), human serum albumin (HSA), and hemopexin (Hx). First, the free heme binds to HDL and LDL, then it moves to HSA, and lastly it is transferred to Hx. The HSA:heme complex [31] is internalized by the CD71 receptor, whereas the Hx:heme complex [32] binds to the CD91 receptor and is then moved into macrophages. The figure has been partially generated through the website Servier Medical Art licensed under a Creative Commons Attribution 3.0 imported license. The three-dimensional structures of Hb (PDB ID: 1JY7) [33], Hp:Hb (PDB ID: 4F4O) [34,35], HSA:heme (PDB ID: 1N5U) [31], and Hx:heme (PDB ID: 1QJS) [32] complexes have been drawn using UCSF-Chimera [36].

2.2. Regulation of the Intracellular and Extracellular Heme Levels
The regulation of the extracellular and intracellular heme levels is crucial for preventing pathological iron or heme accumulation. Heme synthesis and degradation are inhibited and induced, respectively, by heme itself. Heme production is regulated differently in erythroid and non-erythroid cells. In non-erythroid cells, heme reduces its own production by (i) decreasing the activity of ALA Synthase 1 (ALAS1), the rate-limiting enzyme in heme biosynthesis, and (ii) increasing its catabolism [1]. On the contrary, in erythroid cells, heme functions as a positive feedback regulator, promoting its synthesis and inhibiting its breakdown. Importantly, the amount of newly synthesized heme must match the rate at which it is incorporated into apo-hemoproteins. This balance is achieved through the regulation of both heme and apo-hemoprotein synthesis. Indeed, heme can induce the expression of various apo-hemoproteins, such as Hb, myoglobin, neuroglobin, cytochromes. This evolutionary mechanism helps preventing the toxic accumulation of intracellular heme and ensures that the amount of heme synthesized is properly balanced with the amount incorporated into hemoproteins or eventually catabolized [1].
The physiological degradation of heme occurs in a tightly controlled manner through the activity of HO, which cleaves heme in the presence of NADPH + H+ and O2, resulting in the production of carbon monoxide (CO), Fe(II), and biliverdin IX. Fe(II) is either bound to ferritin, which represents the main intracellular iron storage protein, or exported into the bloodstream by ferroportin. Biliverdin IX is degraded by a NADPH-dependent reductase into bilirubin IX, which is finally excreted in the bile and urine [17,19,25,37,38,39]. Biliverdin, bilirubin, HO-1, and CO all display important antioxidant properties [40]. The major site of heme breakdown is the liver, although spleen, brain, and erythropoietic system are also important heme catabolic organs [1,21,39]. When not catabolized, intracellular heme must be either incorporated into apo-proteins or expelled out from cells. The most characterized heme exporters are the Feline Leukemia Virus Subgroup C Receptor 1a (FLVCR1a) [41] and the ATP-binding cassette sub-family G 2 (ABCG2) [42].
On the contrary, when heme is requested for incorporation into apo-proteins, it can be either newly synthesized or imported from the extracellular space. Heme is ubiquitously synthesized through eight enzymatic reactions that take place between the mitochondria and the cytoplasm. The rate-limiting step of this process is represented by the first reaction that is catalyzed by the ALAS1 enzyme, which condensates glycine with succinyl CoA to form aminolevulinic acid (ALA) [1,40].
To date, the only known proteins with a well-established function as heme importers are (i) the Heme-Responsive Gene 1 (HRG1) [43,44] and the FLVSCR 2 (FLVCR2), which are expressed ubiquitously [45], and (ii) the Heme Carrier Protein 1/Proton-Coupled Folate Transporter (HCP1/PCFT), which is expressed in the duodenum, liver, kidney, spleen, and placenta, and to a minor extent in the colon, rectum, ileum, jejunum, cecum, and testis [40,46].
3. Heme Scavenging from HSA
HSA is synthesized in the hepatocytes at the rate of ~0.7 mg/hour/gram of liver (i.e., at ~10–15 g/day) and is then released into the plasma where its concentration ranges from 5.3 × 10−4 M to 1.0 × 10−3 M. The rate of HSA synthesis depends mainly on (i) the blood oncotic pressure, as the HSA concentration is detected by osmoreceptors in the hepatic interstitium, (ii) hormonal stimuli, (iii) nutrition, and (iv) inflammation [47,48,49].
HSA is a monomeric globular protein of 585 amino acids (molecular weight of ~66 kDa) composed of 67% α-helix without β-sheet, and organized in three domains (i.e., I, II, and III) encompassing amino acids 1–195, 196–383, and 384–585, respectively. Each domain includes 10 helices organized in subdomains A and B that are built from six and four α-helices, respectively, connected by a long loop [16,30,50,51]. HSA binds up to nine equivalents of fatty acids (FAs), its primary physiological ligands, at sites FA1 to FA9 [30,52]. HSA also binds heme, metal ions, hormones, and nucleic acids. it affects pharmacokinetics of many drugs, renders potential toxins harmless, accounts for most of the antioxidant capacity of human plasma, and displays (pseudo-)enzymatic activities [16,52,53,54,55].
Heme binding to HSA is a simple process, with K(heme) = 1.3 × 10−8 M, kon (heme) = 7.4 × 105 M−1 s−1, and koff (heme) = 9.6 × 10−3 s−1 [56]. Heme binds HSA at the center of subdomain IB (i.e., at the FA1 site). First, heme binds reversibly to His146 at the surface of HSA to generate an intermediate complex; then, the macrocycle binds to the Tyr161 residue placed within subdomain IB [57]. Interestingly, the HSA:heme complex displays globin-like catalytic properties, including peroxynitrite scavenging functions as well as catalase and peroxidase activities [16,58,59].
Heme binding to HSA is modulated by drug binding to the FA2 and FA7 sites, and vice versa, through competitive and allosteric mechanisms. Indeed, drugs binding to the FA1 site (e.g., rifampicin, isoniazid, and fipronil) directly impairs heme recognition. Moreover, warfarin binding to the FA7 site (i.e., Sudlow’s site I) decreases by about one order of magnitude the values of K(heme) and kon (heme) for heme binding to HSA at the FA1 site. In turn, heme binding to HSA inhibits drug binding to the FA2 and FA7 sites [60,61,62]. Upon drug binding to either the FA2 or the FA7 sites, the reorientation of the Glu131-Arg145 α-helix occurs causing a shift in the Phe149-Tyr150 dyad and the Arg257 residue next to the FA1 cavity. This reduces heme affinity, induces the hexa-coordination of the metal center by the His146 residue, and inhibits heme-based reactivity [30,62,63] (Figure 2). Of note, drugs binding to HSA:heme inhibits the heme-based detoxification of ROS and RNS [16,56,64,65,66,67,68]. As heme scavenging from HSA is a transient process, the heme-based catalytic properties of HSA represent a case of “chronosteric effects” [66].
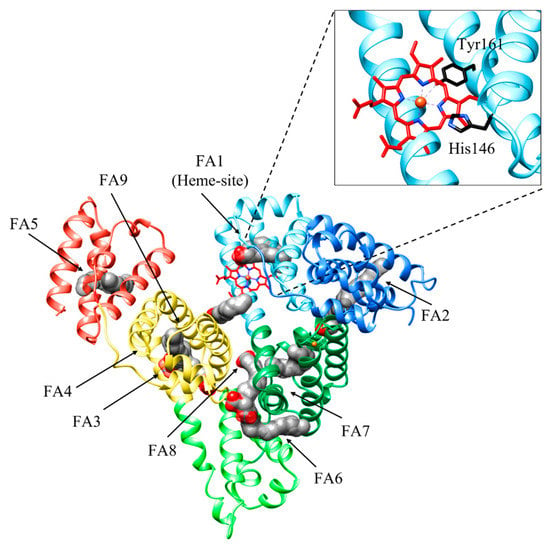
Figure 2.
Three-dimensional structure of HSA bound to fatty acids (FAs) and heme. HSA is organized in three domains: domain IA (light blue), domain IB (sky blue), and domain IA (forest green); domain IIA (light green); and domain IIIA (yellow) and domain IIIB (coral). HSA binds up to nine equivalents of FAs, its primary physiological ligands, at sites FA1 to FA9. FAs are rendered as space-fill (gray), whereas the heme is rendered as sticks (red). His141 and Tyr146 residues coordinating the heme-Fe atom are shown in black. The subdomains of HSA are rendered with different colors. The picture has been drawn using the UCSF Chimera package [36].
The modulation of heme binding to HSA by drugs may be relevant in pharmacotherapy management. Indeed, an increase in heme levels under pathological conditions (e.g., severe hemolytic anemia, crash syndrome, and post-ischemic reperfusion) increases the drug plasma concentration and induces the release of HSA-bound drugs with the consequent patient intoxication [16,30,62,66,69,70,71,72,73,74,75,76,77]. In turn, high levels of heme-albumin due to hemolytic events or pathological states characterized by low HSA levels (i.e., hypoalbuminemia, [HSA] < 5.3 × 10−4 M) may cause a lower availability of circulating albumin. This determines an increase in the unbound fraction of drugs, resulting in a lower efficacy [72]. However, this does not necessary result in potential adverse effects, because many drugs can bind not only HSA, but also lipocalins (e.g., α-1-acid glycoprotein (AGP) and retinol-binding protein 4 (RPB4)) [30,72].
4. HSA: Heme Complex Internalization
Upon secretion from hepatocytes, HSA enters the circulation and translocates to the extracellular space through the pores of sinusoidal or fenestrated endothelium cells of the liver, pancreas, small intestine, and bone marrow [78,79]. HSA can cross the endothelium via active transcytotic mechanisms, including receptor-mediated processes [80]. These receptors can selectively recognize the native or conformationally modified HSA (e.g., gold-labeled HSA, formaldehyde- or maleic anhydride-treated HSA [80,81]). To date, eight membrane-associated HSA binding proteins have been described: albondin/glycoprotein 60 (gp60) [82], glycoprotein 18 (gp18) [83], glycoprotein 30 (gp30) [83], neonatal Fc Receptor (FcRn) [84], heterogeneous nuclear RiboNucleoProteins (hnRNPs) [85], calreticulin [85], cubilin [86,87], megalin [86,87], and Secreted Protein Acidic and Rich in Cysteine (SPARC) [88].
To date, very little is known regarding the HSA:heme internalization mechanisms. Recently, it has been suggested that CD71 (also known as Transferrin Receptor 1, TfR1) acts as a specific cellular receptor for the HSA:heme complex [89]. CD71 is ubiquitously expressed, is bound to two Fe(III) atoms (Tf:Fe(III)2), and internalizes transferrin (Tf), [90,91]. CD71 is a homodimeric type II transmembrane protein composed of a small cytoplasmic domain, a single-pass transmembrane region, and a complex extracellular domain. Each monomer of the ectodomain is composed of (i) a protease-like domain that is in contact with the cell membrane, (ii) a helical domain that comprises the homodimer interface, and (iii) an apical domain. The ectodomain binds several proteins. Indeed, the CD71 basal portion, composed of the protease-like and the helical domains, recognizes Tf; the dimer interface region forms a complex with the hereditary hemochromatosis factor (HFE); the upper part of the apical domain interacts with ferritin as well as with Arenaviruses and with the Plasmodium vivax invasion protein PvRBP2b1, which exploit CD71 for cell invasion [92].
HSA:heme binding to CD71 allows for complex internalization and represents an alternative source of iron to Tf:Fe(III)2. This implies that HSA plays a role in providing iron to cells, which is fundamental to sustain vital processes such as cell metabolism and proliferation [89]. Both the HSA:heme complex and Tf-Fe(III)2 recognize the basal portion of the CD71 ectodomain. The Kd value for HSA:heme binding to CD71 is lower than that of Tf:Fe(III)2 at physiological pH, depending on the species and on the tissues (Kd(HSA:heme) = 7.5 × 10−7 M; Kd(Tf-Fe(III)2) ~ 10−8 M) [89]. The CD71/HSA:heme recognition mechanism appears to be species specific; indeed human CD71 is unable to recognize the bovine serum albumin:heme complex [89]. Upon internalization, the HSA:heme complex can be used as a Fe(III) source by primary human T cells, as well as by immortalized cell lines [89]. Once HSA:heme is internalized, the isoform 1 of HO (i.e., HO-1) is pivotal to utilize heme as a Fe source. Indeed, while supplementation of serum-free medium with HSA:heme supports the growth of lymphoblastoid cells expressing wild-type HO-1, the supplementation is ineffective in HO-1 deficient lymphoblastoid cells. Furthermore, the proliferation of primary human T cells in the presence of HSA:heme is inhibited by the Tin protoporphyrin HO-1 inhibitor [89].
It is noteworthy that the HSA:heme complex shows peroxidase activity, which is a well-known antimicrobial mechanism of the human innate immune response [93,94]. As some viruses causing hemorrhagic fever (e.g., Arenavirus, Machupo virus) use the CD71 receptor to enter human cells, high levels of HSA:heme may exert a protective function towards CD71-mediated virus entry [89].
5. Functional Aspects of the HSA-Dependent Heme Internalization
The differentiation of macrophages of the reticuloendothelial system is modulated by HSA:heme complex internalization. This is pivotal for the modulation of the inflammatory response in patients showing either acute or chronic heme release in the plasma [95]. Heme, and specifically its iron moiety, promotes pronounced changes in macrophages towards an M1-like proinflammatory phenotype. The classically activated M1 macrophages, induced by microbial agents and proinflammatory T-helper (Th) 1 cytokines, can exert inflammatory functions and bactericidal activity, and can induce high levels of proinflammatory cytokines, ROS, and RNS. On the contrary, the alternatively activated M2 macrophages, generally induced by Th2 cytokines, exert immunoregulatory functions, support pathogen clearance, and are involved in cell growth control, matrix remodeling, angiogenesis, and tissue repair [95].
Notably, Hx reduces heme accumulation in macrophages, thus preventing heme-induced proinflammatory phenotypic switching from M2 to M1, both in vitro and in vivo [95,96]. The administration of exogenous Hx in an experimental model of hemolysis and in a mouse model of Sickle cell disease is beneficial in counteracting the heme-driven proinflammatory status of macrophages [97]. On the contrary, heme levels are significantly increased in bone marrow-derived macrophages treated with HSA:heme compared with those treated with Hx:heme. Furthermore, monocyte/macrophage-like cells treated with HSA:heme accumulate higher levels of heme, show increased levels of H- and L-ferritin chains, ferroportin, and HO, and display enhanced HO-1 activity compared with Hx:heme treated cells. In addition, ROS production and IL-6 and TNFα expression increased in cells treated with HSA:heme rather than those supplemented with Hx:heme. These findings indicate that HSA allows for a delivery rate of heme into macrophages that is significantly higher than that of Hx, thus playing a key role in driving the transition from M2 to M1 macrophages [95].
6. HSA, Heme, and COVID-19
The SARS-CoV-2 virus binds to Hb and causes heme release, resulting in impaired O2 supply and ROS generation. In turn, this causes increased oxidative stress, hypoxia, and potential cardiac injury (e.g., heart attack and cardiac arrest) [98,99,100]. Recently, it has been reported that ORF1Ab, ORF3a, and ORF10 SARS-CoV-2 viral proteins can coordinately uptake the heme localized in the β chains of Hb. Both oxygenated and deoxygenated Hb can be attacked, but the latter is more sensitive to the virus [100]. Indeed, COVID-19 patients showed increased heme plasma levels [101], comparable to those reported under hemolytic conditions (~2.0 × 10−5 M) [102]. To properly buffer high levels of the labile heme pool and to counteract the consequent inflammatory response, the production of heme-scavenging proteins such as Hx and HSA is increased [98,103]. Accordingly, hypoalbuminemia is significantly correlated with COVID-19 progression and severity as a predictive index of the disease outcome, independently from patient age and morbidity [98,104,105,106,107,108].
7. Role of HSA in Heme Uptake by Bacteria and Fungi
Heme also constitutes a major iron source for pathogens such as bacteria and fungi. To overcome the scarcity of free iron in the animal host, pathogens have developed systems to extract and uptake heme from host proteins. The strategies of iron acquisition are (i) the activation of the secondary metabolism for the production of small iron-chelating compounds, called siderophores, and (ii) specialized secreted and/or membrane-bound hemophores that are able to acquire both free heme and heme bound to Hb, Hp:Hb, Hx:heme, HSA:heme, and Mb [29,66,109,110,111] in order to deliver heme to a specific outer membrane receptor expressed on the pathogen membrane (see [66], and the references therein). Both siderophores and hemophores contribute to the virulence of many pathogens such as Bordetella, Haemophilus, Brucella, Vibrio, Streptococcus, and Staphylococcus, and many other Gram-positive and Gram-negative species. Interestingly, hosts characterized by high levels of free heme are generally more susceptible to infection. This implies that hemophores preferentially uses the labile free heme compared with the bound fraction available in the host [112].
HSA promotes heme utilization in fungi belonging to the Candida species. This HSA stimulatory activity could reflect the solubilization of free heme that otherwise would aggregate in the solution, with a consequent reduction in its effective concentration [111]. HSA-induced heme utilization was reported for albumin concentrations as low as 5.0 × 10−6 M, which means more than two orders of magnitude lower than the HSA concentration in human plasma. This implies that HSA tissue concentrations (ranging from 6.5 × 10−2 M to 2 × 10−1 M) could be sufficient to provide heme-iron acquisition for fungi that have penetrated tissues [111,113]. As the affinity of HSA for heme might be in the same range as that of bacterial surface receptors for heme, it is likely that heme bound to HSA is recognized by heme/Hb receptors, and then it is passively transferred to them from HSA [111].
8. Clinical Use of HSA in Hemolytic Diseases
From a clinical perspective, the hypoalbuminemic condition is correlated with an increased risk of mortality in several diseases in which hemolytic events occur (e.g., malaria, general systemic inflammation, sepsis, cirrhosis, splenomegaly, portal hypertension, lupus erythematosus, and infectious diseases) [98,104,105,106,107,108,114,115]. A significant association between increased hemolytic markers and both albuminuria and glomerular hyperfiltration has been reported in patients with severe forms of sickle cell anemia and thalassemia [116]. Overall, labile heme exerts pro-inflammatory, vasoactive, and cytotoxic effects that can contribute to the pathogenesis of hemolytic conditions [117,118,119,120,121,122,123,124].
Hepatic dysfunction occurs frequently during sepsis, whose pathogenesis is driven by an inadequate response of the host to infection, leading to dysregulation of iron metabolism and the buildup of labile heme. Of note, heme and Hb scavenging reduces disease severity. The observation that the administration of 4% albumin reduces oxidative stress, mortality, and endothelial and kidney dysfunctions in mice subjected to endotoxemia, which is induced by labile heme, supports the protective role towards sepsis of HSA [125,126], by reducing heme-mediated in vitro cytotoxicity and in vivo heme-mediated vasoconstriction [115].
In a prospective cohort study of 116 septic patients, it was found that those with low concentrations of HSA had a poorer outcome. Furthermore, a subgroup analysis of patients with severe sepsis enrolled in the ALBIOS trial showed that administering HSA might improve survival [127]. Moreover, the results of the study validate the beneficial effects of administering HSA during severe sepsis, which includes increasing the distribution of fluids within the intravascular compartment. Indeed, HSA may act as a scavenger of nitric oxide, leading to peripheral vasodilatation during sepsis [127]. A recent retrospective study conducted on 2829 patients hospitalized between January 2013 and April 2018 with a diagnosis of sepsis/septic shock showed that the use of HSA within 24 h of hospital admission was associated with a shorter time to discharge and a higher rate of discharge with clinical stability, suggesting an improvement in healthcare resource utilization among patients [128].
Malaria is a severe disease caused by parasites of the Plasmodium genus. As part of its life cycle, the parasite consumes Hb from RBCs, leading to the release of heme. At least one third of malaria patients are hypoalbuminemic. Moreover, an association between low serum albumin levels and both a longer parasitemia time and a higher incidence of cerebral malaria has been found [129]. Interestingly, a randomized trial comparing HSA and saline in children with malaria demonstreated that the mortality rate was significantly lower among patients who received albumin than among those who received saline (3.6% vs 18%; 95% CI 1.2–24.8; p = 0.013) [130]. Similarly, a controlled trial in malaria children demonstrated that those who received HSA underwent mortality in the coma with a significantly lower incidence (1/25; 4%) compared with patients who received the colloid Gelofusine (6/23; 26%); however, the underpowered sample size did not allow for solid inferences [131]. As heme toxicity contributes to cerebral malaria pathogenesis, a specific neuroprotective effect of albumin can be hypothesized [131]. However, no further evidence based on clinical trials is still available.
HSA is also of primary importance in the binding and detoxification of bilirubin, the end product of heme catabolism, whose concentration increases during hemolytic events [132]. Unbound bilirubin can cross the blood−brain barrier and cause neurotoxicity [133,134]. The administration of HSA has been proven to be very beneficial in hyperbilirubinemia, which is a common condition in the neonatal period. Indeed, hyperbilirubinemic infants treated with HSA showed reduced levels of circulating unbound bilirubin, thus decreasing the incidence of complications such as fever, allergic reactions, and encephalopathies [135]. Of note, medications that interfere with HSA:bilirubin binding or that inhibit the p-glycoprotein, increase the risk of acute bilirubin encephalopathy [136].
HSA could also be used as a therapeutic adjuvant in major post-operatory complications such as kidney’s ischemia reperfusion injuries, in which high levels of free heme in the kidney are correlated with inflammation after organ transplants [137,138]. In a study involving a mouse model of kidney ischemia, it has been observed that HSA was able to reduce the release of pro-inflammatory cytokines and the expression levels of complement receptors in the renal tissue [138]. In this regard, future studies will be required to develop clinically applicable therapies to reduce the effects of free heme in ischemic organs, which, in turn, may result in more favorable post-transplant outcomes.
9. Conclusions and Perspectives
HSA binds free heme with a high affinity, contributing to its scavenging and to the maintenance of cellular homeostasis, and avoiding free heme-related toxicity. Heme scavenging by HSA could also modulate the bioavailability of this macrocycle to pathogens as an iron source. Therefore, HSA plays a key role in regulating heme metabolism, influencing both eukaryotic and prokaryotic cell growth (Figure 3). However, the overall ability of HSA to facilitate heme-Fe utilization by pathogens needs to be further clarified.
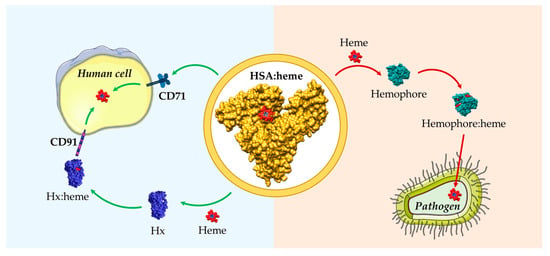
Figure 3.
Schematic representation of heme scavenging and delivery by HSA in both human cells and pathogens. HSA can either act as a heme scavenger in human blood reducing the free heme labile concentrations or as a heme donor to pathogens. HSA:heme (PDB ID: 1N5U) [31], Hx:heme (PDB ID: 1QJS) [32], and the C. albicans Csa2 hemophore in complex with heme (Csa2:heme, PDB ID: 4Y7S) [110] have been drawn with UCSF-Chimera [36]. The figure has been partially generated using the website Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
The high plasma concentration of HSA (~10−4 M) and the high endogenous and exogenous ligands affinity for HSA (Kd < 10−4 M) implies that the fraction of free ligand(s) in the plasma is negligible compared with that bound to HSA. This implies that ligand internalization may occur only by an HSA-dependent mechanism, either through the uptake of the HSA:ligand complex to cell receptors/channels or by ligand transfer from HSA to cell surface proteins. In the last case, a transient trimeric complex built by the HSA:ligand:receptor should occur.
The transfusion of donor blood has become a common and routine practice. However, the requirement for an enhanced level of safety has a significant cost, and blood transmitted infection remains a challenging problem. Additionally, donor blood transfusions necessitate crossmatching and compatibility testing to prevent a hemolytic reaction in the recipient, and the purified RBCs must be stored at 4 °C. Interestingly, the capability of HSA to bind heme at the FA1 site renders HSA:heme functionally similar to other O2-transporter hemoproteins such as Hb and Mb. However HSA:heme lacks the proximal histidine residue necessary for the formation of the fifth coordination bond of the heme-Fe atom, which in Hb and Mb allows for the prosthetic group to reversibly bind O2. Physiological responses to exchange transfusion in acute anemia using recombinant HSA:heme revealed that this synthetic RBC substitute can resuscitate hemorrhagic shock, suggesting its promising future use as a new class of RBC substitute. Engineered HSA:heme may be a viable alternative in hemo transfusions, without the risks deriving from the transmission of pathogenic infections and incompatibilities between blood groups. In the future, further studies are required to explore this intriguing possibility.
Overall, the multifunctional properties of HSA are causing its role to be redefined beyond that of a mere plasma expander. The increasing knowledges on HSA ligand binding properties and protective roles will probably widen the therapeutic indications for this protein. In perspective, therapeutic approaches targeting heme removal via HSA will lead to interesting new concepts for the treatment of medical conditions, with a particular focus on hemolytic diseases [115,118].
Author Contributions
Conceptualization, A.d.M., G.D.S., P.A.; writing—original draft preparation, A.d.M., G.D.S., P.A., R.V., T.F.R.; writing—review and editing, A.d.M., G.D.S., R.V., T.F.R.; supervision, A.d.M., P.A.; project administration, A.d.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Grant Departments of Excellence-2017-legge 232/2016-art.1, commi 314-337 awarded to the Department of Science of Roma Tre University (Rome, Italy) for 2018-2022.
Acknowledgments
We apologize to many authors of the exceptional papers not directly cited due to space limitations. We thank Stefano Di Bella for helpful discussion on the clinical use of HSA in hemolytic diseases.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Tolosano, E.; Fagoonee, S.; Morello, N.; Vinchi, F.; Fiorito, V. Heme scavenging and the other facets of hemopexin. Antioxid. Redox Signal. 2010, 12, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Numazawa, S.; Yoshida, T. Redox regulation of the transcriptional repressor Bach1. Free Radic. Biol. Med. 2005, 38, 1344–1352. [Google Scholar] [CrossRef]
- Kitamuro, T.; Takahashi, K.; Ogawa, K.; Udono-Fujimori, R.; Takeda, K.; Furuyama, K.; Nakayama, M.; Sun, J.; Fujita, H.; Hida, W. Bach1 functions as a hypoxia-inducible repressor for the heme oxygenase-1 gene in human cells. J. Biol. Chem. 2003, 278, 9125–9133. [Google Scholar] [CrossRef]
- Kumar, S.; Bandyopadhyay, U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 2005, 157, 175–188. [Google Scholar] [CrossRef]
- Soares, M.P.; Bozza, M.T. Red alert: Labile heme is an alarmin. Curr. Opin. Immunol. 2016, 38, 94–100. [Google Scholar] [CrossRef]
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliveira, M.F.; Oliveira, P.L.; Graça-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of Toll-like receptor 4. J. Biol. Chem. 2007, 282, 20221–20229. [Google Scholar] [CrossRef] [PubMed]
- Dutra, F.F.; Alves, L.S.; Rodrigues, D.; Fernandez, P.L.; de Oliveira, R.B.; Golenbock, D.T.; Zamboni, D.S.; Bozza, M.T. Hemolysis-induced lethality involves inflammasome activation by heme. Proc. Natl. Acad. Sci. USA. 2014, 111, E4110–E4118. [Google Scholar] [CrossRef]
- Khan, A.A.; Quigley, J.G. Control of intracellular heme levels: Heme transporters and heme oxygenases. Biochim. Biophys. Acta 2011, 1813, 668–682. [Google Scholar] [CrossRef]
- Swenson, S.A.; Moore, C.M.; Marcero, J.R.; Medlock, A.E.; Reddi, A.R.; Khalimonchuk, O. From synthesis to utilization: The ins and outs of mitochondrial heme. Cells 2020, 9, 579. [Google Scholar] [CrossRef]
- Ascenzi, P.; Bocedi, A.; Visca, P.; Altruda, F.; Tolosano, E.; Beringhelli, T.; Fasano, M. Hemoglobin and heme scavenging. IUBMB Life 2005, 57, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Brunori, M. Hemoglobin is an honorary enzyme. Trends Biochem. Sci 1999, 24, 158–161. [Google Scholar] [CrossRef] [PubMed]
- Gow, A.J.; Luchsinger, B.P.; Pawloski, J.R.; Singel, D.J.; Stamler, J.S. The oxyhemoglobin reaction of nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 9027–9032. [Google Scholar] [CrossRef]
- Bunn, H.F.; Forget, B.G. Hemoglobin: Molecular, Genetic, and Clinical Aspects; WB Saunders Co.: Philadelphia, PA, USA, 1986. [Google Scholar]
- Fibach, E. The redox balance and membrane shedding in RBC production, maturation, and senescence. Front. Physiol. 2021, 12, 604738. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; di Masi, A.; Ascenzi, P. Serum albumin: A multifaced enzyme. Int. J. Mol. Sci. 2021, 22, 10086. [Google Scholar] [CrossRef] [PubMed]
- Di Masi, A.; De Simone, G.; Ciaccio, C.; D’Orso, S.; Coletta, M.; Ascenzi, P. Haptoglobin: From hemoglobin scavenging to human health. Mol. Aspects Med. 2020, 73, 100851. [Google Scholar] [CrossRef] [PubMed]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef]
- Wagener, F.; Volk, H.-D.; Willis, D.; Abraham, N.G.; Soares, M.P.; Adema, G.J.; Figdor, C.G. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol. Rev. 2003, 55, 551–571. [Google Scholar] [CrossRef]
- Garby, L.; Noyes, W.D. Studies on hemoglobin metabolism. I. The kinetic properties of the plasma hemoglobin pool in normal man. J. Clin. Invest. 1959, 38, 1479–1483. [Google Scholar] [CrossRef]
- Hvidberg, V.; Maniecki, M.B.; Jacobsen, C.; Højrup, P.; Møller, H.J.; Moestrup, S.K. Identification of the receptor scavenging hemopexin-heme complexes. Blood 2005, 106, 2572–2579. [Google Scholar] [CrossRef]
- Grinshtein, N.; Bamm, V.V.; Tsemakhovich, V.A.; Shaklai, N. Mechanism of low-density lipoprotein oxidation by hemoglobin-derived iron. Biochemistry 2003, 42, 6977–6985. [Google Scholar] [CrossRef]
- Miller, Y.I.; Shaklai, N. Kinetics of hemin distribution in plasma reveals its role in lipoprotein oxidation. Biochim. Biophys. Acta 1999, 1454, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Muller-Eberhard, U.; Javid, J.; Liem, H.; Hanstein, A.; Hanna, M. Plasma concentrations of hemopexin, haptoglobin and heme in patients with various hemolytic diseases. Blood 1968, 32, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Vinchi, F.; De Franceschi, L.; Ghigo, A.; Townes, T.; Cimino, J.; Silengo, L.; Hirsch, E.; Altruda, F.; Tolosano, E. Hemopexin therapy improves cardiovascular function by preventing heme-induced endothelial toxicity in mouse models of hemolytic diseases. Circulation 2013, 127, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Hare, G.M.T.; Mu, A.; Romaschin, A.; Tsui, A.K.Y.; Shehata, N.; Beattie, W.S.; Mazer, C.D. Plasma methemoglobin as a potential biomarker of anemic stress in humans. Can. J. Anaesth. 2012, 59, 348–356. [Google Scholar] [CrossRef]
- Rother, R.P.; Bell, L.; Hillmen, P.; Gladwin, M.T. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: A novel mechanism of human disease. JAMA 2005, 293, 1653–1662. [Google Scholar] [CrossRef]
- Fasano, M.; Mattu, M.; Coletta, M.; Ascenzi, P. The heme-iron geometry of ferrous nitrosylated heme-serum lipoproteins, hemopexin, and albumin: A comparative EPR study. J. Inorg. Biochem. 2002, 91, 487–490. [Google Scholar] [CrossRef]
- Marassi, V.; Giordani, S.; Reschiglian, P.; Roda, B.; Zattoni, A. Tracking heme-protein interactions in healthy and pathological human serum in native conditions by miniaturized FFF-multidetection. Appl. Sci. 2022, 12, 6762. [Google Scholar] [CrossRef]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Aspects Med. 2012, 33, 209–290. [Google Scholar] [CrossRef]
- Wardell, M.; Wang, Z.; Ho, J.X.; Robert, J.; Ruker, F.; Ruble, J.; Carter, D.C. The atomic structure of human methemalbumin at 1.9 Å. Biochem. Biophys. Res. Commun. 2002, 291, 813–819. [Google Scholar] [CrossRef]
- Paoli, M.; Anderson, B.F.; Baker, H.M.; Morgan, W.T.; Smith, A.; Baker, E.N. Crystal structure of hemopexin reveals a novel high-affinity heme site formed between two β-propeller domains. Nat. Struct. Biol. 1999, 6, 926–931. [Google Scholar] [CrossRef]
- Biswal, B.K.; Vijayan, M. Structures of human oxy- and deoxyhaemoglobin at different levels of humidity: Variability in the T state. Acta Crystallogr. Sect. D. Biol. Crystallogr. 2002, 58, 1155–1161. [Google Scholar] [CrossRef]
- Andersen, C.B.F.; Torvund-Jensen, M.; Nielsen, M.J.; de Oliveira, C.L.P.; Hersleth, H.-P.; Andersen, N.H.; Pedersen, J.S.; Andersen, G.R.; Moestrup, S.K. Structure of the haptoglobin–haemoglobin complex. Nature 2012, 489, 456–459. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Pasquadibisceglie, A.; Polticelli, F.; di Masi, A.; Ascenzi, P. Haptoglobin and the related haptoglobin protein: The N-terminus makes the difference. J. Biomol. Struct. Dyn. 2022, 40, 2244–2253. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.J.; Buehler, P.W.; Alayash, A.I.; Belcher, J.D.; Vercellotti, G.M. Hemolysis and free hemoglobin revisited: Exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 2013, 121, 1276–1284. [Google Scholar] [PubMed]
- Wu, B.; Wu, Y.; Tang, W. Heme catabolic pathway in inflammation and immune disorders. Front. Pharmacol. 2019, 10, 825. [Google Scholar] [CrossRef] [PubMed]
- Gullotta, F.; di Masi, A.; Coletta, M.; Ascenzi, P. CO metabolism, sensing, and signaling. BioFactors 2012, 38, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kiening, M.; Lange, N. A recap of heme metabolism towards understanding protoporphyrin IX selectivity in cancer cells. Int. J. Mol. Sci. 2022, 23, 7974. [Google Scholar] [CrossRef]
- Quigley, J.G.; Yang, Z.; Worthington, M.T.; Phillips, J.D.; Sabo, K.M.; Sabath, D.E.; Berg, C.L.; Sassa, S.; Wood, B.L.; Abkowitz, J.L. Identification of a human heme Exporter that is essential for erythropoiesis. Cell 2004, 118, 757–766. [Google Scholar] [CrossRef]
- Zhou, S.; Zong, Y.; Ney, P.A.; Nair, G.; Stewart, C.F.; Sorrentino, B.P. Increased expression of the Abcg2 transporter during erythroid maturation plays a role in decreasing cellular protoporphyrin IX levels. Blood 2005, 105, 2571–2576. [Google Scholar] [CrossRef]
- Rajagopal, A.; Rao, A.U.; Amigo, J.; Tian, M.; Upadhyay, S.K.; Hall, C.; Uhm, S.; Mathew, M.K.; Fleming, M.D.; Paw, B.H.; et al. Haem homeostasis is regulated by the conserved and concerted functions of HRG-1 proteins. Nature 2008, 453, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Yuan, X.; Schmidt, P.J.; Bresciani, E.; Samuel, T.K.; Campagna, D.; Hall, C.; Bishop, K.; Calicchio, M.L.; Lapierre, A.; et al. HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab. 2013, 17, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.P.; Shing, J.; Saraon, P.; Berger, L.C.; Eiden, M.V.; Wilde, A.; Tailor, C.S. The Fowler syndrome-associated Protein FLVCR2 is an importer of heme. Mol. Cell. Biol. 2010, 30, 5318–5324. [Google Scholar] [CrossRef] [PubMed]
- Shayeghi, M.; Latunde-Dada, G.O.; Oakhill, J.S.; Laftah, A.H.; Takeuchi, K.; Halliday, N.; Khan, Y.; Warley, A.; McCann, F.E.; Hider, R.C.; et al. Identification of an intestinal heme transporter. Cell 2005, 122, 789–801. [Google Scholar] [CrossRef]
- Levitt, D.G.; Levitt, M.D. Human serum albumin homeostasis: A new look at the roles of synthesis, catabolism, renal and gastrointestinal excretion, and the clinical value of serum albumin measurements. Int. J. Gen. Med. 2016, 9, 229. [Google Scholar] [CrossRef]
- Rothschild, M.A.; Oratz, M. Albumin metabolism: A brief review. Mt. Sinai, J. Med. 1992, 59, 155–156. [Google Scholar]
- Rothschild, M.A.; Oratz, M.; Schreiber, S.S. Ethanol effects on albumin sythesis. In Biological Effects of Alcohol; Springer: Boston, MA, USA, 1980; Volume 126, pp. 385–396. [Google Scholar]
- Ascenzi, P.; Fasano, M. Allostery in a monomeric protein: The case of human serum albumin. Biophys. Chem. 2010, 148, 16–22. [Google Scholar] [CrossRef]
- Quinlan, G.J.; Martin, G.S.; Evans, T.W. Albumin: Biochemical properties and therapeutic potential. Hepatology 2005, 41, 1211–1219. [Google Scholar] [CrossRef]
- Leboffe, L.; di Masi, A.; Trezza, V.; Pasquadibisceglie, A.; Macari, G.; Polticelli, F.; Ascenzi, P. Neonicotinoid trapping by the FA1 site of human serum albumin. IUBMB Life 2019, 72, 716–723. [Google Scholar] [CrossRef]
- Di Masi, A.; Leboffe, L.; Polticelli, F.; Tonon, F.; Zennaro, C.; Caterino, M.; Stano, P.; Fischer, S.; Hägele, M.; Müller, M. Human serum albumin is an essential component of the host defense mechanism against Clostridium difficile intoxication. J. Infect. Dis. 2018, 218, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Sudlow, G.; Birkett, D.; Wade, D. The characterization of two specific drug binding sites on human serum albumin. Mol. Pharmacol. 1975, 11, 824–832. [Google Scholar]
- Vita, G.M.; De Simone, G.; Leboffe, L.; Montagnani, F.; Mariotti, D.; Di Bella, S.; Luzzati, R.; Gori, A.; Ascenzi, P.; di Masi, A. Human serum albumin binds Streptolysin O (SLO) toxin produced by Group a Streptococcus and inhibits its cytotoxic and hemolytic effects. Front. Immunol. 2020, 11, 507092. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Fasano, M. Serum heme-albumin: An allosteric protein. IUBMB Life 2009, 61, 1118–1122. [Google Scholar] [CrossRef] [PubMed]
- Kamal, J.A.; Behere, D.V. Binding of heme to human serum albumin: Steady-state fluorescence, circular dichroism and optical difference spectroscopic studies. Indian J. Biochem. Biophys. 2005, 42, 7–12. [Google Scholar] [PubMed]
- Cha, M.-K.; Kim, I.-H. Glutathione-linked thiol peroxidase activity of human serum albumin: A possible antioxidant role of serum albumin in blood plasma. Biochem. Biophys. Res. Commun. 1996, 222, 619–625. [Google Scholar] [CrossRef]
- Kamal, J.A.; Behere, D.V. Spectroscopic studies on human serum albumin and methemalbumin: Optical, steady-state, and picosecond time-resolved fluorescence studies, and kinetics of substrate oxidation by methemalbumin. J. Biol. Inorg. Chem. 2002, 7, 273–283. [Google Scholar] [CrossRef]
- Ascenzi, P.; Bolli, A.; di Masi, A.; Tundo, G.R.; Fanali, G.; Coletta, M.; Fasano, M. Isoniazid and rifampicin inhibit allosterically heme binding to albumin and peroxynitrite isomerization by heme–albumin. J. Biol. Inorg. Chem. 2011, 16, 97–108. [Google Scholar] [CrossRef]
- Ascenzi, P.; Leboffe, L.; Toti, D.; Polticelli, F.; Trezza, V. Fipronil recognition by the FA1 site of human serum albumin. J. Mol. Recognit. 2018, 31, e2713. [Google Scholar] [CrossRef]
- Leboffe, L.; di Masi, A.; Polticelli, F.; Trezza, V.; Ascenzi, P. Structural basis of drug recognition by human serum albumin. Curr. Med. Chem. 2020, 27, 4907–4931. [Google Scholar] [CrossRef]
- di Masi, A.; Leboffe, L.; Trezza, V.; Fanali, G.; Coletta, M.; Fasano, M.; Ascenzi, P. Drugs modulate allosterically heme-Fe-recognition by human serum albumin and heme-Fe-mediated reactivity. Curr. Pharm. Des. 2015, 21, 1837–1847. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Bocedi, A.; Gioia, M.; Fanali, G.; Fasano, M.; Coletta, M. Warfarin inhibits allosterically the reductive nitrosylation of ferric human serum heme-albumin. J. Inorg. Biochem. 2017, 177, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; di Masi, A.; De Sanctis, G.; Coletta, M.; Fasano, M. Ibuprofen modulates allosterically NO dissociation from ferrous nitrosylated human serum heme-albumin by binding to three sites. Biochem. Biophys. Res. Commun. 2009, 387, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; di Masi, A.; Fanali, G.; Fasano, M. Heme-based catalytic properties of human serum albumin. Cell Death Discov. 2015, 1, 15025. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Fasano, M. Abacavir modulates peroxynitrite-mediated oxidation of ferrous nitrosylated human serum heme–albumin. Biochem. Biophys. Res. Commun. 2007, 353, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Imperi, F.; Coletta, M.; Fasano, M. Abacavir and warfarin modulate allosterically kinetics of NO dissociation from ferrous nitrosylated human serum heme-albumin. Biochem. Biophys. Res. Commun. 2008, 369, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Bocedi, A.; Notari, S.; Menegatti, E.; Fanali, G.; Fasano, M.; Ascenzi, P. Allosteric modulation of anti-HIV drug and ferric heme binding to human serum albumin. FEBS J. 2005, 272, 6287–6296. [Google Scholar] [CrossRef]
- Bocedi, A.; Notari, S.; Narciso, P.; Bolli, A.; Fasano, M.; Ascenzi, P. Binding of anti-HIV drugs to human serum albumin. IUBMB Life 2004, 56, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Chuang, V.T.G.; Otagiri, M. How do fatty acids cause allosteric binding of drugs to human serum albumin? Pharm. Res. 2002, 19, 1458–1464. [Google Scholar] [CrossRef]
- di Masi, A.; Trezza, V.; Leboffe, L.; Ascenzi, P. Human plasma lipocalins and serum albumin: Plasma alternative carriers? J. Control. Release 2016, 228, 191–205. [Google Scholar] [CrossRef]
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005, 57, 787–796. [Google Scholar] [CrossRef]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Seedher, N.; Kanojia, M. Fluorescence spectroscopic study for competitive binding of antidiabetic drugs and endogenous substances on serum albumin. Drug Metabol. Drug Interact. 2013, 28, 107–114. [Google Scholar] [CrossRef]
- Sułkowska, A.; Bojko, B.; Równicka, J.; Sułkowski, W. Competition of drugs to serum albumin in combination therapy. Biopolymers 2004, 74, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Chuang, V.T.G.; Maruyama, T.; Otagiri, M. Albumin–drug interaction and its clinical implication. Biochim. Biophys. Acta 2013, 1830, 5435–5443. [Google Scholar] [CrossRef]
- Moman, R.N.; Gupta, N.; Varacallo, M. Physiology, Albumin; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Peters Jr, T. All about Albumin: Biochemistry, Genetics, and Medical Applications; Academic Press: San Diego, CA, USA, 1995. [Google Scholar]
- Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Unraveling the mysteries of serum albumin—More than just a serum protein. Front. Physiol. 2014, 5, 299. [Google Scholar] [CrossRef]
- Vita, G.M.; De Simone, G.; De Marinis, E.; Nervi, C.; Ascenzi, P.; di Masi, A. Serum albumin and nucleic acids biodistribution: From molecular aspects to biotechnological applications. IUBMB Life 2022. [Google Scholar] [CrossRef] [PubMed]
- Schnitzer, J.E.; Carley, W.W.; Palade, G.E. Albumin interacts specifically with a 60-kDa microvascular endothelial glycoprotein. Proc. Natl. Acad. Sci. USA 1988, 85, 6773–6777. [Google Scholar] [CrossRef] [PubMed]
- Ghinea, N.; Fixman, A.; Alexandru, D.; Popov, D.; Hasu, M.; Ghitescu, L.; Eskenasy, M.; Simionescu, M.; Simionescu, N. Identification of albumin-binding proteins in capillary endothelial cells. J. Cell Biol. 1988, 107, 231–239. [Google Scholar] [CrossRef]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Fritzsche, T.; Schnölzer, M.; Fiedler, S.; Weigand, M.; Wiessler, M.; Frei, E. Isolation and identification of heterogeneous nuclear ribonucleoproteins (hnRNP) from purified plasma membranes of human tumour cell lines as albumin-binding proteins. Biochem. Pharmacol. 2004, 67, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Amsellem, S.; Gburek, J.; Hamard, G.; Nielsen, R.; Willnow, T.E.; Devuyst, O.; Nexo, E.; Verroust, P.J.; Christensen, E.I.; Kozyraki, R. Cubilin is essential for albumin reabsorption in the renal proximal tubule. J. Am. Soc. Nephrol. 2010, 21, 1859–1867. [Google Scholar] [CrossRef]
- Zhai, X.Y.; Nielsen, R.; Birn, H.; Drumm, K.; Mildenberger, S.; Freudinger, R.; Moestrup, S.K.; Verroust, P.J.; Christensen, E.I.; Gekle, M. Cubilin- and megalin-mediated uptake of albumin in cultured proximal tubule cells of opossum kidney. Kidney Int. 2000, 58, 1523–1533. [Google Scholar] [CrossRef] [PubMed]
- Schnitzer, J.E.; Oh, P. Antibodies to SPARC inhibit albumin binding to SPARC, gp60, and microvascular endothelium. Am. J. Physiol. Heart Circ. Physiol. 1992, 263, H1872–H1879. [Google Scholar] [CrossRef]
- Jennifer, B.; Berg, V.; Modak, M.; Puck, A.; Seyerl-Jiresch, M.; Künig, S.; Zlabinger, G.J.; Steinberger, P.; Chou, J.; Geha, R.S.; et al. Transferrin receptor 1 is a cellular receptor for human heme-albumin. Commun. Biol. 2020, 3, 621. [Google Scholar] [CrossRef]
- Kleven, M.D.; Jue, S.; Enns, C.A. Transferrin receptors TfR1 and TfR2 bind transferrin through differing mechanisms. Biochemistry 2018, 57, 1552–1559. [Google Scholar] [CrossRef] [PubMed]
- Richard, C.; Verdier, F. Transferrin receptors in erythropoiesis. Int. J. Mol. Sci. 2020, 21, 9713. [Google Scholar] [CrossRef]
- Montemiglio, L.C.; Testi, C.; Ceci, P.; Falvo, E.; Pitea, M.; Savino, C.; Arcovito, A.; Peruzzi, G.; Baiocco, P.; Mancia, F.; et al. Cryo-EM structure of the human ferritin–transferrin receptor 1 complex. Nat. Commun. 2019, 10, 1121. [Google Scholar] [CrossRef]
- Tonoyan, L.; Fleming, G.T.A.; Mc Cay, P.H.; Friel, R.; O’Flaherty, V. Antibacterial potential of an antimicrobial agent inspired by peroxidase-catalyzed systems. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef]
- Vlasova, I.I. Peroxidase activity of human hemoproteins: Keeping the fire under control. Molecules 2018, 23, 2561. [Google Scholar] [CrossRef]
- Vinchi, F.; Costa da Silva, M.; Ingoglia, G.; Petrillo, S.; Brinkman, N.; Zuercher, A.; Cerwenka, A.; Tolosano, E.; Muckenthaler, M.U. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood 2016, 127, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Gammella, E.; Buratti, P.; Cairo, G.; Recalcati, S. Macrophages: Central regulators of iron balance. Metallomics 2014, 6, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Win, N.; Lee, E.; Needs, M.; Chia, L.W.; Stasi, R. Measurement of macrophage marker in hyperhaemolytic transfusion reaction: A case report. Transfus. Med. 2012, 22, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.S.; Fatemi, R.; Winlow, W. SARS-CoV-2 bound human serum albumin and systemic septic shock. Front. Cardiovasc. Med. 2020, 7, 153. [Google Scholar] [CrossRef]
- Lippi, G.; Mattiuzzi, C. Hemoglobin value may be decreased in patients with severe coronavirus disease 2019. Hematol. Transfus. Cell Ther. 2020, 42, 116–117. [Google Scholar] [CrossRef]
- Liu, W.; Li, H. COVID-19: Attacks the 1-beta chain of hemoglobin and captures the porphyrin to inhibit human heme metabolism. ChemRxiv 2022. [Google Scholar] [CrossRef]
- Su, W.-L.; Lin, C.-P.; Hang, H.-C.; Wu, P.-S.; Cheng, C.-F.; Chao, Y.-C. Desaturation and heme elevation during COVID-19 infection: A potential prognostic factor of heme oxygenase-1. J. Microbiol. Immunol. Infect. 2021, 54, 113–116. [Google Scholar] [CrossRef]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O.; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef]
- Hopp, M.-T.; Domingo-Fernández, D.; Gadiya, Y.; Detzel, M.S.; Graf, R.; Schmalohr, B.F.; Kodamullil, A.T.; Imhof, D.; Hofmann-Apitius, M. Linking COVID-19 and heme-driven pathophysiologies: A combined computational–experimental approach. Biomolecules 2021, 11, 644. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, Y.; Zhao, X.; Tao, M.; Yan, W.; Fu, Y. Hypoalbuminemia - an indicator of the severity and prognosis of COVID-19 patients: A multicentre retrospective analysis. Infect. Drug Resist. 2021, 14, 3699–3710. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, R.; Xu, Y.; Gong, P. Clinical characteristics of 36 non-survivors with COVID-19 in Wuhan, China. medRxiv 2020. [Google Scholar] [CrossRef]
- Sharma, Y.P.; Panda, P.; Uppal, L. Albumin and its potential role in COVID-19. Rapid response to “Preventing a COVID-19 epidemic” by John Watkins. BMJ 2020, 368, m810. [Google Scholar] [CrossRef]
- Violi, F.; Ceccarelli, G.; Cangemi, R.; Alessandri, F.; D’Ettorre, G.; Oliva, A.; Pastori, D.; Loffredo, L.; Pignatelli, P.; Ruberto, F. Hypoalbuminemia, coagulopathy, and vascular disease in COVID-19. Circ. Res. 2020, 127, 400–401. [Google Scholar] [CrossRef] [PubMed]
- Zerbato, V.; Sanson, G.; De Luca, M.; Di Bella, S.; di Masi, A.; Caironi, P.; Marini, B.; Ippodrino, R.; Luzzati, R. The impact of serum albumin levels on COVID-19 mortality. Infect. Dis. Rep. 2022, 14, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Kotsiliti, E. Siderophore cross-feeding between fungi and Salmonella. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 65. [Google Scholar] [CrossRef]
- Nasser, L.; Weissman, Z.; Pinsky, M.; Amartely, H.; Dvir, H.; Kornitzer, D. Structural basis of haem-iron acquisition by fungal pathogens. Nat. Microbiol. 2016, 1, 16156. [Google Scholar] [CrossRef] [PubMed]
- Pinsky, M.; Roy, U.; Moshe, S.; Weissman, Z.; Kornitzer, D. Human serum albumin facilitates heme-iron utilization by fungi. mBio 2020, 11, e00607-20. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J.; Kolnagou, A.; Skiada, A.; Petrikkos, G. The role of iron and chelators on infections in iron overload and non iron loaded conditions: Prospects for the design of new antimicrobial therapies. Hemoglobin 2010, 34, 227–239. [Google Scholar] [CrossRef]
- Ellmerer, M.; Schaupp, L.; Brunner, G.A.; Sendlhofer, G.; Wutte, A.; Wach, P.; Pieber, T.R. Measurement of interstitial albumin in human skeletal muscle and adipose tissue by open-flow microperfusion. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E352–E356. [Google Scholar] [CrossRef]
- de Villiers, K.A.; Egan, T.J. Heme detoxification in the malaria parasite: A target for antimalarial drug development. Acc. Chem. Res. 2021, 54, 2649–2659. [Google Scholar] [CrossRef]
- Englert, F.A.; Seidel, R.A.; Galler, K.; Gouveia, Z.; Soares, M.P.; Neugebauer, U.; Clemens, M.G.; Sponholz, C.; Heinemann, S.H.; Pohnert, G.; et al. Labile heme impairs hepatic microcirculation and promotes hepatic injury. Arch. Biochem. Biophys. 2019, 672, 108075. [Google Scholar] [CrossRef] [PubMed]
- Day, T.G.; Drasar, E.R.; Fulford, T.; Sharpe, C.C.; Thein, S.L. Association between hemolysis and albuminuria in adults with sickle cell anemia. Haematologica 2012, 97, 201–205. [Google Scholar] [CrossRef]
- Ferreira, A.; Marguti, I.; Bechmann, I.; Jeney, V.; Chora, Â.; Palha, N.R.; Rebelo, S.; Henri, A.; Beuzard, Y.; Soares, M.P. Sickle hemoglobin confers tolerance to Plasmodium infection. Cell 2011, 145, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Larsen, R.; Gozzelino, R.; Jeney, V.; Tokaji, L.; Bozza, F.A.; Japiassú, A.M.; Bonaparte, D.; Cavalcante, M.M.; Chora, Â.; Ferreira, A.; et al. A central role for free heme in the pathogenesis of severe sepsis. Sci. Transl. Med. 2010, 2, 51ra71. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Quattrocchi, A.; Mancini, B.; di Masi, A.; Nervi, C.; Ascenzi, P. Thalassemias: From gene to therapy. Mol. Aspects Med. 2022, 84, 101028. [Google Scholar] [CrossRef] [PubMed]
- Effenberger-Neidnicht, K.; Brencher, L.; Broecker-Preuss, M.; Hamburger, T.; Petrat, F.; de Groot, H. Immune stimulation by exogenous melatonin during experimental endotoxemia. Inflammation 2014, 37, 738–744. [Google Scholar] [CrossRef]
- Lansink, M.O.; Görlinger, K.; Hartmann, M.; de Groot, H.; Effenberger-Neidnicht, K. Melatonin does not affect disseminated intravascular coagulation but diminishes decreases in platelet count during subacute endotoxaemia in rats. Thromb. Res. 2016, 139, 38–43. [Google Scholar] [CrossRef]
- Adamzik, M.; Hamburger, T.; Petrat, F.; Peters, J.; de Groot, H.; Hartmann, M. Free hemoglobin concentration in severe sepsis: Methods of measurement and prediction of outcome. Crit. Care 2012, 16, R125. [Google Scholar] [CrossRef]
- Hartmann, M.; de Groot, H. Cell-free hemoglobin: A new player in sepsis pathophysiology. Crit. Care Med. 2013, 41, e186–e189. [Google Scholar] [CrossRef]
- Effenberger-Neidnicht, K.; Hartmann, M. Mechanisms of hemolysis during sepsis. Inflammation 2018, 41, 1569–1581. [Google Scholar] [CrossRef]
- Kremer, H.; Baron-Menguy, C.; Tesse, A.; Gallois, Y.; Mercat, A.; Henrion, D.; Andriantsitohaina, R.; Asfar, P.; Meziani, F. Human serum albumin improves endothelial dysfunction and survival during experimental endotoxemia: Concentration-dependent properties. Crit. Care Med. 2011, 39, 1414–1422. [Google Scholar] [CrossRef] [PubMed]
- Larsen, R.; Gouveia, Z.; Soares, M.; Gozzelino, R. Heme cytotoxicity and the pathogenesis of immune-mediated inflammatory diseases. Front. Pharmacol. 2012, 3. [Google Scholar] [CrossRef]
- Caironi, P.; Tognoni, G.; Masson, S.; Fumagalli, R.; Pesenti, A.; Romero, M.; Fanizza, C.; Caspani, L.; Faenza, S.; Grasselli, G.; et al. Albumin replacement in patients with severe sepsis or septic shock. N. Engl. J. Med. 2014, 370, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Raghunathan, K.; Kempker, J.A.; Davis, E.A.; Sindhwani, N.S.; Telang, S.; Lodaya, K.; Martin, G.S. Early albumin infusion is associated with greater survival to discharge among patients with sepsis/septic shock who develop severe acute kidney injury. Crit. Care Explor. 2022, 4, e0793. [Google Scholar] [PubMed]
- Bi, D.; Lin, J.; Luo, X.; Lin, L.; Tang, X.; Luo, X.; Lu, Y.; Huang, X. Biochemical characteristics of patients with imported malaria. Front. Cell. Infect. Microbiol. 2022, 12. [Google Scholar] [CrossRef]
- Maitland, K.; Pamba, A.; English, M.; Peshu, N.; Marsh, K.; Newton, C.; Levin, M. Randomized trial of volume expansion with albumin or saline in children with severe malaria: Preliminary evidence of albumin benefit. Clin. Infect. Dis. 2005, 40, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Akech, S.; Gwer, S.; Idro, R.; Fegan, G.; Eziefula, A.C.; Newton, C.R.J.C.; Levin, M.; Maitland, K. Volume expansion with albumin compared to gelofusine in children with severe malaria: Results of a controlled trial. PLoS Clin. Trials 2006, 1, e21. [Google Scholar] [CrossRef]
- Kratz, F. A clinical update of using albumin as a drug vehicle—A commentary. J. Control. Release 2014, 190, 331–336. [Google Scholar] [CrossRef]
- Funato, M.; Tamai, H.; Shimada, S.; Nakamura, H. Vigintiphobia, unbound bilirubin, and auditory brainstem responses. Pediatrics 1994, 93, 50–53. [Google Scholar] [CrossRef]
- Bratlid, D. How bilirubin gets into the brain. Clin. Perinatol. 1990, 17, 449–465. [Google Scholar] [CrossRef]
- Hosono, S.; Ohno, T.; Kimoto, H.; Nagoshi, R.; Shimizu, M.; Nozawa, M. Effects of albumin infusion therapy on total and unbound bilirubin values in term infants with intensive phototherapy. Pediatr. Int. 2001, 43, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Watchko, J.F.; Daood, M.J.; Biniwale, M. Understanding neonatal hyperbilirubinaemia in the era of genomics. Semin. Neonatol. 2002, 7, 143–152. [Google Scholar] [CrossRef]
- Biglarnia, A.-R.; Huber-Lang, M.; Mohlin, C.; Ekdahl, K.N.; Nilsson, B. The multifaceted role of complement in kidney transplantation. Nat. Rev. Nephrol. 2018, 14, 767–781. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Vijayan, V.; Jang, M.-S.; Thorenz, A.; Greite, R.; Rong, S.; Chen, R.; Shushakova, N.; Tudorache, I.; Derlin, K.; et al. Labile Heme Aggravates Renal Inflammation and Complement Activation After Ischemia Reperfusion Injury. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).