Abstract
Diabetes mellitus, a well-established risk factor for stroke, is related to higher mortality and poorer outcomes following the stroke event. Advanced glycation end products(AGEs), their receptors RAGEs, other ligands, and several other processes contribute to the cerebrovascular pathomechanism interaction in the diabetes–ischemic stroke combination. Critical reappraisal of molecular targets and therapeutic agents to mitigate them is required to identify key elements for therapeutic interventions that may improve patient outcomes. This scoping review maps evidence on the key roles of AGEs, RAGEs, other ligands such as Leukotriene B4 (LTB4), High-mobility group box 1 (HMGB1) nuclear protein, brain–kidney–muscle crosstalk, alternate pathomechanisms in neurodegeneration, and cognitive decline related to diabetic ischemic stroke. RAGE, HMGB1, nitric oxide, and polyamine mechanisms are important therapeutic targets, inflicting common consequences of neuroinflammation and oxidative stress. Experimental findings on a number of existing–emerging therapeutic agents and natural compounds against key targets are promising. The lack of large clinical trials with adequate follow-up periods is a gap that requires addressing to validate the emerging therapeutic agents. Five therapeutic components, which include agents to mitigate the AGE–RAGE axis, improved biomarkers for risk stratification, better renal dysfunction management, adjunctive anti-inflammatory–antioxidant therapies, and innovative neuromuscular stimulation for rehabilitation, are identified. A comprehensive therapeutic strategy that features all the identified components is needed for outcome improvement in diabetic stroke patients.
1. Introduction
Diabetes mellitus, currently considered one of the largest epidemics, is associated with cardio-cerebrovascular complications. Together, they present a major public health burden in terms of healthcare economics, enhanced morbidity, and mortality, with the prevalence rising at an exponential rate worldwide [1,2]. A 26% increase in global stroke deaths was indicated during the past two decades [1]. Two-thirds of all strokes are estimated to occur in developing countries, which, despite their preventable nature, continue to occur increasingly [3,4]. Stroke-related deaths are expected to nearly double in North Africa and the Middle East by 2030 [4]. Stroke survivors experience motor impairments requisite to locomotion and post-stroke dysfunction of nearly 75%. Severe functional disability of 15–30% has been reported by the American Heart Association, which warrants more attention towards improving disease management strategies [5].
Stroke is defined as “Central nervous system (CNS) infarction is brain, spinal cord, or retinal cell death attributable to ischemia, based on 1. pathological, imaging, or other objective evidence of cerebral, spinal cord, or retinal focal ischemic injury in a defined vascular distribution; or 2. clinical evidence of cerebral, spinal cord, or retinal focal ischemic injury based on symptoms persisting ≥24 h or until death, and other etiologies excluded” [5]. Ischemic stroke due to thrombosis, embolism, or systemic hypoperfusion is one of the two broad categories of stroke, with the other being hemorrhagic stroke due to intracerebral hemorrhage or subarachnoid hemorrhage [5]. Strokes due to ischemic cerebral infarction comprise approximately 80% of strokes [6]. The ischemic cascade of activated biochemical reactions initiated in the brain and other aerobic tissues during and after ischemia causes neuronal death and usually lasts for 2-3 h but can last for days, even after normal blood flow returns [7]. The Trial of Org 10,172 in Acute Stroke Treatment (TOAST) system guides proper management decisions and categorizes ischemic stroke subtypes [8].
Diabetes mellitus, a metabolic disease, is also a major risk factor for cardiovascular and ischemic stroke disease development [9,10,11]. Enhanced generation and accumulation of advanced glycation end products (AGEs) occurring due to chronic hyperglycemia of diabetes has been implicated in the pathogenesis of macrovascular and microvascular complications leading to end-organ damage and cardio- and cerebrovascular diseases, including stroke [11,12,13,14,15]. Non-enzymatic glycation and oxidation of proteins, nucleic acids, and lipids produce AGEs [16,17]. Receptors for AGEs or RAGEs, which are multiligand receptors of the immunoglobulin cell surface molecule superfamily, modulate atherosclerotic processes, trigger oxidative stress, inflammation, and apoptosis, and enhance tissue damage in focal cerebral ischemia [14,18].
Subsets of AGEs known as toxic AGEs (TAGEs) exert cytotoxic effects, and neurotoxicity has been reported in diabetic ischemic stroke although the exact molecular mechanisms are not known [18,19,20,21,22]. Interestingly, a link between the G protein-coupled receptor (GPCR) and RAGE in ischemic stroke pathology was revealed recently when RAGE was reported to modulate the signaling of a high-affinity GPCR for Leukotriene B4(LTB4), a potent proinflammatory lipid mediator [23,24,25]. RAGE has been proposed as a novel GPCR class of modulators and a target for future studies on therapeutic interventions. Intriguing organ crosstalk between the brain and kidney, renal dysfunction, and decreased renal clearance of AGEs cause their accumulation in circulation to occur in ischemic stroke [26,27]. A low glomerular filtration rate (GFR) in stroke was found to increase all-cause mortality and recurrent stroke risks [28]. This evidence indicates the interplay between AGEs, RAGE ligands, GPCR, and neurotoxicity in diabetic ischemic stroke [13,14,15,18,24,26].
This scoping review systematically maps the current and previous findings on AGEs, RAGE ligands, Leukotriene B4GPCR, organ crosstalk, and pathomechanisms in diabetic ischemic stroke to decipher the intricate interplay of molecular mechanisms. This will explicate the close interaction between diabetes mellitus and ischemic stroke as two complementary disorders associated with cerebrovascular complications. Experimental and clinical findings on several potentially useful therapeutic agents that target these mechanisms are suitably presented. The appraisal of key findings on all these components is required for the identification of key targets and to aid the development of effective comprehensive therapeutic strategies for improving patient outcomes in diabetic ischemic stroke.
2. AGEs, Toxic AGEs, and RAGEs
AGEs are irreversible covalent adducts formed endogenously through non-enzymatic glycation and glycoxidation of proteins, nucleic acids, and lipid biomolecules. Dietary AGE sources also exist, such as processed and barbequed foods [16,17,29]. AGEs such as carboxymethyllysine, pentosidine, and pyrraline, which do not exert direct cytotoxic effects, are considered non-toxic. They form a defense mechanism to trap aldehyde/carbonyl compounds with high reactivity and detoxify them [17,18,21]. The enhanced generation of AGEs in diabetes contributes to the development of micro- and macro-vascular complications, end-organ damage, and cardio-cerebrovascular diseases including stroke, as AGEs crosslink intracellular and extracellular matrix proteins, thus causing modifications in the structure, function, and mechanical properties of tissues [29,30]. Toxic AGEs (TAGEs), a subset of AGEs, exert toxic effects on certain cells and tissues through their interaction with receptors for AGEs (RAGEs) [18,19,20,21]. TAGEs, which bind to RAGEs, have been implicated in diabetes mellitus and its associated vascular complication pathogenesis [17,18,21]. Based on their effects and properties such as fluorescence, different types of AGEs exist, as shown in Table 1 [16,17,29,30].

Table 1.
AGEs and Toxic AGEs: Types and examples.
For AGE measurements in human subjects, a noninvasive and effective tool termed skin autofluorescence (SAF) is available, which detects AGEs in the dermis layer [31,32]. SAF is a tool to establish the risk of chronic complications of diabetes mellitus for prompt intervention. Another investigation, namely, Blood AGE levels by ELISA, an enzyme-linked immunosorbent assay, employs the AGE antibodies [31,33]. Serum TAGE levels are also promising novel biomarkers to monitor the onset and progression of diseases related to lifestyle, measured using a specific antibody and competitive ELISA methods [17]. Tissue, plasma fluorescence, and SAF have been used to estimate AGE accumulation, and point-of-care testing (POCT) devices to monitor serum AGEs are currently available [31,32]. In both chronic cerebral infarction and silent brain infarction patients, higher SAF levels of AGEs have been reported. Significantly higher serum levels of fluorescent AGE pentosidine were found in acute ischemic stroke in comparison with healthy controls using the ELISA method [33].
Accumulated AGEs primarily interact with their receptors—RAGEs—to produce persistent vascular inflammation. Excessive downstream inflammatory indicators linked to the AGE–RAGE interaction have been linked to post-stroke inflammation, which is known to worsen ischemic brain damage and lead to cardiac injury [34]. By triggering signalling cascades through RAGE, AGEs mediate their detrimental effects. Numerous cell types in the peripheral and central nervous systems, including neurons, microglia, astrocytes, epithelial cells, mononuclear phagocytes, and endothelial cells, express RAGE, which is a 45 kDa immunoglobulin-type transmembrane receptor [35]. Due to the presence of multiple domains, isoforms, and polymorphisms, RAGEs bind to a discrete repertoire of ligands. The ligands for RAGEs also include many members of the S100/calgranulin family, oligomeric forms of A, high-mobility group box 1 (HMGB1), phosphatidylserine (PS), and lysophosphatidic acid. Interactions between AGEs and RAGEs result in diverse cell responses such as altered gene expression, migration, proliferation, and the activation of signaling pathways that cause oxidative stress, inflammation, apoptosis, atherosclerosis, and other vascular complications [35,36]. One of the numerous isoforms of RAGE is the soluble form of RAGE (sRAGE). Soluble RAGE also consists of several forms, including a spliced variant of RAGE, called endogenous secretory RAGE (esRAGE). These RAGE isoforms have extracellular domains but lack the intracytoplasmic and transmembrane domains. They can bind to ligands, including AGEs, and antagonize RAGE signalling. Scavenging receptors such as SR-AI are involved in the removal of AGEs. The pathophysiological roles of these RAGE isoforms in cardiovascular disease and the usefulness of soluble RAGE as disease biomarkers and therapeutic targets have all been indicated. ELISA techniques are used to assess the plasma levels of esRAGE and total sRAGE [34,35,36].
3. Diabetic Ischemic Stroke and RAGE-Mediated Ischemic Brain Damage
Stroke is a frequent, severe neurovascular disorder that is a major cause of disability and mortality. As per the TOAST classification, subtype 1 of ischemic stroke is associated with large-artery atherosclerosis [8] as follows: “Patients having clinical and brain imaging findings of either significant (>50%) stenosis or occlusion of a major brain artery or branch cortical artery, presumably due to atherosclerosis. Clinical findings include those of cerebral cortical impairment (aphasia, neglect, restricted motor involvement, etc.) or brain stem or cerebellar dysfunction. History of intermittent claudication, transient ischemic attacks (TIAs) in the same vascular territory, a carotid bruit, or diminished pulses helps support the clinical diagnosis. Cortical or cerebellar lesions and brain stem or subcortical hemispheric infarcts greater than 1.5cm in diameter on Computed Tomography(CT) or Magnetic Resonance Imaging (MRI) are considered to be of potential large-artery atherosclerotic origin. Supportive evidence by duplex imaging or arteriography of stenosis of greater than 50% of an appropriate intracranial or extracranial artery is needed. Diagnostic studies should exclude potential sources of cardiogenic embolism. Stroke diagnosis secondary to large-artery atherosclerosis cannot be made if duplex or arteriographic studies are normal or show only minimal changes“ [8]. Diabetes mellitus (DM) is associated with ischemic stroke subtype 1–large-artery atherosclerosis as it is the main cause of atherosclerosis. DM is also associated with subtype 3 of ischemic stroke—small vessel occlusion or lacunae—as the clinical diagnosis is supported by DM or hypertension history [8].
An increasing body of evidence shows the involvement of RAGE and its ligands in the pathogenesis of cardio-cerebrovascular, neurodegenerative, neuroinflammatory, and autoimmune disorders. sRAGE levels are elevated in the acute phase in the serum of stroke patients [37]. Neurons and glial cells in the brain express RAGE [38,39]. RAGE levels have been found to be elevated in biopsy samples taken from human patients who had a unilateral cerebral infarction as well as in the ischemic brain hemisphere of mice one day following an experimental stroke [40]. It is crucial to underscore the key roles of AGEs in oxidative stress, lipid oxidation, atherosclerotic processes, triggering of inflammation, and dysfunctions of mitochondria, as well as endothelial cells, in chronic cerebral ischemia. The interaction between AGE–RAGE and oxidative stress is also crucial when discussing the cardio-cerebrovascular consequences of diabetes. The carotid intima-media thickness (IMT) is employed as a measure of subclinical atherosclerosis since atherosclerosis is characterized by intramural thickening of the sub-intima. It is interesting to note that carotid IMT progression is a surrogate marker for risk stratification of both cerebrovascular and cardiovascular disease [41]. Following focal cerebral ischemia, the amount of irreversible damage and necrosis is increased by AGE-modified proteins and peptides [22]. In this respect, the number of skin AGEs measured via skin autofluorescence (SAF) is a helpful metric in the treatment of acute stroke as a predictor of the outcome of ischemic stroke in patients with diabetes mellitus [34].
Numerous variables in diabetes patients, such as hyperglycemia, vascular risk factors such as hypertension and dyslipidemia, and genetic, demographic, and lifestyle variables all play a role in the overall cardio-cerebrovascular risk to varying degrees [42,43,44]. The importance of secondary prevention methods against debilitating strokes in diabetic individuals is highlighted by the fact that their risk of ischemic stroke is roughly double that of people without diabetes [10]. The relationship between diabetes and ischemic stroke is reciprocal, and diabetes may cause more subtle brain damage indicated by lacunar infarcts, which raises the risk of dementia and causes a faster loss of cognitive function [9]. In an ischemic stroke, the necrotic core is surrounded by an inflammatory zone. The initial injury is aggravated by delayed cell death. As a mediator of ischemic brain damage, a sensor of necrotic cell death, and a contributor to both inflammation and ischemic brain damage, RAGE plays key roles. The high-mobility group box 1 (HMGB1) ligand released from necrotic cells seems to be the most obvious RAGE activator in stroke. In experimental models of cerebral ischemia, RAGE was shown to be a key mediator. An independent predictor of poor outcomes for stroke in type 2 diabetes is an elevated blood RAGE level [45]. After a stroke, RAGE may play a role in a more vigorous inflammatory response that is mediated by HMGB1 [46].
HMGB1, a non-histone nuclear protein, is a crucial proinflammatory alarmin in cerebrovascular ischemic disorders [43]. RAGE and the toll-like receptors TLR-2 and-4 are the main HMGB1 receptors investigated in brain injury. These receptors are ubiquitously expressed on microglia, astrocytes, and neurons in the CNS [47,48]. In the early stages of stroke, several neurons undergo sustained oxidative toxicity and hypoxia. HMGB1 modifications occur via acetylation and phosphorylation, which decrease its affinity for DNA when microglia and astrocytes are activated. As neuron cell membranes are destroyed, HMGB1 loosely bound to chromosomes is passively released into the extracellular space within 2–4 h after ischemia–reperfusion [46]. Hyperglycemia causes early extracellular HMGB1 secretion from ischemic brain tissue and also increases the extracellular accumulation of glutamate, an excitatory amino acid that plays a central role in neuronal death. This increases infarct volume, neurological deficits, cerebral edema, and blood–brain barrier (BBB) disruption, which is a critical event in the growth of cerebral edema in the early stages of ischemic brain injury [46,49]
The following molecular processes occur after HMGB1 release during ischemic events [46]:
- HMGB1 interacts with the receptors TLR-2 and TLR-4, which are expressed on monocytes through the adaptor protein myeloid differentiation factor 88 (MyD88) and elevates serum levels of TNF-α, IL-6, and IL-1β l, which leads to cerebral vessel occlusion.
- TNF-α and IL-1β, which are HMGB1-induced cytokines, can indirectly promote the upregulation of matrix metalloproteinase MMP-9. MMP upregulation causes a rise in infarct size, brain edema, and recombinant tissue plasminogen activator-induced bleeding, which hastens damage to the tight junction protein Occludin and other neurovascular substrates.
- HMGB1 stimulation of TNF-α, IL-1, IL-6, and IL-8 production induces the expression of inducible NOS (iNOS) during ischemic brain damage. The induction of iNOS and TNF-α occurs mainly in microglia. This produces an inflammatory response and BBB disruption, leading to brain infarction aggravation.
- RAGE expression, which is low in cells under physiological conditions, rises in response to an increase in HMGB1 ligand molecules, for which RAGE has a strong affinity. HMGB1 binding to upregulated RAGE leads to the activation of several signal-transduction pathways including Mitogen-activated protein kinases (MAPKs), phosphatidylinositol 3 kinase/protein kinase B (PI3K/Akt)-p38 kinase, SAPK/JNK, extracellular regulated protein kinases1/2 (ERK 1/2), Akt, Ras-related C3 botulinum toxin substrate (Rac), Cell division cycle 42 (Cdc42), and Just another kinase/signal transducer and activator of transcription 1 (JAK/STAT1)-mediated signal transduction pathways. Finally, these processes result in the translocation of nuclear factor kappa-light-chain-enhancer (NF-κB), which triggers the expression of inflammatory cytokines and chemokines that help immune cells mature, migrate, and express surface receptors, and cause neuritis [50].
RAGE has been linked to neuronal cell death following ischemia and is expressed by endothelial and microglial cells in the human brain. Endothelial RAGE performs two distinct functions in ischemia: It transports endogenous secretory RAGE (esRAGE), a neuroprotectant, to the brain and also induces vascular injury and neuronal damage [37]. The interaction between toxic AGEs (TAGE) and RAGE elicits vascular inflammatory processes and oxidative stress in many cell types, including endothelial, causing microvascular circulation derangement [18]. Previous studies in mice models of distal permanent middle cerebral artery blockage, which generate focal ischemic stroke, showed a critical role of neuronal and microglial RAGE in ischemia-induced neuronal death and inflammation [51]. In a mouse model of cerebral ischemia, HMGB1 was found to be released from ischemic brain tissue and increased in the serum of stroke victims. Ischemic brain damage was lessened by a neutralizing anti-HMGB1 antibody and HMGB1 box A, an antagonist of HMGB1 at the receptor RAGE. Decoy receptor soluble RAGE and genetic RAGE deficiency caused a reduction in infarct size [52].In vitro expression of RAGE mediated the toxic effect of HMGB1 on microglial cells. Brain macrophages mediate the HMGB1–RAGE-induced injury. Data from in vivo animal model experiments using chimeric mice strongly indicated that for the development of ischemic brain damage, RAGE expressed by immigrant macrophages is required, possibly for binding the HMGB1 released from necrotic cells in the ischemia core [52]. HMGB1–RAGE signaling in stroke links macrophage activation with necrosis, providing a therapeutic target. The HMGB1 antagonist box A, a neutralizing anti-HMGB1 antibody or soluble RAGE, may be a specific tool to target inflammation and delayed cell death [52].
4. AGE-RAGE System, Blood–Brain Barrier Dysfunction, Neuroinflammation and Neurodegeneration in Ischemic Stroke
Ischemic stroke contributes to the disintegration of the neurovascular unit, which consists of endothelial cells of the blood–brain barrier (BBB), neurons, astrocytes, myocytes, pericytes, and extracellular matrix components. BBB, a highly selective semipermeable border of endothelial cells, is the blood–brain interface, mediating communication between central and peripheral nervous systems. Synaptic-neuronal dysfunction, damage, and loss are caused by a vicious cycle of degeneration driven by an irreversible BBB breakdown, chemical imbalance in the internal milieu of neurons, and neurovascular inflammatory immunological responses [36,53]. The molecular mechanisms involved in the neurodegenerative processes of ischemic stroke that have been studied are explained as follows:
- During thrombosis, amyloid beta (Aβ) peptides produced in blood vessels are discharged into the brain and momentarily accumulate there. During an ischemic stroke, they act as additional damaging factors by forming ion channels. RAGE is implicated in the neurotoxic immunoinflammatory cascades and in the amyloidogenic pathway. RAGE is the primary influx transporter for Aβ across the BBB. RAGE-soluble Aβ binding mediates the pathophysiologic cellular responses. The RAGE–Aβ interaction causes oxidative damage to RAGE-expressing neurons and activates microglia, which both directly and indirectly contributes to neuron death. RAGE inhibitors can prevent the production of cytokines and chemokines, oxidative stress, and Aβ BBB transport by blocking the pathophysiological effects of the RAGE–Aβ interaction in the afflicted vasculature [36,53].
- RAGE is connected to both independent neurotoxic immunoinflammatory cascades and the amyloidogenic pathway in neurodegenerative diseases [48]. RAGE is overexpressed in neurons, microglia, astrocytes, and the BBB vasculature when endogenous ligands such as AGE, S100, or Aβ bind to physiologically expressed RAGE [36,48]. RAGE is more prevalent at the BBB, which causes an influx of monocytes and Aβ into the brain. However, RAGE is more active in neurons, where it enhances Aβ-producing β-secretase enzyme (BACE1) activity, tau hyperphosphorylation, and neuroinflammation and impairs neuronal function. Ischemia-induced Aβ/tau pathology, similar to Alzheimer’s disease, is reported to be involved in post-stroke cognitive impairment [36].
- Hyperglycemia-induced overexpression of mitochondrial superoxide in endothelial cells causes microvascular injury in diabetes mellitus. Superoxide overproduction can activate AGEs formation and protein kinase C (PKC) signaling. PKC induces BBB damage through the disruption of tight junction (TJ) proteins, phosphorylation of cytoplasmic adaptor zona occuldens-1 (ZO1), and enhanced expression of vascular endothelial growth factor. Upregulated and activated RAGEs induce oxidative stress and activate the NF-κB pathway. TNF-α, IL-6, and IL-1 transcription are increased when the NF-κB pathway is activated in vascular cells [18,36].
- AGEs cause neurodegeneration by pathways unrelated to RAGE, such as protein modification and cross-linking, contributing to toxic effects of Aβ, affecting cellular damage, tissue stiffness, vascular pathological processes, and the formation of aggregates [54,55,56].
- The AGE–RAGE system may also participate in Apolipoprotein E-ε4 (APOE-ε4 allele)-associated pathological processes of dementia. Associations between higher skin autofluorescence due to AGEs, lower cognitive function, and APOE-ε4 status have been reported [57]. APOE-ε4 impacts the risk of dementia associated with stroke in patients, and both pre- and post-event dementia were found to be linked to APOE-ε4 homozygosity. The relationships were not related to the burden of the cerebrovascular system and may be explained by increased neurodegenerative disease or damage susceptibility [58].
5. Interplay of Leukotriene B4 Receptor 1 (BLT1) and RAGE in Ischemic Stroke
The potent, short-lived, pro-inflammatory lipid mediators Leukotrienes (LTs) are expressed in macrophages, neutrophils, and mast cells. LTs have a part to play in cerebrovascular disorders, as well as innate immune responses. They are reported to have roles in the pathogenesis of ischemic stroke and other atherosclerosis processes in the cardio-cerebral vasculature [23,24]. Leukotriene B4 (LTB4) is a group of LTs with the specific receptor BLT1 [23]. Early and sustained increases in LTB4 levels have been associated with poor clinical outcomes in ischemic stroke patients [25,59].
Leukotriene B4 receptor 1 (BLT1), a G protein-coupled receptor (GPCR) with a high affinity for LTB4,has important roles in inflammatory and immune reactions. The LTB4–BLT1 axis was found to enhance inflammation, but the binding proteins that modulate LTB4–BLT1 signalling were unidentified [23,24]. Recently, RAGE was shown to be a BLT1-binding protein that regulated BLT1 signalling. RAGE acts as a molecular switch for BLT1 by inhibiting BLT1-dependent activation of NF-κB and stimulating BLT1-dependent chemotaxis. RAGE was identified as a new class of GPCR modulator because of its capacity to bind to GPCRs other than BLT1 [23,24]. Due to these recent findings on leukotriene receptors, they are currently viewed as new drug targets [59,60].
6. Neurotoxicity of AGEs Demonstrated by Animal and Human Models
The proper functioning of both glial cells and neurons in the grey matter of the brain is of paramount importance in preventing neurodegenerative diseases. AGEs were reported to have neurotoxicity-potentiating effects causing brain damage during diabetic ischemic stroke in an animal study [22]. The maximal neurotoxicity window was within a few hours after AGE exposure with downregulation. Aminoguanidine (AG), a dicarbonyl scavenger and AGE crosslink breaker/inhibitor, attenuated the infarct volume in AGE-treated animals [22]. Among the TAGEs, AA-AGE and AGE-2 appeared to be particularly neurotoxic [20]. In cortical neuronal cells, AGE-2 exhibited neurotoxic effects, which were more potent than that of CML and Glu-AGEs. The neurotoxic effects of serum AGE fractions were attenuated entirely by anti-GA-AGE antibody addition but not by other types of AGE antibodies in another line of evidence wherein GA-AGE involvement in neurodegeneration was shown [20,22]. Assessment of the effects of AGE-2 and AGE-3 on cultured Schwann cells in the white matter of the brain showed that the production and proliferation of proinflammatory cytokines and cell viability were significantly affected by TAGE treatments [20,61].
The mechanisms for AGE neurotoxicity are possibly related to nitric oxide and polyamine metabolisms [22]. Excessive production of nitric oxide (NO) by brain parenchyma is a characteristic of ischemic stroke [62,63,64]. Endothelium-derived NO bioavailability and activity are decreased by AGEs. This effect of AGEs on NO may be significant to atherogenesis since NO inhibits several of the processes that lead to atherosclerosis, including leukocyte adherence to the artery wall, vascular smooth muscle development, and platelet adhesion-aggregation. The antiproliferative actions of NO are inhibited by matrix-bound and soluble AGEs [62,63,64]. Impaired vasodilation in diabetes may be a result of the reduction in NO activity by AGEs [65].By speeding up mRNA degradation and decreasing endothelial NO synthase (eNOS) activity, AGEs shorten the half-life of eNOS mRNA. Through the binding of CML to endothelial RAGE, AGEs hinder NO generation by reducing the phosphorylation of serine residues in eNOS, which deactivates the enzyme [62,63,64]. AGEs may also quench and inactivate NO produced by the endothelium [65]. The flavanoid Quercetin greatly inhibited total AGEs and high-molecular-weight AGE in vitro [66]. The highly favorable therapeutic potential of quercetin and its analogues against NO synthase from natural sources for ischemic stroke treatment has been reported recently using molecular docking analysis scores involving ligand–protein interactions [67].
There is a lack of clarity on in vivo mechanisms directly linking AGEs to polyamine metabolism in diabetic ischemic stroke. However, in diabetes mellitus, in vitro formation of the AGE Pyralline was inhibited by polyamines–spermine, spermidine, and L-Arginine, the biological precursor of NO [64]. Polyamine oxidation is enhanced in cerebral ischemia and the production of their toxic metabolites may enhance neuronal death [68]. Therefore, the AGEs polyamine and NO are possibly linked by unknown mechanisms. Polyamine mechanisms have been described as possible pathomechanisms in diabetic ischemic stroke. The polyamine process in eukaryotes produces spermidine, which triggers the hypusination of eukaryotic initiation factor 5A (eIF5A), a translation factor. Inhibition of the eIF5A hypusination pathway is a new pharmacological target for stroke therapy [69]. The polyamine inhibitor was also reported to ameliorate brain infarction size in a mouse model [70].
The multiple molecular mechanisms involving the AGE–RAGE axis and other pathomechanisms involved in diabetic ischemic stroke are represented in Figure 1.
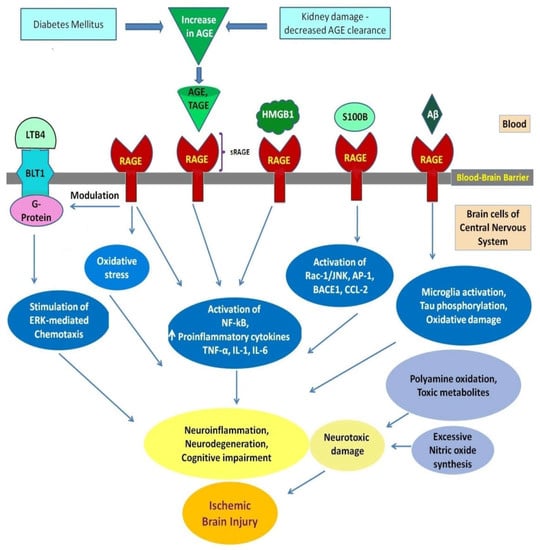
Figure 1.
AGE–RAGE, G-protein coupled Leukotriene B4 receptor BLT-mediated molecular and pathological mechanisms in neurons, astrocytes, microglia, endothelial cells, and blood–brain barrier of central nervous system in diabetic ischemic stroke: (1) Modulation of G protein-coupled Leukotriene B4 (LTB4) receptor BLT1 by RAGE. (2) Binding of AGE to RAGE. sRAGE, Soluble RAGE—the extracellular/cytosolic domain of RAGE. (3) Binding of RAGE with ligands: AGE/TAGE, Advanced Glycation End Product/Toxic Advanced Glycation End Product; extracellular HMGB1, High-Mobility Group Box 1 protein released from necrotic cells; S100B protein from S100/Calgranulin family of proteins; Aβ, Amyloid beta peptides. (4) Molecular, pathological mechanisms involving ERK, the Extracellular-Signal-Regulated Kinase; NF-κB, Nuclear factor kappa-light-chain-enhancer; TNF-α, Tumor Necrosis Factor-α; IL-1, IL- 6, Interleukins 1,6; Rac-1/JNK, Rac-1 G protein/Jun N-terminal kinase; BACE1, Beta-site APP Cleaving Enzyme 1; AP-1, Activation Protein 1 and CCL2, Chemokine (C-C motif) ligand 2.
7. Brain–Kidney Organ Crosstalk, Renal Dysfunction, and Plasma AGEs
The brain and kidneys are inextricably linked through complex interactions and shared risk factors such as diabetes and hypertension [26,27]. Knowing how the brain and kidney communicate bidirectionally after cerebral injury will have a practical impact on therapeutic strategies for improving stroke patient outcomes. Post-stroke brain–kidney crosstalk has been suggested in both experimental and observational studies. After a stroke, the roles of the central autonomic network, autoregulation, inflammatory and immunological responses, extracellular vesicles, and their cargo microRNA have been studied [26]. Yet, current evidence is mainly associative, and evidence on mechanisms underlying direct brain–kidney interaction is limited. Patients with type 2 diabetes who also had early chronic kidney disease (CKD) in deteriorating stages had a greater risk of incident stroke [71]. Renal dysfunction influenced stroke prognosis as a low estimated glomerular filtration rate (eGFR) <45 mL/min/1.73 m2 in stroke patients was associated with increased all-cause mortality risk and recurrent stroke [28]. In patients with decreased renal function, fluorescent AGE levels were reportedly elevated due to reduced clearance and increased carbonyl stress. Fluorescent AGE levels were inversely related to eGFR in those studies [72,73,74]. Interestingly, one of those studies also reported the presence of a new class of AGEs called Melibiose-derived glycation products (MAGEs, derived from melibiose, a disaccharide of glucose and galactose), which were significantly higher in patients with hypertension [74]. The quality of dialysis water and infusion fluid during renal replacement therapy modalities influences plasma AGE levels in end-stage renal disease patients. The highest AGE fluorescence and CML levels were found in hemodialysis patients treated with standard dialysis fluid rather than those with ultrapure fluid and hemofiltration [75].
8. Therapeutic Agents and Their Effects against the AGE–RAGE Axis and Other Key Targets in Diabetic Cardio-Cerebrovascular Complications
Several therapeutic agents/drugs, including several investigational/candidate therapeutic agents targeting the crucial molecular mechanisms, have been reported to be effective against diabetic cardio-cerebrovascular complications including ischemic stroke [76,77,78]. They are elaborated on in Table 2, Table 3, Table 4, Table 5 and Table 6. Some of the agents can be obtained from natural sources or diets. Several vitamins, including antioxidant vitamins E and C, which can reduce oxidative stress, have also been studied for their possible antiglycation effects and impact on AGE levels, as detailed in Table 4.
Table 2.
Experimental findings on investigational/candidate therapeutic agents against AGE–RAGE axis in diabetic cardio-cerebrovascular complications.
Table 3.
Experimental and clinical findings on therapeutic drugs against AGE–RAGE axis in diabetic cardio-cerebrovascular complications.
Table 4.
Experimental and clinical findings on vitamins as therapeutic agents against AGEs in diabetic patients with and without renal complications.
Table 5.
Experimental findings on natural compound-based AGE–RAGE inhibitors against diabetic, cardio-, and cerebrovascular complications.
Table 6.
Experimental findings on investigational/candidate therapeutic agents against targets other than AGE–RAGE Axis in ischemic stroke.
Metformin therapy in type 2 diabetes mellitus patients was found to be associated with lower risk of acute ischemic stroke during COVID-19 in a retrospective cohort study [100]. Iminosugars and other sugar derivatives form an additional avenue for therapeutic agent development, which involves the precise modification of carbohydrate structures and the modulation of glycosidase activity. They offer broad therapeutic applications including anti-diabetic and immunosuppressive activities. Iminosugar analogs, which have nitrogen atoms substituted for oxygen atoms in the sugar ring structures, are potent inhibitors of the enzyme glucosidase that catalyzes the hydrolysis of glycosidic bonds and glycosyltransferases that catalyze glycosidic bond formation using sugar donors, in vivo [101,102,103,104,105].
Although there is a host of potential therapeutic agents against diabetes–ischemic stroke conditions, clinical evidence of most of the agents listed in Table 2, Table 3, Table 4, Table 5 and Table 6 is limited. In terms of clinical efficacy and practicality, mixed results were yielded in some of the conducted trials. The limitation of aminoguanidine for clinical usage was that it interfered with important regulatory systems and yielded some toxic side effects. The small size and short follow-up periods are common limitations of most of the clinical trials conducted. Yet, AGE–RAGE antagonists remain interesting therapeutic options for diabetes–stroke-associated complications [76,77,78]. An important factor that remains for consideration in stroke patients is better management of their motor impairments. Therefore, in addition to the employment of suitable molecular therapeutic targets, improved protocols for motoring recovery becomes an important component in the therapeutic strategies for stroke patients.
9. Myokines, Muscle-Organ Crosstalk, Neuromuscular Electrical Stimulation, and Improved Motor Recovery in Stroke Patients
Although the underlying mechanisms are not fully understood, electrical stimulation techniques have demonstrated good therapeutic potential in post-stroke motor rehabilitation. A recently developed promising method for restoring upper limb control and embodiment in stroke patients with long-term deficits combines a carefully calibrated, innovative neuromuscular electrical stimulation (NMES) technology with conventional rehabilitation techniques [106]. NMES induces circulating myokine secretion by repeated skeletal muscle contraction [107]. These recent advances are indicative of the emerging roles of myokines and their therapeutic potential. Evidence shows myokine production from skeletal muscle in response to exercise or neuromuscular electrical stimulation, which allows for crosstalk between the muscle and other organs, including the brain.
Myokines, which are cytokines or other small proteins and peptides produced and released by skeletal muscle, exert autocrine, paracrine, or endocrine effects [108]. The myokines IL-6, Brain-derived neurotrophic factor (BDNF), and Irisin modulate vital processes such as brain neuroplasticity, hypertrophy, angiogenesis, and inflammatory and extracellular matrix regulation with exercise-induced myokines having therapeutic potential for muscle wasting [109]. The potential mechanism for Irisin’s therapeutic effect on vascular complications of diabetes has been investigated. Irisin significantly alleviated inflammasome signaling, AGE-induced oxidative stress, endothelial dysfunction, and increased eNOS and NO production in a dose-dependent manner, thus demonstrating the Irisin–AGE interplay [110]. By increasing levels of BDNF, which guards nerve cells, Irisin can reduce brain damage in ischemic stroke [111]. Low serum Irisin levels were found to be a predictor of poor early functional outcome in ischemic stroke patients [112] adding value to the potential of its development as a prognostic biomarker, as was Chemokine (C-C motif) ligand 2 (CCL2) levels in blood or cerebrospinal fluid, which were found to correlate with the clinical symptoms of stroke patients [113]. Finding new myokines, their specific receptors, and their neuroprotective functions may reveal novel therapeutic targets and biomarkers and open up new avenues for clinical interventions.
10. Plausible Comprehensive Therapeutic Strategies for Improved Management of Diabetic Ischemic Stroke
Diabetes, a well-established risk factor for ischemic stroke, has also been associated with the worsening of short- and long-term outcomes after the stroke event, making diabetes–ischemic stroke a deadly combination. Risk of cardiovascular or non-cardiovascular events, stroke recurrence, and death were at higher rates in patients with diabetes as compared to those without in a large study [114]. Finding an effective recipe of strategies for diabetic ischemic stroke patient management is, therefore, pivotal to combating the associated morbidity–mortality. The extensiveness of molecular mechanisms and the diversity of associated factors and their interplay in diabetic ischemic stroke thathave been described so far plausibly necessitate comprehensiveness in therapeutic strategies to improve patient outcomes. The key underlying target processes to focus on in diabetic ischemic stroke management can be categorized as neuroinflammation, oxidative stress, and improved neuromuscular stimulation for rehabilitation. An improved system for continual blood-level monitoring of promising biomarkers and the administration of inhibitor drugs/nutrients to target key axes are the other components required. Clinical laboratories will need to validate the novel biomarker testing in the current scenario to enable improved monitoring of the patients. Five key therapeutic/neuroprotective components for effective diabetic ischemic stroke management are identified and shown in Figure 2.
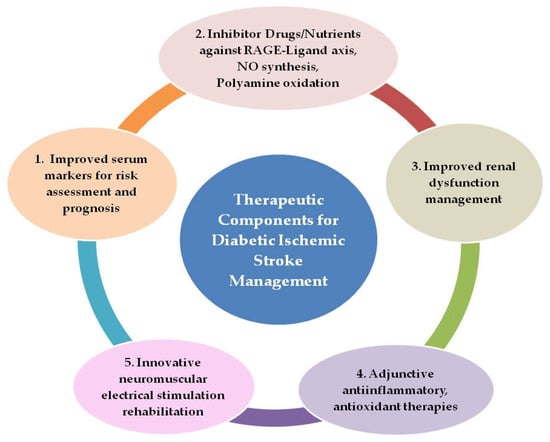
Figure 2.
Components for comprehensive outcome-improvement therapeutic strategy in diabetic ischemic stroke patients.
These five components show potential for application in future therapeutic interventions of diabetic ischemic stroke after the conduction of suitable large-sized clinical trials with longer follow-up periods and appropriate validation:
- Rapid screening of patients for serum AGEs, TAGEs, and risk stratification with point-of-care testing (POCT) devices validated for routine clinical use for monitoring disease progression and treatment effectiveness, as well as the employment of HMGB1 and CCL2 blood levels as prognostic markers for stroke patients.
- Administration of inhibitor drugs and nutrients against RAGE–ligand axes, NO synthesis, and polyamine oxidation as adjunctive therapy along with primary therapy.
- Improved management of renal dysfunction, the use of appropriate dialysate fluid to clear plasma AGEs more efficiently, and the formulation of renal dialysis modalities to improve clearance of low-molecular-weight fluorescent AGEs
- Anti-inflammatory therapy with agents from natural sources such as the plant flavanoid Quercetin or its analogues, which can target HMGB1–RAGE, LTB4–BLT1, and NO synthesis for selective neutralization of pathogenic immune signaling, tissue preservation, and neurological recovery. In addition, antioxidant therapy, which can combat not only the oxidative stress associated with hyperglycemia but also stress due to polyamine oxidation and NO synthesis, offering a key node in the preventive and therapeutic strategies for diabetes-related fatal cardio-cerebrovascular events.
- Neuromuscular stimulation interventions in diabetic ischemic stroke patients with possible employment of myokine Irisin as a biomarker for monitoring the impact of exercise type and amount.
11. Conclusions
Critical reappraisal of the myriad of molecular cascade events involving AGEs and RAGE-ligands reveals the intricate interplay in diabetes–ischemic stroke pathogenesis and resultant poor disease outcomes. AGEs, RAGE–ligand interactions, nitric oxide, and polyamines are pivotal molecular targets in the explication of associated key pathomechanisms. Inflammatory proliferative processes, oxidative damage, and cognitive decline can all be attributed to derangement in these key molecular mechanisms. An effective therapeutic strategy for diabetic ischemic stroke needs comprehensiveness to combat neurodegenerative damage and enhance motor recovery to improve the quality of outcomes in diabetic ischemic stroke patients. Agents to mitigate the AGE–RAGE axis, improved biomarkers for risk stratification, better renal dysfunction management, adjunctive anti-inflammatory–antioxidant therapies, and innovative neuromuscular stimulation for rehabilitation therefore become essential components to feature in the comprehensive therapeutic strategy for diabetic ischemic stroke.
Author Contributions
Conceptualization, design of the study, writing and editing, N.L.R.; interpretation of data, writing, review and editing, N.L.R., B.P.S., J.K.S., M.K.D., S.P.S. and S.D.A.; creation of figures and tables, N.L.R. and J.K.S.; review and editing, G.B.K., S.R.S. and E.C.L. All authors have read and agreed to the published version of the manuscript.
Funding
No external funding was received.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Whiting, D.R.; Guariguata, L.; Weil, C.; Shaw, J. IDF Diabetes Atlas: Global Estimates of the Prevalence of Diabetes for 2011 and 2030. Diabetes Res. Clin. Pract. 2011, 94, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Vos, T.; Abajobir, A.A.; Abbafati, C.; Abbas, K.M.; Abate, K.H.; Abd-Allah, F.; Abdulle, A.M.; Abebo, T.A.; Abera, S.F.; Aboyans, V.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef]
- Kamalakannan, S.; Gudlavalleti, A.S.V.; Gudlavalleti, V.S.M.; Goenka, S.; Kuper, H. Incidence & Prevalence of Stroke in India: A Systematic Review. Indian J. Med. Res. 2017, 146, 175. [Google Scholar] [CrossRef] [PubMed]
- Tran, J.; Mirzaei, M.; Anderson, L.; Leeder, S.R. The Epidemiology of Stroke in the Middle East and North Africa. J. Neurol. Sci. 2010, 295, 38–40. [Google Scholar] [CrossRef] [PubMed]
- Sacco, R.L.; Kasner, S.E.; Broderick, J.P.; Caplan, L.R.; Connors, J.J.B.; Culebras, A.; Elkind, M.S.V.; George, M.G.; Hamdan, A.D.; Higashida, R.T.; et al. An Updated Definition of Stroke for the 21st Century: A Statement for Healthcare Professionals from the American Heart Association/American Stroke Association. Stroke 2013, 44, 2064–2089. [Google Scholar] [CrossRef]
- Boehme, A.K.; Esenwa, C.; Elkind, M.S.V. Stroke Risk Factors, Genetics, and Prevention. Circ. Res. 2017, 120, 472–495. [Google Scholar] [CrossRef]
- Donnan, G.A.; Fisher, M.; Macleod, M.; Davis, S.M. Stroke. Lancet 2008, 371, 1612–1623. [Google Scholar] [CrossRef]
- Adams, H.P.; Bendixen, B.H.; Kappelle, L.J.; Biller, J.; Love, B.B.; Gordon, D.L.; Marsh, E.E. Classification of Subtype of Acute Ischemic Stroke. Definitions for Use in a Multicenter Clinical Trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 1993, 24, 35–41. [Google Scholar] [CrossRef]
- Tuttolomondo, A. Relationship between Diabetes and Ischemic Stroke: Analysis of Diabetes- Related Risk Factors for Stroke and of Specific Patterns of Stroke Associated with Diabetes Mellitus. J. Diabetes Metab. 2015, 6. [Google Scholar] [CrossRef]
- Einarson, T.R.; Acs, A.; Ludwig, C.; Panton, U.H. Prevalence of Cardiovascular Disease in Type 2 Diabetes: A Systematic Literature Review of Scientific Evidence from across the World in 2007–2017. Cardiovasc. Diabetol. 2018, 17. [Google Scholar] [CrossRef]
- Rangel, É.B.; Rodrigues, C.O.; de Sá, J.R. Micro- and Macrovascular Complications in Diabetes Mellitus: Preclinical and Clinical Studies. J. Diabetes Res. 2019, 2019, 2161085. [Google Scholar] [CrossRef]
- Khan, R.; Yee Ooi, X.; Parvus, M.; Valdez, L.; Tsin, A. Advanced Glycation End Products: Formation, Role in Diabetic Complications, and Potential in Clinical Applications. Eye Foot Diabetes 2020. [Google Scholar] [CrossRef]
- Guerin-Dubourg, A.; Cournot, M.; Planesse, C.; Debussche, X.; Meilhac, O.; Rondeau, P.; Bourdon, E. Association between Fluorescent Advanced Glycation End-Products and Vascular Complications in Type 2 Diabetic Patients. BioMed Res. Int. 2017, 2017, 7989180. [Google Scholar] [CrossRef]
- Yamagishi, S.; Matsui, T. Role of Hyperglycemia-Induced Advanced Glycation End Product (AGE) Accumulation in Atherosclerosis. Ann. Vasc. Dis. 2018, 11, 253–258. [Google Scholar] [CrossRef]
- Ahmad, M.N.; Farah, A.I.; Al-Qirim, T.M. The Cardiovascular Complications of Diabetes: A Striking Link through Protein Glycation. Rom. J. Intern. Med. 2020, 58, 188–198. [Google Scholar] [CrossRef]
- Perrone, A.; Giovino, A.; Benny, J.; Martinelli, F. Advanced Glycation End Products (AGEs): Biochemistry, Signaling, Analytical Methods, and Epigenetic Effects. Oxidative Med. Cell. Longev. 2020, 2020, 3818196. [Google Scholar] [CrossRef]
- Takeuchi, M. Serum Levels of Toxic AGEs (TAGE) May Be a Promising Novel Biomarker for the Onset/Progression of Lifestyle-Related Diseases. Diagnostics 2016, 6, 23. [Google Scholar] [CrossRef]
- Takeuchi, M.; Takino, J.; Yamagishi, S. Involvement of the Toxic AGEs (TAGE)-RAGE System in the Pathogenesis of Diabetic Vascular Complications: A Novel Therapeutic Strategy. Curr. Drug Targets 2010, 11, 1468–1482. [Google Scholar] [CrossRef]
- Sato, T.; Shimogaito, N.; Wu, X.; Kikuchi, S.; Yamagishi, S.; Takeuchi, M. Toxic Advanced Glycation End Products (TAGE) Theory in Alzheimer’s Disease. Am. J. Alzheimer’s Dis. Other Dement. 2006, 21, 197–208. [Google Scholar] [CrossRef]
- Kuzan, A. Toxicity of Advanced Glycation End Products (Review). Biomed. Rep. 2021, 14. [Google Scholar] [CrossRef]
- Takeuchi, M.; Sakasai-Sakai, A.; Takata, T.; Takino, J.; Koriyama, Y.; Kikuchi, C.; Furukawa, A.; Nagamine, K.; Hori, T.; Matsunaga, T. Intracellular Toxic AGEs (TAGE) Triggers Numerous Types of Cell Damage. Biomolecules 2021, 11, 387. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, G.A.; Meistrell, M.; Bloom, O.; Cockroft, K.M.; Bianchi, M.; Risucci, D.; Broome, J.; Farmer, P.; Cerami, A.; Vlassara, H. Neurotoxicity of Advanced Glycation End products during Focal Stroke and Neuroprotective Effects of Aminoguanidine. Proc. Natl. Acad. Sci. USA 1995, 92, 3744–3748. [Google Scholar] [CrossRef] [PubMed]
- Ichiki, T.; Koga, T.; Okuno, T.; Saeki, K.; Yamamoto, Y.; Yamamoto, H.; Sakaguchi, M.; Yokomizo, T. Modulation of Leukotriene B4Receptor 1 Signaling by Receptor for Advanced Glycation End Products (RAGE). FASEB J. 2016, 30, 1811–1822. [Google Scholar] [CrossRef] [PubMed]
- Ichiki, T.; Koga, T.; Yokomizo, T. Receptor for Advanced Glycation End Products Regulates Leukotriene B4 Receptor 1 Signaling. DNA Cell Biol. 2016, 35, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.J.; Ng, M.P.E.; Zhao, H.; Ng, G.J.L.; De Foo, C.; Wong, P.T.-H.; Seet, R.C.S. Early and Sustained Increases in Leukotriene B4 Levels Are Associated with Poor Clinical Outcome in Ischemic Stroke Patients. Neurotherapeutics 2019, 17, 282–293. [Google Scholar] [CrossRef]
- Zhao, Q.; Yan, T.; Chopp, M.; Venkat, P.; Chen, J. Brain-Kidney Interaction: Renal Dysfunction Following Ischemic Stroke. J. Cereb. Blood Flow Metab. 2020, 40, 246–262. [Google Scholar] [CrossRef]
- Shah, B.; Jagtap, P.; Sarmah, D.; Datta, A.; Raut, S.; Sarkar, A.; Bohra, M.; Singh, U.; Baidya, F.; Kalia, K.; et al. Cerebro-Renal Interaction and Stroke. Eur. J. Neurosci. 2020, 53, 1279–1299. [Google Scholar] [CrossRef]
- Liu, H.; Zheng, H.; Wu, P.; Liu, C.-F.; Wang, D.; Li, H.; Meng, X.; Wang, Y.; Cao, Y.; Wang, Y.; et al. Estimated Glomerular Filtration Rate, Anemia and Outcomes in Patients with Ischemic Stroke. Ann. Transl. Med. 2020, 8, 2. [Google Scholar] [CrossRef]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced Glycoxidation and Lipoxidation End Products (AGEs and ALEs): An Overview of Their Mechanisms of Formation. Free. Radic. Res. 2013, 47, 3–27. [Google Scholar] [CrossRef]
- Sajithlal, G.B.; Chithra, P.; Chandrakasan, G. Advanced Glycation End Products Induce Crosslinking of Collagen in Vitro. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 1998, 1407, 215–224. [Google Scholar] [CrossRef]
- Münch, G.; Keis, R.; Weßels, A.; Riederer, P.; Bahner, U.; Heidland, A.; Niwa, T.; Lemke, H.-D.; Schinzel, R. Determination of Advanced Glycation End Products in Serum by Fluorescence Spectroscopy and Competitive ELISA. Clin. Chem. Lab. Med. 1997, 35. [Google Scholar] [CrossRef]
- MohdAshraf, J.; Ahmad, S.; Choi, I.; Ahmad, N.; Farhan, M.; Tatyana, G.; Shahab, U. Recent Advances in Detection of AGEs: Immunochemical, Bioanalytical and Biochemical Approaches. IUBMB Life 2015, 67, 897–913. [Google Scholar] [CrossRef]
- Leńska-Mieciek, M.; Korczak-Kowalska, G.; Bocian, K.; Fiszer, U. Pentosidine, Advanced Glycation End Product, in Acute Ischaemic Stroke Patients with and without Atrial Rhythm Disturbances. Neurol. Neurochir. Pol. 2020, 54, 323–328. [Google Scholar] [CrossRef]
- Filipov, A.; Fuchshuber, H.; Kraus, J.; Ebert, A.D.; Sandikci, V.; Alonso, A. Measuring of Advanced Glycation End Products in Acute Stroke Care: Skin Autofluorescence as a Predictor of Ischemic Stroke Outcome in Patients with Diabetes Mellitus. J. Clin. Med. 2022, 11, 1625. [Google Scholar] [CrossRef]
- Cheng, C.; Tsuneyama, K.; Kominami, R.; Shinohara, H.; Sakurai, S.; Yonekura, H.; Watanabe, T.; Takano, Y.; Yamamoto, H.; Yamamoto, Y. Expression Profiling of Endogenous Secretory Receptor for Advanced Glycation End Products in Human Organs. Mod. Pathol. 2005, 18, 1385–1396. [Google Scholar] [CrossRef][Green Version]
- Zhang, T.; Pan, B.-S.; Sun, G.-C.; Sun, X.; Sun, F.-Y. Diabetes Synergistically Exacerbates Poststroke Dementia and Tau Abnormality in Brain. Neurochem. Int. 2010, 56, 955–961. [Google Scholar] [CrossRef]
- Tang, S.C.; Wang, Y.C.; Li, Y.I.; Li, H.C.; Manzanero, S.; Hsieh, Y.H.; Phipps, S.; Hu, C.J.; Chiou, H.Y.; Huang, Y.S.; et al. Functional role of soluble receptor for advanced glycation end products in stroke. Arter. Thromb. Vasc. Biol. 2013, 33, 585–594. [Google Scholar] [CrossRef]
- Arancio, O.; Zhang, H.P.; Chen, X.; Lin, C.; Trinchese, F.; Puzzo, D.; Liu, S.; Hegde, A.; Yan, S.F.; Stern, A.; et al. Rage Potentiates Aβ-Induced Perturbation of Neuronal Function in Transgenic Mice. EMBO J. 2004, 23, 4096–4105. [Google Scholar] [CrossRef]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the Receptor for Advanced Glycation End Products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Zhai, D.-X.; Kong, Q.-F.; Xu, W.-S.; Bai, S.-S.; Peng, H.-S.; Zhao, K.; Li, G.-Z.; Wang, D.-D.; Sun, B.; Wang, J.-H.; et al. Rage Expression Is up-Regulated in Human Cerebral Ischemia and pMCAO Rats. Neurosci. Lett. 2008, 445, 117–121. [Google Scholar] [CrossRef]
- Willeit, P.; Tschiderer, L.; Allara, E.; Reuber, K.; Seekircher, L.; Gao, L.; Liao, X.; Lonn, E.; Gerstein, H.C.; Yusuf, S.; et al. Carotid intima-media thickness progression as surrogate marker for cardiovascular risk: Meta-analysis of 119 clinical trials involving 100 667 patients. Circulation 2020, 142, 621–642. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, Y.; Ohkubo, T. Hypertension with Diabetes Mellitus: Significance from an Epidemiological Perspective for Japanese. Hypertens. Res. 2017, 40, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, M. Hypertension with Diabetes Mellitus: Physiology and Pathology. Hypertens. Res. 2018, 41, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.; Mishra, M.; Tiwari, A.; Bisen, P.S.; Goswamy, H.M.; Prasad, G.B.K.S. Prevalence of Dyslipidemia and Hypertension in Indian Type 2 Diabetic Patients with Metabolic Syndrome and Its Clinical Significance. Osong Public Health Res. Perspect. 2014, 5, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Liu, B.; Zhao, Q.; Jin, P.; Hua, F.; Zhang, Z.; Liu, Y.; Zan, K.; Cui, G.; Ye, X. Bone Marrow Stromal Cells Inhibits HMGB1-Mediated Inflammation after Stroke in Type 2 Diabetic Rats. Neuroscience 2016, 324, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Richard Seidu, A.; Sackey, M.; Su, Z.; Xu, H. Pivotal Neuroinflammatory and Therapeutic Role of High Mobility Group Box 1 in Ischemic Stroke. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef]
- Huang, J.; Liu, B.; Yang, C.; Chen, H.; Eunice, D.; Yuan, Z. Acute Hyperglycemia Worsens Ischemic Stroke-Induced Brain Damage via High Mobility Group Box-1 in Rats. Brain Res. 2013, 1535, 148–155. [Google Scholar] [CrossRef]
- Carty, M.; Bowie, A.G. Evaluating the Role of Toll-like Receptors in Diseases of the Central Nervous System. Biochem. Pharmacol. 2011, 81, 825–837. [Google Scholar] [CrossRef]
- Ding, Q.; Keller, J.N. Evaluation of Rage Isoforms, Ligands, and Signaling in the Brain. Biochim. Et Biophys. Acta (BBA) Mol. Cell Res. 2005, 1746, 18–27. [Google Scholar] [CrossRef]
- Ye, Y.; Zeng, Z.; Jin, T.; Zhang, H.; Xiong, X.; Gu, L. The Role of High Mobility Group Box 1 in Ischemic Stroke. Front. Cell. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Shichita, T.; Sugiyama, Y.; Ooboshi, H.; Sugimori, H.; Nakagawa, R.; Takada, I.; Iwaki, T.; Okada, Y.; Iida, M.; Cua, D.J.; et al. Pivotal Role of Cerebral Interleukin-17–Producing ΓδT Cells in the Delayed Phase of Ischemic Brain Injury. Nat. Med. 2009, 15, 946–950. [Google Scholar] [CrossRef]
- Muhammad, S.; Barakat, W.; Stoyanov, S.; Murikinati, S.; Yang, H.; Tracey, K.J.; Bendszus, M.; Rossetti, G.; Nawroth, P.P.; Bierhaus, A.; et al. The HMGB1 Receptor RAGE Mediates Ischemic Brain Damage. J. Neurosci. 2008, 28, 12023–12031. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Blood–Brain Barrier and Neurovascular Mechanisms of Neurodegeneration and Injury. Encycl. Neurosci. 2009, 265–271. [Google Scholar] [CrossRef]
- Horie, K.; Miyata, T.; Yasuda, T.; Takeda, A.; Yasuda, Y.; Maeda, K.; Sobue, G.; Kurokawa, K. Immunohistochemical Localization of Advanced Glycation End Products, Pentosidine, and Carboxymethyllysine in Lipofuscin Pigments of Alzheimer’s Disease and Aged Neurons. Biochem. Biophys. Res. Commun. 1997, 236, 327–332. [Google Scholar] [CrossRef]
- Sasaki, N.; Fukatsu, R.; Tsuzuki, K.; Hayashi, Y.; Yoshida, T.; Fujii, N.; Koike, T.; Wakayama, I.; Yanagihara, R.; Garruto, R.; et al. Advanced Glycation End Products in Alzheimer’s Disease and Other Neurodegenerative Diseases. Am. J. Pathol. 1998, 153, 1149–1155. [Google Scholar] [CrossRef]
- Li, X.-H.; Du, L.-L.; Cheng, X.-S.; Jiang, X.; Zhang, Y.; Lv, B.-L.; Liu, R.; Wang, J.-Z.; Zhou, X.-W. Glycation Exacerbates the Neuronal Toxicity of β-Amyloid. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef]
- Chen, J.; Mooldijk, S.S.; Licher, S.; Waqas, K.; Ikram, M.K.; Uitterlinden, A.G.; Zillikens, M.C.; Ikram, M.A. Assessment of Advanced Glycation End Products and Receptors and the Risk of Dementia. JAMA Netw. Open 2021, 4, e2033012. [Google Scholar] [CrossRef]
- Pendlebury, S.T.; Poole, D.; Burgess, A.; Duerden, J.; Rothwell, P.M. APOE-ε4 Genotype and Dementia before and after Transient Ischemic Attack and Stroke. Stroke 2020, 51, 751–758. [Google Scholar] [CrossRef]
- Hao, J.; Feng, Y.; Xu, X.; Li, L.; Yang, K.; Dai, G.; Gao, W.; Zhang, M.; Fan, Y.; Yin, T.; et al. Plasma Lipid Mediators Associate with Clinical Outcome after Successful Endovascular Thrombectomy in Patients with Acute Ischemic Stroke. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef]
- Yokomizo, T.; Nakamura, M.; Shimizu, T. Leukotriene Receptors as Potential Therapeutic Targets. J. Clin. Investig. 2018, 128, 2691–2701. [Google Scholar] [CrossRef]
- Takeuchi, M.; Yamagishi, S. TAGE (Toxic AGEs) Hypothesis in Various Chronic Diseases. Med. Hypotheses 2004, 63, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced Glycation End Products. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Q.; Mou, R.T.; Feng, D.X.; Wang, Z.; Chen, G. The role of nitric oxide in stroke. Med. Gas Res. 2017, 7, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Wierońska, J.M.; Cieślik, P.; Kalinowski, L. Nitric Oxide-dependent pathways as critical factors in the consequences and recovery after brain ischemic hypoxia. Biomolecules 2021, 11, 1097. [Google Scholar] [CrossRef]
- Bucala, R.; Tracey, K.J.; Cerami, A. Advanced Glycosylation Products Quench Nitric Oxide and Mediate Defective Endothelium-Dependent Vasodilatation in Experimental Diabetes. J. Clin. Investig. 1991, 87, 432–438. [Google Scholar] [CrossRef]
- Grzebyk, E.; Piwowar, A. Inhibitory Actions of Selected Natural Substances on Formation of Advanced Glycation Endproducts and Advanced Oxidation Protein Products. BMC Complement. Altern. Med. 2016, 16. [Google Scholar] [CrossRef]
- Alshehri, B.; Vijayakumar, R.; Senthilkumar, S.; Ismail, A.; Abdel-Hadi, A.; Choudhary, R.K.; Albenasy, K.S.; Banawas, S.; Alaidarous, M.A.; Manikandan, P. Therapeutic Potential of Nitric Oxide Synthase Inhibitor from Natural Sources for the Treatment of Ischemic Stroke. Saudi J. Biol. Sci. 2022, 29, 984–991. [Google Scholar] [CrossRef]
- Murray, S.T.; Dunston, T.T.; Woster, P.M.; Casero, R.A. Polyamine Catabolism and Oxidative Damage. J. Biol. Chem. 2018, 293, 18736–18745. [Google Scholar] [CrossRef]
- Bourourou, M.; Gouix, E.; Melis, N.; Friard, J.; Heurteaux, C.; Tauc, M.; Blondeau, N. Inhibition of EIF5A Hypusination Pathway as a New Pharmacological Target for Stroke Therapy. J. Cereb. Blood Flow Metab. 2020, 41, 1080–1090. [Google Scholar] [CrossRef]
- Masuko, T.; Takao, K.; Samejima, K.; Shirahata, A.; Igarashi, K.; Casero, R.A.; Kizawa, Y.; Sugita, Y. N1-Nonyl-1,4-Diaminobutane Ameliorates Brain Infarction Size in Photochemically Induced Thrombosis Model Mice. Neurosci. Lett. 2018, 672, 118–122. [Google Scholar] [CrossRef]
- Kaze, A.D.; Jaar, B.G.; Fonarow, G.C.; Echouffo-Tcheugui, J.B. Diabetic Kidney Disease and Risk of Incident Stroke among Adults with Type 2 Diabetes. BMC Med. 2022, 20. [Google Scholar] [CrossRef] [PubMed]
- Kratochvilová, M.; Zakiyanov, O.; Kalousová, M.; Kříha, V.; Zima, T.; Tesař, V. Associations of Serum Levels of Advanced. Glycation End Products with Nutrition Markers and Anemia in Patients with Chronic Kidney Disease. Ren. Fail. 2011, 33, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Dozio, E.; Vettoretti, S.; Caldiroli, L.; Nerini-Molteni, S.; Tacchini, L.; Ambrogi, F.; Messa, P.; Corsi Romanelli, M.M. Advanced Glycation End Products (AGE) and Soluble Forms of AGE Receptor: Emerging Role as Mortality Risk Factors in CKD. Biomedicines 2020, 8, 638. [Google Scholar] [CrossRef] [PubMed]
- Indyk, D.; Bronowicka-Szydełko, A.; Gamian, A.; Kuzan, A. Advanced Glycation End Products and Their Receptors in Serum of Patients with Type 2 Diabetes. Sci. Rep. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Gerdemann, A.; Wagner, Z.; Solf, A.; Bahner, U.; Heidland, A.; Vienken, J.; Schinzel, R. Plasma levels of advanced glycation end products during haemodialysis, haemodiafiltration and haemofiltration: Potential importance of dialysate quality. Nephrol. Dial. Transplant. 2002, 17, 1045–1049. [Google Scholar] [CrossRef][Green Version]
- Hong Sheng, C.; Ha, T.S.; Khalid, A.K. Therapeutic Agents Targeting at AGE-RAGE Axis for the Treatment of Diabetes and Cardiovascular Disease: A Review of Clinical Evidence. Clin. Diabetes Res. 2017, 1. [Google Scholar] [CrossRef]
- Yang, P.; Feng, J.; Peng, Q.; Liu, X.; Fan, Z. Advanced Glycation End Products: Potential Mechanism and Therapeutic Target in Cardiovascular Complications under Diabetes. Oxid. Med. Cell. Longev. 2019, 2019, 9570616. [Google Scholar] [CrossRef]
- Bongarzone, S.; Savickas, V.; Luzi, F.; Gee, A.D. Targeting the Receptor for Advanced Glycation Endproducts (RAGE): A Medicinal Chemistry Perspective. J. Med. Chem. 2017, 60, 7213–7232. [Google Scholar] [CrossRef]
- Forbes, J.M.; Yee, L.T.L.; Thallas, V.; Lassila, M.; Candido, R.; Jandeleit-Dahm, K.A.; Thomas, M.C.; Burns, W.C.; Deemer, E.K.; Thorpe, S.R.; et al. Advanced Glycation End Product Interventions Reduce Diabetes-Accelerated Atherosclerosis. Diabetes 2004, 53, 1813–1823. [Google Scholar] [CrossRef]
- Ha, C.H.; Kim, S.; Chung, J.; An, S.H.; Park, S.; Choi, D.; Kwon, K. Inhibitory effect of soluble RAGE in disturbed flow-induced atherogenesis. Int. J. Mol. Med. 2013, 32, 373–380. [Google Scholar] [CrossRef]
- Shimizu, Y.; Harashima, A.; Munesue, S.; Oishi, M.; Hattori, T.; Hori, O.; Kitao, Y.; Yamamoto, H.; Leerach, N.; Nakada, M.; et al. Neuroprotective Effects of Endogenous Secretory Receptor for Advanced Glycation End-Products in Brain Ischemia. Aging Dis. 2020, 11, 547. [Google Scholar] [CrossRef]
- Mirmiranpour, H.; Mousavizadeh, M.; Noshad, S.; Ghavami, M.; Ebadi, M.; Ghasemiesfe, M.; Nakhjavani, M.; Esteghamati, A. Comparative Effects of Pioglitazone and Metformin on Oxidative Stress Markers in Newly Diagnosed Type 2 Diabetes Patients: A Randomized Clinical Trial. J. Diabetes Complicat. 2013, 27, 501–507. [Google Scholar] [CrossRef]
- Sakata, K.; Hayakawa, M.; Yano, Y.; Tamaki, N.; Yokota, N.; Eto, T.; Watanabe, R.; Hirayama, N.; Matsuo, T.; Kuroki, K.; et al. Efficacy of alogliptin, a dipeptidyl peptidase-4 inhibitor, on glucose parameters, the activity of the advanced glycation end product (AGE)—Receptor for AGE (RAGE) axis and albuminuria in Japanese type 2 diabetes. Diabetes Metab. Res. Rev. 2013, 29, 624–630. [Google Scholar] [CrossRef]
- Jinnouchi, Y.; Yamagishi, S.; Takeuchi, M.; Ishida, S.; Jinnouchi, Y.; Jinnouchi, J.; Imaizumi, T. Atorvastatin Decreases Serum Levels of Advanced Glycation End Products (AGEs) in Patients with Type 2 Diabetes. Clin. Exp. Med. 2006, 6, 191–193. [Google Scholar] [CrossRef]
- Kawai, T.; Takei, I.; Tokui, M.; Funae, O.; Miyamoto, K.; Tabata, M.; Hirata, T.; Saruta, T.; Shimada, A.; Itoh, H. Effects of Epalrestat, an Aldose Reductase Inhibitor, on Diabetic Peripheral Neuropathy in Patients with Type 2 Diabetes, in Relation to Suppression of Nɛ-Carboxymethyl Lysine. J. Diabetes Its Complicat. 2010, 24, 424–432. [Google Scholar] [CrossRef]
- Contreras, C.L.; Guzman-Rosiles, I.; Del Castillo, D.; Gomez-Ojeda, A.; Garay-Sevilla, M.E. Advanced glycation end products (AGEs) and sRAGE levels after benfotiamine treatment in diabetes mellitus type 2. FASEB J. 2017, 31, 646.32. [Google Scholar] [CrossRef]
- Williams, M.E.; Bolton, W.K.; Khalifah, R.G.; Degenhardt, T.P.; Schotzinger, R.J.; McGill, J.B. Effects of Pyridoxamine in Combined Phase 2 Studies of Patients with Type 1 and Type 2 Diabetes and Overt Nephropathy. Am. J. Nephrol. 2007, 27, 605–614. [Google Scholar] [CrossRef]
- Noori, N.; Tabibi, H.; Hosseinpanah, F.; Hedayati, M.; Nafar, M. Effects of Combined Lipoic Acid and Pyridoxine on Albuminuria, Advanced Glycation End-Products, and Blood Pressure in Diabetic Nephropathy. Int. J. Vitam. Nutr. Res. 2013, 83, 77–85. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, W.; Jia, Q.; Feng, Z.; Guo, J.; Han, X.; Liu, Y.; Shang, H.; Wang, Y.; Liu, W.J. High Dose Vitamin E Attenuates Diabetic Nephropathy via Alleviation of Autophagic Stress. Front. Physiol. 2019, 9, 1939. [Google Scholar] [CrossRef]
- Zafar, H.; Sheikh, M.A.; Hussain, F.; Maan, M.A. Inhibition of protein glycation and advanced glycation end products by ascorbic acid. Afr. J. Biotechnol. 2012, 11, 11309–11314. [Google Scholar] [CrossRef]
- Umadevi, S.; Gopi, V.; Vellaichamy, E. Inhibitory Effect of Gallic Acid on Advanced Glycation End Products Induced Up-Regulation of Inflammatory Cytokines and Matrix Proteins in H9C2 (2-1) Cells. Cardiovasc. Toxicol. 2013, 13, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, L.-S.; Dong, L.-L. Inhibitory Effect of Polysaccharides from Pumpkin on Advanced Glycation End-Products Formation and Aldose Reductase Activity. Food Chem. 2012, 130, 821–825. [Google Scholar] [CrossRef]
- Rao, A.R.; Veeresham, C.; Asres, K. In vitro and in vivo inhibitory activities of four indian medicinal plant extracts and their major components on rat aldose reductase and generation of advanced glycation endproducts. Phytother. Res. 2013, 27, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Ni, M.; Zhang, G.; Liao, Y.; Hu, X.; Zhang, Y.; Gong, D. The Inhibition of Oleanolic Acid on Protein Non-Enzymatic Glycation. LWT 2020, 125, 109253. [Google Scholar] [CrossRef]
- Wu, D.; Wen, W.; Qi, C.-L.; Zhao, R.-X.; Lü, J.-H.; Zhong, C.-Y.; Chen, Y.-Y. Ameliorative Effect of Berberine on Renal Damage in Rats with Diabetes Induced by High-Fat Diet and Streptozotocin. Phytomedicine 2012, 19, 712–718. [Google Scholar] [CrossRef]
- Bae, O.-N.; Serfozo, K.; Baek, S.-H.; Lee, K.Y.; Dorrance, A.; Rumbeiha, W.; Fitzgerald, S.D.; Farooq, M.U.; Naravelta, B.; Bhatt, A.; et al. Safety and Efficacy Evaluation of Carnosine, an Endogenous Neuroprotective Agent for Ischemic Stroke. Stroke 2013, 44, 205–212. [Google Scholar] [CrossRef]
- Agustin, A.; Safitri, A.; Fatchiyah, F. An in Silico Approach Reveals the Potential Function of Cyanidin-3-o-Glucoside of Red Rice in Inhibiting the Advanced Glycation End Products (AGES)-Receptor (RAGE) Signaling Pathway. Acta Inform. Med. 2020, 28, 170. [Google Scholar] [CrossRef]
- Ivanova, S.; Batliwalla, F.; Mocco, J.; Kiss, S.; Huang, J.; Mack, W.; Coon, A.; Eaton, J.W.; Al-Abed, Y.; Gregersen, P.K.; et al. Neuroprotection in cerebral ischemia by neutralization of 3-aminopropanal. PNAS 2002, 99, 5579–5584. [Google Scholar] [CrossRef]
- Danielisova, V.; Nemethova, M.; Burda, J. The Protective Effect of Aminoguanidine on Cerebral Ischemic Damage in the Rat Brain. Physiol. Res. 2004, 53, 533–540. [Google Scholar]
- Al-kuraishy, H.M.; Al-Gareeb, A.I.; Alblihed, M.; Cruz-Martins, N.; Batiha, G.E.-S. COVID-19 and Risk of Acute Ischemic Stroke and Acute Lung Injury in Patients with Type II Diabetes Mellitus: The Anti-Inflammatory Role of Metformin. Front. Med. 2021, 8, 644295. [Google Scholar] [CrossRef]
- Nash, R.J.; Kato, A.; Yu, C.-Y.; Fleet, G.W. Iminosugars as Therapeutic Agents: Recent Advances and Promising Trends. Future Med. Chem. 2011, 3, 1513–1521. [Google Scholar] [CrossRef]
- Yang, L.-F.; Shimadate, Y.; Kato, A.; Li, Y.-X.; Jia, Y.-M.; Fleet, G.W.J.; Yu, C.-Y. Synthesis and Glycosidase Inhibition of N-Substituted Derivatives of 1,4-Dideoxy-1,4-Imino-D-Mannitol (DIM). Org. Biomol. Chem. 2020, 18, 999–1011. [Google Scholar] [CrossRef]
- Chennaiah, A.; Dahiya, A.; Dubbu, S.; Vankar, Y.D. A Stereoselective Synthesis of an IminoGlycal: Application in the Synthesis of (-)-1-Epi -Adenophorine and a Homoimindosugar. Eur. J. Org. Chem. 2019, 2019, 2089. [Google Scholar] [CrossRef]
- Chennaiah, A.; Bhowmick, S.; Vankar, Y.D. Conversion of Glycals into Vicinal-1,2-Diazides and 1,2-(or 2,1)-Azidoacetates Using Hypervalent Iodine Reagents and Me3SiN3. Application in the Synthesis of N-Glycopeptides, Pseudo-Trisaccharides and an Iminosugar. RSC Adv. 2017, 7, 41755–41762. [Google Scholar] [CrossRef]
- Rajasekaran, P.; Ande, C.; Vankar, Y.D. Synthesis of (5,6 & 6,6)-Oxa-Oxa Annulated Sugars as Glycosidase Inhibitors from 2-Formyl Galactal Using Iodocyclization as a Key Step. Arkivoc 2022, 2022, 5–23. [Google Scholar] [CrossRef]
- Crema, A.; Bassolino, M.; Guanziroli, E.; Colombo, M.; Blanke, O.; Serino, A.; Micera, S.; Molteni, F. Neuromuscular Electrical Stimulation Restores Upper Limb Sensory-Motor Functions and Body Representations in Chronic Stroke Survivors. Med 2022, 3, 58–74.e10. [Google Scholar] [CrossRef]
- Sanchis-Gomar, F.; Lopez-Lopez, S.; Romero-Morales, C.; Maffulli, N.; Lippi, G.; Pareja-Galeano, H. Neuromuscular Electrical Stimulation: A New Therapeutic Option for Chronic Diseases Based on Contraction-Induced Myokine Secretion. Front. Physiol. 2019, 10, 1463. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Febbraio, M.A. Muscles, Exercise and Obesity: Skeletal Muscle as a Secretory Organ. Nat. Rev. Endocrinol. 2012, 8, 457–465. [Google Scholar] [CrossRef]
- Piccirillo, R. Exercise-Induced Myokines with Therapeutic Potential for Muscle Wasting. Front. Physiol. 2019, 10, 289. [Google Scholar] [CrossRef]
- Deng, X.; Huang, W.; Peng, J.; Zhu, T.-T.; Sun, X.-L.; Zhou, X.-Y.; Yang, H.; Xiong, J.-F.; He, H.-Q.; Xu, Y.-H.; et al. Irisin Alleviates Advanced Glycation End Products-Induced Inflammation and Endothelial Dysfunction via Inhibiting ROS-NLRP3 Inflammasome Signaling. Inflammation 2017, 41, 260–275. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, C.; Guo, J.; Chen, Y.; Meng, C. The Neuroprotective Effect of Irisin in Ischemic Stroke. Front. Aging Neurosci. 2020, 12, 588958. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Guo, P.; Jin, Z.; Li, X.; Yang, X.; Tang, C.; Wang, Y.; Ke, J. Serum Levels of Irisin Predict Short-Term Outcomes in Ischemic Stroke. Cytokine 2019, 122, 154303. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.; Chen, L.; Tang, J.; Chen, Y.; Wang, L. The Role of CCL2/CCR2 Axis in Cerebral Ischemia-Reperfusion Injury and Treatment: From Animal Experiments to Clinical Trials. Int. J. Mol. Sci. 2022, 23, 3485. [Google Scholar] [CrossRef] [PubMed]
- Echouffo-Tcheugui, J.B.; Xu, H.; Matsouaka, R.A.; Xian, Y.; Schwamm, L.H.; Smith, E.E.; Bhatt, D.L.; Hernandez, A.F.; Heidenreich, P.A.; Fonarow, G.C. Diabetes and Long-Term Outcomes of Ischaemic Stroke: Findings from Get with the Guidelines-Stroke. Eur. Heart J. 2018, 39, 2376–2386. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).