
Calcium-Permeable Channels Cooperation for Rheumatoid Arthritis: Therapeutic Opportunities
 , ,
, ,
Abstract
1. Introduction
2. Pathology of Rheumatoid Arthritis (RA)
3. Acid-Sensitive Ion Channels (ASIC)
3.1. ASIC1a
3.2. ASIC2a
3.3. ASIC3
4. Transient Receptor Potential (TRP) Channels
4.1. TRPM7 Channel
4.2. TRPC Channel
4.3. TRPV Channel
4.3.1. TRPV1
4.3.2. TRPV2
4.3.3. TRPV4
4.3.4. TRPV5
5. P2X7 Receptor (P2X7R)
6. Other Channels
6.1. Orai1 Channel
6.2. Piezo1 Channel
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, D.M.; Weinblatt, M.E. Rheumatoid arthritis. Lancet 2001, 358, 903–911. [Google Scholar] [CrossRef]
- McQueen, F.M.; Stewart, N.; Crabbe, J.; Robinson, E.; Yeoman, S.; Tan, P.L.; McLean, L. Magnetic resonance imaging of the wrist in early rheumatoid arthritis reveals a high prevalence of erosions at four months after symptom onset. Ann. Rheum. Dis. 1998, 57, 350–356. [Google Scholar] [CrossRef]
- McGonagle, D.; Conaghan, P.G.; O’Connor, P.; Gibbon, W.; Green, M.; Wakefield, R.; Ridgway, J.; Emery, P. The relationship between synovitis and bone changes in early untreated rheumatoid arthritis: A controlled magnetic resonance imaging study. Arthritis Rheum. 1999, 42, 1706–1711. [Google Scholar] [CrossRef]
- Brzustewicz, E.; Henc, I.; Daca, A.; Szarecka, M.; Sochocka-Bykowska, M.; Witkowski, J.; Bryl, E. Autoantibodies, C-reactive protein, erythrocyte sedimentation rate and serum cytokine profiling in monitoring of early treatment. Cent. Eur. J. Immunol. 2017, 42, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Smolen, J.S. Diagnosis and Management of Rheumatoid Arthritis: A Review. JAMA 2018, 320, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Sayah, A.; English, J.C., 3rd. Rheumatoid arthritis: A review of the cutaneous manifestations. J. Am. Acad. Dermatol. 2005, 53, 191–209; quiz 210-192. [Google Scholar] [CrossRef]
- Alunno, A.; Gerli, R.; Giacomelli, R.; Carubbi, F. Clinical, Epidemiological, and Histopathological Features of Respiratory Involvement in Rheumatoid Arthritis. Biomed. Res. Int. 2017, 2017, 7915340. [Google Scholar] [CrossRef]
- Schjerning, A.M.; McGettigan, P.; Gislason, G. Cardiovascular effects and safety of (non-aspirin) NSAIDs. Nat. Rev. Cardiol. 2020, 17, 574–584. [Google Scholar] [CrossRef]
- Harirforoosh, S.; Asghar, W.; Jamali, F. Adverse effects of nonsteroidal antiinflammatory drugs: An update of gastrointestinal, cardiovascular and renal complications. J. Pharm. Pharm. Sci. 2013, 16, 821–847. [Google Scholar] [CrossRef]
- Solomon, D.H.; Glynn, R.J.; Karlson, E.W.; Lu, F.; Corrigan, C.; Colls, J.; Xu, C.; MacFadyen, J.; Barbhaiya, M.; Berliner, N.; et al. Adverse Effects of Low-Dose Methotrexate: A Randomized Trial. Ann. Intern. Med. 2020, 172, 369–380. [Google Scholar] [CrossRef]
- Morand, E.F.; McCloud, P.I.; Littlejohn, G.O. Life table analysis of 879 treatment episodes with slow acting antirheumatic drugs in community rheumatology practice. J. Rheumatol. 1992, 19, 704–708. [Google Scholar] [PubMed]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S. Calcium Signaling: From Basic to Bedside. Adv. Exp. Med. Biol. 2020, 1131, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Alm, E.; Bloom, G.D. Cyclic nucleotide involvement in histamine release from mast cells—a reevaluation. Life Sci. 1982, 30, 213–218. [Google Scholar] [CrossRef]
- Ye, Z.; Shen, Y.; Jin, K.; Qiu, J.; Hu, B.; Jadhav, R.R.; Sheth, K.; Weyand, C.M.; Goronzy, J.J. Arachidonic acid-regulated calcium signaling in T cells from patients with rheumatoid arthritis promotes synovial inflammation. Nat. Commun. 2021, 12, 907. [Google Scholar] [CrossRef]
- Liang, H.Y.; Chen, Y.; Wei, X.; Ma, G.G.; Ding, J.; Lu, C.; Zhou, R.P.; Hu, W. Immunomodulatory functions of TRPM7 and its implications in autoimmune diseases. Immunology 2021, 165, 3–21. [Google Scholar] [CrossRef]
- So, S.Y.; Ip, M.; Lam, W.K. Calcium channel blockers and asthma. Lung 1986, 164, 1–16. [Google Scholar] [CrossRef]
- Sueta, D.; Tabata, N.; Hokimoto, S. Clinical roles of calcium channel blockers in ischemic heart diseases. Hypertens. Res. 2017, 40, 423–428. [Google Scholar] [CrossRef]
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H.T. Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 2017, 120, 1969–1993. [Google Scholar] [CrossRef]
- Clem, J.R. Pharmacotherapy of ischemic heart disease. AACN Clin. Issues 1995, 6, 404–417; quiz 493-404. [Google Scholar] [CrossRef]
- Lee, S.; Jo, S.; Talbot, S.; Zhang, H.B.; Kotoda, M.; Andrews, N.A.; Puopolo, M.; Liu, P.W.; Jacquemont, T.; Pascal, M.; et al. Novel charged sodium and calcium channel inhibitor active against neurogenic inflammation. Elife 2019, 8, e48118. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhu, C.J.; Zhu, F.; Dai, B.B.; Song, S.J.; Wang, Z.Q.; Feng, Y.B.; Ge, J.F.; Zhou, R.P.; Chen, F.H. Necrostatin-1 ameliorates adjuvant arthritis rat articular chondrocyte injury via inhibiting ASIC1a-mediated necroptosis. Biochem. Biophys. Res. Commun. 2018, 504, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.P.; Dai, B.B.; Xie, Y.Y.; Wu, X.S.; Wang, Z.S.; Li, Y.; Wang, Z.Q.; Zu, S.Q.; Ge, J.F.; Chen, F.H. Interleukin-1beta and tumor necrosis factor-alpha augment acidosis-induced rat articular chondrocyte apoptosis via nuclear factor-kappaB-dependent upregulation of ASIC1a channel. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Anzaghe, M.; Schulke, S. Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis. Cells 2020, 9, 880. [Google Scholar] [CrossRef]
- Henderson, B.; Revell, P.A.; Edwards, J.C. Synovial lining cell hyperplasia in rheumatoid arthritis: Dogma and fact. Ann. Rheum. Dis. 1988, 47, 348–349. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef]
- Suto, T.; Tosevska, A.; Dalwigk, K.; Kugler, M.; Dellinger, M.; Stanic, I.; Platzer, A.; Niederreiter, B.; Sevelda, F.; Bonelli, M.; et al. TNFR2 is critical for TNF-induced rheumatoid arthritis fibroblast-like synoviocyte inflammation. Rheumatology (Oxford) 2022. [Google Scholar] [CrossRef]
- Luz-Crawford, P.; Hernandez, J.; Djouad, F.; Luque-Campos, N.; Caicedo, A.; Carrere-Kremer, S.; Brondello, J.M.; Vignais, M.L.; Pene, J.; Jorgensen, C. Mesenchymal stem cell repression of Th17 cells is triggered by mitochondrial transfer. Stem Cell Res. Ther. 2019, 10, 232. [Google Scholar] [CrossRef]
- Pap, T.; Muller-Ladner, U.; Gay, R.E.; Gay, S. Fibroblast biology. Role of synovial fibroblasts in the pathogenesis of rheumatoid arthritis. Arthritis Res. 2000, 2, 361–367. [Google Scholar] [CrossRef][Green Version]
- McInnes, I.B.; Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef]
- Saeki, N.; Imai, Y. Reprogramming of synovial macrophage metabolism by synovial fibroblasts under inflammatory conditions. Cell Commun. Signal. 2020, 18, 188. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Gravallese, E. Bone erosion in rheumatoid arthritis: Mechanisms, diagnosis and treatment. Nat. Rev. Rheumatol. 2012, 8, 656–664. [Google Scholar] [CrossRef]
- McGonagle, D.; Tan, A.L.; Moller Dohn, U.; Ostergaard, M.; Benjamin, M. Microanatomic studies to define predictive factors for the topography of periarticular erosion formation in inflammatory arthritis. Arthritis Rheum. 2009, 60, 1042–1051. [Google Scholar] [CrossRef]
- Hayer, S.; Redlich, K.; Korb, A.; Hermann, S.; Smolen, J.; Schett, G. Tenosynovitis and osteoclast formation as the initial preclinical changes in a murine model of inflammatory arthritis. Arthritis Rheum. 2007, 56, 79–88. [Google Scholar] [CrossRef]
- Teitelbaum, S.L. Bone resorption by osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Stolina, M.; Bolon, B.; Middleton, S.; Adlam, M.; Brown, H.; Zhu, L.; Feige, U.; Zack, D.J. Analysis of the kinetics of osteoclastogenesis in arthritic rats. Arthritis Rheum. 2005, 52, 3192–3201. [Google Scholar] [CrossRef] [PubMed]
- Aeberli, D.; Eser, P.; Bonel, H.; Widmer, J.; Caliezi, G.; Varisco, P.A.; Moller, B.; Villiger, P.M. Reduced trabecular bone mineral density and cortical thickness accompanied by increased outer bone circumference in metacarpal bone of rheumatoid arthritis patients: A cross-sectional study. Arthritis Res. Ther. 2010, 12, R119. [Google Scholar] [CrossRef]
- Sharp, J.T.; Tsuji, W.; Ory, P.; Harper-Barek, C.; Wang, H.; Newmark, R. Denosumab prevents metacarpal shaft cortical bone loss in patients with erosive rheumatoid arthritis. Arthritis Care Res. 2010, 62, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Tournis, S.; Samdanis, V.; Psarelis, S.; Liakou, C.; Antoniou, J.; Georgoulas, T.; Dontas, I.; Papaioannou, N.; Gazi, S.; Lyritis, G.P. Effect of rheumatoid arthritis on volumetric bone mineral density and bone geometry, assessed by peripheral quantitative computed tomography in postmenopausal women treated with bisphosphonates. J. Rheumatol. 2012, 39, 1215–1220. [Google Scholar] [CrossRef]
- Hoff, M.; Haugeberg, G.; Odegard, S.; Syversen, S.; Landewe, R.; van der Heijde, D.; Kvien, T.K. Cortical hand bone loss after 1 year in early rheumatoid arthritis predicts radiographic hand joint damage at 5-year and 10-year follow-up. Ann. Rheum. Dis. 2009, 68, 324–329. [Google Scholar] [CrossRef]
- Haugeberg, G.; Lodder, M.C.; Lems, W.F.; Uhlig, T.; Orstavik, R.E.; Dijkmans, B.A.; Kvien, T.K.; Woolf, A.D. Hand cortical bone mass and its associations with radiographic joint damage and fractures in 50-70 year old female patients with rheumatoid arthritis: Cross sectional Oslo-Truro-Amsterdam (OSTRA) collaborative study. Ann. Rheum. Dis. 2004, 63, 1331–1334. [Google Scholar] [CrossRef]
- Wang, R.; Liu, J.; Wang, Z.; Wu, X.; Guo, H.; Jiao, X.; Zhang, H.; Qi, C.; Li, X. Mangiferin exert protective effects on joints of adjuvant-induced arthritis rats by regulating the MAPKs/NF-kappaB pathway of fibroblast-like synoviocytes. Int. Immunopharmacol. 2021, 101, 108352. [Google Scholar] [CrossRef]
- Wang, W.; Deng, Z.; Liu, G.; Yang, J.; Zhou, W.; Zhang, C.; Shen, W.; Zhang, Y. Platelet-derived extracellular vesicles promote the migration and invasion of rheumatoid arthritis fibroblast-like synoviocytes via CXCR2 signaling. Exp. Ther. Med. 2021, 22, 1120. [Google Scholar] [CrossRef]
- Page, G.; Miossec, P. Paired synovium and lymph nodes from rheumatoid arthritis patients differ in dendritic cell and chemokine expression. J. Pathol. 2004, 204, 28–38. [Google Scholar] [CrossRef]
- Krishtal, O.A.; Pidoplichko, V.I. A receptor for protons in the nerve cell membrane. Neuroscience 1980, 5, 2325–2327. [Google Scholar] [CrossRef]
- Waldmann, R.; Champigny, G.; Bassilana, F.; Heurteaux, C.; Lazdunski, M. A proton-gated cation channel involved in acid-sensing. Nature 1997, 386, 173–177. [Google Scholar] [CrossRef]
- Zhou, R.P.; Wu, X.S.; Wang, Z.S.; Xie, Y.Y.; Ge, J.F.; Chen, F.H. Novel Insights into Acid-Sensing Ion Channels: Implications for Degenerative Diseases. Aging Dis. 2016, 7, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Niu, R.; Hang, X.; Feng, Y.; Zhang, Y.; Qian, X.; Song, S.; Wang, C.; Tao, J.; Peng, X.; Chen, F. ASIC1a promotes synovial invasion of rheumatoid arthritis via Ca(2+)/Rac1 pathway. Int. Immunopharmacol. 2020, 79, 106089. [Google Scholar] [CrossRef]
- Zhang, Y.; Qian, X.; Yang, X.; Niu, R.; Song, S.; Zhu, F.; Zhu, C.; Peng, X.; Chen, F. ASIC1a induces synovial inflammation via the Ca(2+)/NFATc3/ RANTES pathway. Theranostics 2020, 10, 247–264. [Google Scholar] [CrossRef]
- Tao, J.; Lu, Z.; Su, J.; Qian, X.; Zhang, Y.; Xu, Y.; Song, S.; Hang, X.; Peng, X.; Chen, F. ASIC1a promotes the proliferation of synovial fibroblasts via the ERK/MAPK pathway. Lab. Invest. 2021, 101, 1353–1362. [Google Scholar] [CrossRef]
- Geborek, P.; Saxne, T.; Pettersson, H.; Wollheim, F.A. Synovial fluid acidosis correlates with radiological joint destruction in rheumatoid arthritis knee joints. J. Rheumatol. 1989, 16, 468–472. [Google Scholar]
- Yuan, F.L.; Chen, F.H.; Lu, W.G.; Li, X.; Li, J.P.; Li, C.W.; Xu, R.S.; Wu, F.R.; Hu, W.; Zhang, T.Y. Inhibition of acid-sensing ion channels in articular chondrocytes by amiloride attenuates articular cartilage destruction in rats with adjuvant arthritis. Inflamm. Res. 2010, 59, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, S.; Hu, W. Acid-sensing ion channel 1a mediates acid-induced inhibition of matrix metabolism of rat articular chondrocytes via the MAPK signaling pathway. Mol. Cell Biochem. 2018, 443, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Rong, C.; Chen, F.H.; Jiang, S.; Hu, W.; Wu, F.R.; Chen, T.Y.; Yuan, F.L. Inhibition of acid-sensing ion channels by amiloride protects rat articular chondrocytes from acid-induced apoptosis via a mitochondrial-mediated pathway. Cell Biol. Int. 2012, 36, 635–641. [Google Scholar] [CrossRef]
- Gong, W.; Kolker, S.J.; Usachev, Y.; Walder, R.Y.; Boyle, D.L.; Firestein, G.S.; Sluka, K.A. Acid-sensing ion channel 3 decreases phosphorylation of extracellular signal-regulated kinases and induces synoviocyte cell death by increasing intracellular calcium. Arthritis Res. Ther. 2014, 16, R121. [Google Scholar] [CrossRef]
- Zu, S.Q.; Feng, Y.B.; Zhu, C.J.; Wu, X.S.; Zhou, R.P.; Li, G.; Dai, B.B.; Wang, Z.S.; Xie, Y.Y.; Li, Y.; et al. Acid-sensing ion channel 1a mediates acid-induced pyroptosis through calpain-2/calcineurin pathway in rat articular chondrocytes. Cell Biol. Int. 2020, 44, 2140–2152. [Google Scholar] [CrossRef]
- Wu, X.; Ren, G.; Zhou, R.; Ge, J.; Chen, F.H. The role of Ca(2+) in acid-sensing ion channel 1a-mediated chondrocyte pyroptosis in rat adjuvant arthritis. Lab. Invest. 2019, 99, 499–513. [Google Scholar] [CrossRef]
- Zhou, R.; Wu, X.; Wang, Z.; Ge, J.; Chen, F. Interleukin-6 enhances acid-induced apoptosis via upregulating acid-sensing ion channel 1a expression and function in rat articular chondrocytes. Int. Immunopharmacol. 2015, 29, 748–760. [Google Scholar] [CrossRef]
- Udagawa, N.; Takahashi, N.; Jimi, E.; Matsuzaki, K.; Tsurukai, T.; Itoh, K.; Nakagawa, N.; Yasuda, H.; Goto, M.; Tsuda, E.; et al. Osteoblasts/stromal cells stimulate osteoclast activation through expression of osteoclast differentiation factor/RANKL but not macrophage colony-stimulating factor: Receptor activator of NF-kappa B ligand. Bone 1999, 25, 517–523. [Google Scholar] [CrossRef]
- Komarova, S.V.; Pereverzev, A.; Shum, J.W.; Sims, S.M.; Dixon, S.J. Convergent signaling by acidosis and receptor activator of NF-kappaB ligand (RANKL) on the calcium/calcineurin/NFAT pathway in osteoclasts. Proc. Natl. Acad. Sci. USA 2005, 102, 2643–2648. [Google Scholar] [CrossRef]
- Li, X.; Ye, J.X.; Xu, M.H.; Zhao, M.D.; Yuan, F.L. Evidence that activation of ASIC1a by acidosis increases osteoclast migration and adhesion by modulating integrin/Pyk2/Src signaling pathway. Osteoporos. Int. 2017, 28, 2221–2231. [Google Scholar] [CrossRef]
- Negishi-Koga, T.; Takayanagi, H. Ca2+-NFATc1 signaling is an essential axis of osteoclast differentiation. Immunol. Rev. 2009, 231, 241–256. [Google Scholar] [CrossRef]
- Li, X.; Xu, R.S.; Jiang, D.L.; He, X.L.; Jin, C.; Lu, W.G.; Su, Q.; Yuan, F.L. Acid-sensing ion channel 1a is involved in acid-induced osteoclastogenesis by regulating activation of the transcription factor NFATc1. FEBS Lett. 2013, 587, 3236–3242. [Google Scholar] [CrossRef]
- Zhou, R.P.; Ni, W.L.; Dai, B.B.; Wu, X.S.; Wang, Z.S.; Xie, Y.Y.; Wang, Z.Q.; Yang, W.J.; Ge, J.F.; Hu, W.; et al. ASIC2a overexpression enhances the protective effect of PcTx1 and APETx2 against acidosis-induced articular chondrocyte apoptosis and cytotoxicity. Gene 2018, 642, 230–240. [Google Scholar] [CrossRef]
- Wemmie, J.A.; Price, M.P.; Welsh, M.J. Acid-sensing ion channels: Advances, questions and therapeutic opportunities. Trends Neurosci. 2006, 29, 578–586. [Google Scholar] [CrossRef]
- Wemmie, J.A.; Taugher, R.J.; Kreple, C.J. Acid-sensing ion channels in pain and disease. Nat. Rev. Neurosci. 2013, 14, 461–471. [Google Scholar] [CrossRef]
- Ikeuchi, M.; Kolker, S.J.; Burnes, L.A.; Walder, R.Y.; Sluka, K.A. Role of ASIC3 in the primary and secondary hyperalgesia produced by joint inflammation in mice. Pain 2008, 137, 662–669. [Google Scholar] [CrossRef]
- Kolker, S.J.; Walder, R.Y.; Usachev, Y.; Hillman, J.; Boyle, D.L.; Firestein, G.S.; Sluka, K.A. Acid-sensing ion channel 3 expressed in type B synoviocytes and chondrocytes modulates hyaluronan expression and release. Ann. Rheum. Dis. 2010, 69, 903–909. [Google Scholar] [CrossRef]
- Sluka, K.A.; Rasmussen, L.A.; Edgar, M.M.; O’Donnell, J.M.; Walder, R.Y.; Kolker, S.J.; Boyle, D.L.; Firestein, G.S. Acid-sensing ion channel 3 deficiency increases inflammation but decreases pain behavior in murine arthritis. Arthritis Rheum. 2013, 65, 1194–1202. [Google Scholar] [CrossRef]
- Ikeuchi, M.; Kolker, S.J.; Sluka, K.A. Acid-sensing ion channel 3 expression in mouse knee joint afferents and effects of carrageenan-induced arthritis. J. Pain 2009, 10, 336–342. [Google Scholar] [CrossRef]
- Yuan, F.L.; Chen, F.H.; Lu, W.G.; Li, X. Acid-sensing ion channels 3: A potential therapeutic target for pain treatment in arthritis. Mol. Biol. Rep. 2010, 37, 3233–3238. [Google Scholar] [CrossRef]
- Yu, G.M.; Liu, D.; Yuan, N.; Liu, B.H. Dual role of acid-sensing ion channels 3 in rheumatoid arthritis: Destruction or protection? Immunopharmacol. Immunotoxicol. 2018, 40, 273–277. [Google Scholar] [CrossRef]
- Cosens, D.J.; Manning, A. Abnormal electroretinogram from a Drosophila mutant. Nature 1969, 224, 285–287. [Google Scholar] [CrossRef]
- Hardie, R.C.; Minke, B. Novel Ca2+ channels underlying transduction in Drosophila photoreceptors: Implications for phosphoinositide-mediated Ca2+ mobilization. Trends Neurosci. 1993, 16, 371–376. [Google Scholar] [CrossRef]
- Wes, P.D.; Chevesich, J.; Jeromin, A.; Rosenberg, C.; Stetten, G.; Montell, C. TRPC1, a human homolog of a Drosophila store-operated channel. Proc. Natl. Acad. Sci. USA 1995, 92, 9652–9656. [Google Scholar] [CrossRef]
- Li, H. TRP Channel Classification. Adv. Exp. Med. Biol. 2017, 976, 1–8. [Google Scholar] [CrossRef]
- Meng, S.; Alanazi, R.; Ji, D.; Bandura, J.; Luo, Z.W.; Fleig, A.; Feng, Z.P.; Sun, H.S. Role of TRPM7 kinase in cancer. Cell Calcium 2021, 96, 102400. [Google Scholar] [CrossRef]
- Ciurtin, C.; Majeed, Y.; Naylor, J.; Sukumar, P.; English, A.A.; Emery, P.; Beech, D.J. TRPM3 channel stimulated by pregnenolone sulphate in synovial fibroblasts and negatively coupled to hyaluronan. BMC Musculoskelet. Disord. 2010, 11, 111. [Google Scholar] [CrossRef]
- Zhu, S.; Wang, Y.; Pan, L.; Yang, S.; Sun, Y.; Wang, X.; Hu, F. Involvement of transient receptor potential melastatin-8 (TRPM8) in menthol-induced calcium entry, reactive oxygen species production and cell death in rheumatoid arthritis rat synovial fibroblasts. Eur. J. Pharmacol. 2014, 725, 1–9. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Wang, Y.; Li, X.; Huang, C.; Li, J. Inhibition of transient receptor potential melastatin 7 (TRPM7) channel induces RA FLSs apoptosis through endoplasmic reticulum (ER) stress. Clin. Rheumatol. 2014, 33, 1565–1574. [Google Scholar] [CrossRef]
- Lu, D.; Qu, J.; Sun, L.; Li, Q.; Ling, H.; Yang, N.; Ma, T.; Wang, Q.; Li, M.; Zhang, K.; et al. Ca2+/Mg2+ homeostasisrelated TRPM7 channel mediates chondrocyte hypertrophy via regulation of the PI3KAkt signaling pathway. Mol. Med. Rep. 2017, 16, 5699–5705. [Google Scholar] [CrossRef]
- Ma, G.; Yang, Y.; Chen, Y.; Wei, X.; Ding, J.; Zhou, R.P.; Hu, W. Blockade of TRPM7 Alleviates Chondrocyte Apoptosis and Articular Cartilage Damage in the Adjuvant Arthritis Rat Model Through Regulation of the Indian Hedgehog Signaling Pathway. Front. Pharmacol. 2021, 12, 655551. [Google Scholar] [CrossRef]
- Zhou, R.; Chen, Y.; Li, S.; Wei, X.; Hu, W.; Tang, S.; Ding, J.; Fu, W.; Zhang, H.; Chen, F. TRPM7 channel inhibition attenuates rheumatoid arthritis articular chondrocyte ferroptosis by suppression of the PKCalpha-NOX4 axis. Redox Biol. 2022, 55, 102411. [Google Scholar] [CrossRef]
- Abed, E.; Moreau, R. Importance of melastatin-like transient receptor potential 7 and cations (magnesium, calcium) in human osteoblast-like cell proliferation. Cell Prolif. 2007, 40, 849–865. [Google Scholar] [CrossRef]
- Abed, E.; Moreau, R. Importance of melastatin-like transient receptor potential 7 and magnesium in the stimulation of osteoblast proliferation and migration by platelet-derived growth factor. Am. J. Physiol. Cell Physiol. 2009, 297, C360–C368. [Google Scholar] [CrossRef]
- Yu, M.; Wang, Y.; Zhang, Y.; Cui, D.; Gu, G.; Zhao, D. Gallium ions promote osteoinduction of human and mouse osteoblasts via the TRPM7/Akt signaling pathway. Mol. Med. Rep. 2020, 22, 2741–2752. [Google Scholar] [CrossRef]
- Qiao, W.; Wong, K.H.M.; Shen, J.; Wang, W.; Wu, J.; Li, J.; Lin, Z.; Chen, Z.; Matinlinna, J.P.; Zheng, Y.; et al. TRPM7 kinase-mediated immunomodulation in macrophage plays a central role in magnesium ion-induced bone regeneration. Nat. Commun. 2021, 12, 2885. [Google Scholar] [CrossRef]
- Zierler, S.; Hampe, S.; Nadolni, W. TRPM channels as potential therapeutic targets against pro-inflammatory diseases. Cell Calcium 2017, 67, 105–115. [Google Scholar] [CrossRef]
- Wang, C.H.; Rong, M.Y.; Wang, L.; Ren, Z.; Chen, L.N.; Jia, J.F.; Li, X.Y.; Wu, Z.B.; Chen, Z.N.; Zhu, P. CD147 up-regulates calcium-induced chemotaxis, adhesion ability and invasiveness of human neutrophils via a TRPM-7-mediated mechanism. Rheumatology 2014, 53, 2288–2296. [Google Scholar] [CrossRef]
- Nadolni, W.; Immler, R.; Hoelting, K.; Fraticelli, M.; Ripphahn, M.; Rothmiller, S.; Matsushita, M.; Boekhoff, I.; Gudermann, T.; Sperandio, M.; et al. TRPM7 Kinase Is Essential for Neutrophil Recruitment and Function via Regulation of Akt/mTOR Signaling. Front. Immunol. 2020, 11, 606893. [Google Scholar] [CrossRef]
- He, Z. TRPC Channel Downstream Signaling Cascades. Adv. Exp. Med. Biol. 2017, 976, 25–33. [Google Scholar] [CrossRef]
- Ong, E.C.; Nesin, V.; Long, C.L.; Bai, C.X.; Guz, J.L.; Ivanov, I.P.; Abramowitz, J.; Birnbaumer, L.; Humphrey, M.B.; Tsiokas, L. A TRPC1 protein-dependent pathway regulates osteoclast formation and function. J. Biol Chem. 2013, 288, 22219–22232. [Google Scholar] [CrossRef]
- Klein, S.; Mentrup, B.; Timmen, M.; Sherwood, J.; Lindemann, O.; Fobker, M.; Kronenberg, D.; Pap, T.; Raschke, M.J.; Stange, R. Modulation of Transient Receptor Potential Channels 3 and 6 Regulates Osteoclast Function with Impact on Trabecular Bone Loss. Calcif. Tissue Int. 2020, 106, 655–664. [Google Scholar] [CrossRef]
- Alawi, K.M.; Russell, F.A.; Aubdool, A.A.; Srivastava, S.; Riffo-Vasquez, Y.; Baldissera, L., Jr.; Thakore, P.; Saleque, N.; Fernandes, E.S.; Walsh, D.A.; et al. Transient receptor potential canonical 5 (TRPC5) protects against pain and vascular inflammation in arthritis and joint inflammation. Ann. Rheum. Dis. 2017, 76, 252–260. [Google Scholar] [CrossRef]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [CrossRef]
- Kameda, T.; Zvick, J.; Vuk, M.; Sadowska, A.; Tam, W.K.; Leung, V.Y.; Bolcskei, K.; Helyes, Z.; Applegate, L.A.; Hausmann, O.N. Expression and Activity of TRPA1 and TRPV1 in the Intervertebral Disc: Association with Inflammation and Matrix Remodeling. Int. J. Mol. Sci. 2019, 20, 1767. [Google Scholar] [CrossRef]
- Laragione, T.; Cheng, K.F.; Tanner, M.R.; He, M.; Beeton, C.; Al-Abed, Y.; Gulko, P.S. The cation channel Trpv2 is a new suppressor of arthritis severity, joint damage, and synovial fibroblast invasion. Clin. Immunol. 2015, 158, 183–192. [Google Scholar] [CrossRef]
- Montell, C. The TRP superfamily of cation channels. Sci. STKE 2005, 2005, re3. [Google Scholar] [CrossRef]
- Engler, A.; Aeschlimann, A.; Simmen, B.R.; Michel, B.A.; Gay, R.E.; Gay, S.; Sprott, H. Expression of transient receptor potential vanilloid 1 (TRPV1) in synovial fibroblasts from patients with osteoarthritis and rheumatoid arthritis. Biochem. Biophys. Res. Commun. 2007, 359, 884–888. [Google Scholar] [CrossRef]
- Hu, F.; Sun, W.W.; Zhao, X.T.; Cui, Z.J.; Yang, W.X. TRPV1 mediates cell death in rat synovial fibroblasts through calcium entry-dependent ROS production and mitochondrial depolarization. Biochem. Biophys. Res. Commun. 2008, 369, 989–993. [Google Scholar] [CrossRef]
- Idris, A.I.; Landao-Bassonga, E.; Ralston, S.H. The TRPV1 ion channel antagonist capsazepine inhibits osteoclast and osteoblast differentiation in vitro and ovariectomy induced bone loss in vivo. Bone 2010, 46, 1089–1099. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Bellini, G.; Torella, M.; Tortora, C.; Manzo, I.; Giordano, C.; Guida, F.; Luongo, L.; Papale, F.; Rosso, F.; et al. The genetic ablation or pharmacological inhibition of TRPV1 signalling is beneficial for the restoration of quiescent osteoclast activity in ovariectomized mice. Br. J. Pharmacol. 2014, 171, 2621–2630. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.; Singh, A.; Mittal, M.; Sharan, K.; Singh, N.; Dixit, P.; Sanyal, S.; Maurya, R.; Chattopadhyay, N. [6]-Gingerol induces bone loss in ovary intact adult mice and augments osteoclast function via the transient receptor potential vanilloid 1 channel. Mol. Nutr. Food Res. 2012, 56, 1860–1873. [Google Scholar] [CrossRef]
- Yan, S.; Miao, L.; Lu, Y.; Wang, L. Sirtuin 1 inhibits TNF-alpha-mediated osteoclastogenesis of bone marrow-derived macrophages through both ROS generation and TRPV1 activation. Mol. Cell Biochem. 2019, 455, 135–145. [Google Scholar] [CrossRef]
- Rossi, F.; Bellini, G.; Tortora, C.; Bernardo, M.E.; Luongo, L.; Conforti, A.; Starc, N.; Manzo, I.; Nobili, B.; Locatelli, F.; et al. CB(2) and TRPV(1) receptors oppositely modulate in vitro human osteoblast activity. Pharmacol. Res. 2015, 99, 194–201. [Google Scholar] [CrossRef]
- Bellini, G.; Torella, M.; Manzo, I.; Tortora, C.; Luongo, L.; Punzo, F.; Colacurci, N.; Nobili, B.; Maione, S.; Rossi, F. PKCbetaII-mediated cross-talk of TRPV1/CB2 modulates the glucocorticoid-induced osteoclast overactivity. Pharmacol. Res. 2017, 115, 267–274. [Google Scholar] [CrossRef]
- He, L.H.; Liu, M.; He, Y.; Xiao, E.; Zhao, L.; Zhang, T.; Yang, H.Q.; Zhang, Y. TRPV1 deletion impaired fracture healing and inhibited osteoclast and osteoblast differentiation. Sci. Rep. 2017, 7, 42385. [Google Scholar] [CrossRef]
- Hsieh, W.S.; Kung, C.C.; Huang, S.L.; Lin, S.C.; Sun, W.H. TDAG8, TRPV1, and ASIC3 involved in establishing hyperalgesic priming in experimental rheumatoid arthritis. Sci. Rep. 2017, 7, 8870. [Google Scholar] [CrossRef]
- Logashina, Y.A.; Palikova, Y.A.; Palikov, V.A.; Kazakov, V.A.; Smolskaya, S.V.; Dyachenko, I.A.; Tarasova, N.V.; Andreev, Y.A. Anti-Inflammatory and Analgesic Effects of TRPV1 Polypeptide Modulator APHC3 in Models of Osteo- and Rheumatoid Arthritis. Mar. Drugs 2021, 19, 39. [Google Scholar] [CrossRef]
- Laragione, T.; Harris, C.; Gulko, P.S. TRPV2 suppresses Rac1 and RhoA activation and invasion in rheumatoid arthritis fibroblast-like synoviocytes. Int. Immunopharmacol. 2019, 70, 268–273. [Google Scholar] [CrossRef]
- Kajiya, H.; Okamoto, F.; Nemoto, T.; Kimachi, K.; Toh-Goto, K.; Nakayana, S.; Okabe, K. RANKL-induced TRPV2 expression regulates osteoclastogenesis via calcium oscillations. Cell Calcium 2010, 48, 260–269. [Google Scholar] [CrossRef]
- Bai, H.; Zhu, H.; Yan, Q.; Shen, X.; Lu, X.; Wang, J.; Li, J.; Chen, L. TRPV2-induced Ca(2+)-calcineurin-NFAT signaling regulates differentiation of osteoclast in multiple myeloma. Cell Commun. Signal. 2018, 16, 68. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Hui, Z.; Wei, W.; Yang, J.; Chen, Z.; Guo, B.; Xing, F.; Zhang, X.; Pan, L.; Xu, J. Hypotonic stress promotes ATP release, reactive oxygen species production and cell proliferation via TRPV4 activation in rheumatoid arthritis rat synovial fibroblasts. Biochem Biophys Res. Commun. 2017, 486, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Xing, R.; Huang, Z.; Yin, S.; Li, X.; Zhang, L.; Ding, L.; Wang, P. Excessive mechanical stress induces chondrocyte apoptosis through TRPV4 in an anterior cruciate ligament-transected rat osteoarthritis model. Life Sci. 2019, 228, 158–166. [Google Scholar] [CrossRef]
- McNulty, A.L.; Leddy, H.A.; Liedtke, W.; Guilak, F. TRPV4 as a therapeutic target for joint diseases. Naunyn. Schmiedebergs Arch. Pharmacol. 2015, 388, 437–450. [Google Scholar] [CrossRef]
- Nishimura, H.; Kawasaki, M.; Tsukamoto, M.; Menuki, K.; Suzuki, H.; Matsuura, T.; Baba, K.; Motojima, Y.; Fujitani, T.; Ohnishi, H.; et al. Transient receptor potential vanilloid 1 and 4 double knockout leads to increased bone mass in mice. Bone Rep. 2020, 12, 100268. [Google Scholar] [CrossRef]
- Masuyama, R.; Vriens, J.; Voets, T.; Karashima, Y.; Owsianik, G.; Vennekens, R.; Lieben, L.; Torrekens, S.; Moermans, K.; Vanden Bosch, A.; et al. TRPV4-mediated calcium influx regulates terminal differentiation of osteoclasts. Cell Metab. 2008, 8, 257–265. [Google Scholar] [CrossRef]
- Cao, B.; Dai, X.; Wang, W. Knockdown of TRPV4 suppresses osteoclast differentiation and osteoporosis by inhibiting autophagy through Ca(2+) -calcineurin-NFATc1 pathway. J. Cell Physiol. 2019, 234, 6831–6841. [Google Scholar] [CrossRef]
- Segond von Banchet, G.; Boettger, M.K.; Konig, C.; Iwakura, Y.; Brauer, R.; Schaible, H.G. Neuronal IL-17 receptor upregulates TRPV4 but not TRPV1 receptors in DRG neurons and mediates mechanical but not thermal hyperalgesia. Mol. Cell Neurosci. 2013, 52, 152–160. [Google Scholar] [CrossRef]
- Wei, Y.; Jin, Z.; Zhang, H.; Piao, S.; Lu, J.; Bai, L. The Transient Receptor Potential Channel, Vanilloid 5, Induces Chondrocyte Apoptosis via Ca2+ CaMKII-Dependent MAPK and Akt/ mTOR Pathways in a Rat Osteoarthritis Model. Cell Physiol. Biochem. 2018, 51, 2309–2323. [Google Scholar] [CrossRef]
- Atobe, M. Activation of Transient Receptor Potential Vanilloid (TRPV) 4 as a Therapeutic Strategy in Osteoarthritis. Curr. Top. Med. Chem. 2019, 19, 2254–2267. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, H.; Katanosaka, Y.; Chijimatsu, R.; Mori, D.; Xuan, F.; Yano, F.; Omata, Y.; Maenohara, Y.; Murahashi, Y.; Kawaguchi, K.; et al. Involvement of Transient Receptor Potential Vanilloid Channel 2 in the Induction of Lubricin and Suppression of Ectopic Endochondral Ossification in Mouse Articular Cartilage. Arthritis Rheumatol. 2021, 73, 1441–1450. [Google Scholar] [CrossRef] [PubMed]
- O’Conor, C.J.; Ramalingam, S.; Zelenski, N.A.; Benefield, H.C.; Rigo, I.; Little, D.; Wu, C.L.; Chen, D.; Liedtke, W.; McNulty, A.L.; et al. Cartilage-Specific Knockout of the Mechanosensory Ion Channel TRPV4 Decreases Age-Related Osteoarthritis. Sci. Rep. 2016, 6, 29053. [Google Scholar] [CrossRef]
- van der Eerden, B.C.; Hoenderop, J.G.; de Vries, T.J.; Schoenmaker, T.; Buurman, C.J.; Uitterlinden, A.G.; Pols, H.A.; Bindels, R.J.; van Leeuwen, J.P. The epithelial Ca2+ channel TRPV5 is essential for proper osteoclastic bone resorption. Proc. Natl. Acad. Sci. USA 2005, 102, 17507–17512. [Google Scholar] [CrossRef]
- Yan, P.; Li, T.; Bo, M.; Die, L.; Xing, L. Inhibition of bone resorption by econazole in rat osteoclast-like cells through suppressing TRPV5. Arch. Pharm. Res. 2011, 34, 1007–1013. [Google Scholar] [CrossRef]
- Nijenhuis, T.; van der Eerden, B.C.; Hoenderop, J.G.; Weinans, H.; van Leeuwen, J.P.; Bindels, R.J. Bone resorption inhibitor alendronate normalizes the reduced bone thickness of TRPV5(-/-) mice. J. Bone Miner. Res. 2008, 23, 1815–1824. [Google Scholar] [CrossRef]
- Chamoux, E.; Bisson, M.; Payet, M.D.; Roux, S. TRPV-5 mediates a receptor activator of NF-kappaB (RANK) ligand-induced increase in cytosolic Ca2+ in human osteoclasts and down-regulates bone resorption. J. Biol. Chem. 2010, 285, 25354–25362. [Google Scholar] [CrossRef]
- van der Eerden, B.C.; Koek, W.N.; Roschger, P.; Zillikens, M.C.; Waarsing, J.H.; van der Kemp, A.; Schreuders-Koedam, M.; Fratzl-Zelman, N.; Leenen, P.J.; Hoenderop, J.G.; et al. Lifelong challenge of calcium homeostasis in male mice lacking TRPV5 leads to changes in bone and calcium metabolism. Oncotarget 2016, 7, 24928–24941. [Google Scholar] [CrossRef]
- Chen, F.; Ouyang, Y.; Ye, T.; Ni, B.; Chen, A. Estrogen inhibits RANKL-induced osteoclastic differentiation by increasing the expression of TRPV5 channel. J. Cell Biochem. 2014, 115, 651–658. [Google Scholar] [CrossRef]
- Song, T.; Lin, T.; Ma, J.; Guo, L.; Zhang, L.; Zhou, X.; Ye, T. Regulation of TRPV5 transcription and expression by E2/ERalpha signalling contributes to inhibition of osteoclastogenesis. J. Cell Mol. Med. 2018, 22, 4738–4750. [Google Scholar] [CrossRef]
- Illes, P.; Muller, C.E.; Jacobson, K.A.; Grutter, T.; Nicke, A.; Fountain, S.J.; Kennedy, C.; Schmalzing, G.; Jarvis, M.F.; Stojilkovic, S.S.; et al. Update of P2X receptor properties and their pharmacology: IUPHAR Review 30. Br. J. Pharmacol. 2021, 178, 489–514. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Burnstock, G. Biology of purinergic signalling: Its ancient evolutionary roots, its omnipresence and its multiple functional significance. Bioessays 2014, 36, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Hu, L.Q.; Yao, S.R.; Hu, Y.; Wang, D.G.; Fan, Y.G.; Pan, G.X.; Tao, S.S.; Zhang, Q.; Pan, H.F.; et al. P2X7 receptor: A potential therapeutic target for autoimmune diseases. Autoimmun. Rev. 2019, 18, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Tsutsuki, H.; Islam, W.; Ono, K.; Takeda, K.; Akaike, T.; Sawa, T. ATP exposure stimulates glutathione efflux as a necessary switch for NLRP3 inflammasome activation. Redox Biol. 2021, 41, 101930. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; Burnstock, G.; Kennedy, C.; King, B.F.; North, R.A.; Seguela, P.; Voigt, M.; Humphrey, P.P. International union of pharmacology. XXIV. Current status of the nomenclature and properties of P2X receptors and their subunits. Pharmacol. Rev. 2001, 53, 107–118. [Google Scholar]
- Abbracchio, M.P.; Burnstock, G.; Boeynaems, J.M.; Barnard, E.A.; Boyer, J.L.; Kennedy, C.; Knight, G.E.; Fumagalli, M.; Gachet, C.; Jacobson, K.A.; et al. International Union of Pharmacology LVIII: Update on the P2Y G protein-coupled nucleotide receptors: From molecular mechanisms and pathophysiology to therapy. Pharmacol. Rev. 2006, 58, 281–341. [Google Scholar] [CrossRef]
- Baroja-Mazo, A.; Pelegrin, P. Modulating P2X7 Receptor Signaling during Rheumatoid Arthritis: New Therapeutic Approaches for Bisphosphonates. J. Osteoporos. 2012, 2012, 408242. [Google Scholar] [CrossRef]
- Burnstock, G.; Kennedy, C. P2X receptors in health and disease. Adv. Pharmacol. 2011, 61, 333–372. [Google Scholar] [CrossRef]
- Al-Shukaili, A.; Al-Kaabi, J.; Hassan, B.; Al-Araimi, T.; Al-Tobi, M.; Al-Kindi, M.; Al-Maniri, A.; Al-Gheilani, A.; Al-Ansari, A. P2X7 receptor gene polymorphism analysis in rheumatoid arthritis. Int. J. Immunogenet. 2011, 38, 389–396. [Google Scholar] [CrossRef]
- Portales-Cervantes, L.; Nino-Moreno, P.; Salgado-Bustamante, M.; Garcia-Hernandez, M.H.; Baranda-Candido, L.; Reynaga-Hernandez, E.; Barajas-Lopez, C.; Gonzalez-Amaro, R.; Portales-Perez, D.P. The His155Tyr (489C>T) single nucleotide polymorphism of P2RX7 gene confers an enhanced function of P2X7 receptor in immune cells from patients with rheumatoid arthritis. Cell Immunol. 2012, 276, 168–175. [Google Scholar] [CrossRef]
- Al-Shukaili, A.; Al-Kaabi, J.; Hassan, B. A comparative study of interleukin-1beta production and p2x7 expression after ATP stimulation by peripheral blood mononuclear cells isolated from rheumatoid arthritis patients and normal healthy controls. Inflammation 2008, 31, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Portales-Cervantes, L.; Nino-Moreno, P.; Doniz-Padilla, L.; Baranda-Candido, L.; Garcia-Hernandez, M.; Salgado-Bustamante, M.; Gonzalez-Amaro, R.; Portales-Perez, D. Expression and function of the P2X(7) purinergic receptor in patients with systemic lupus erythematosus and rheumatoid arthritis. Hum. Immunol. 2010, 71, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Su, B.; Shang, M. [Diagnostic value of P2X7 receptor and its role in inflammatory reaction in rheumatoid arthritis]. Nan Fang Yi Ke Da Xue Xue Bao 2018, 38, 1453–1458. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Huang, Z.; Zhang, H.; Lu, J.; Wei, Y.; Yang, Y.; Bai, L. IRE1-mTOR-PERK Axis Coordinates Autophagy and ER Stress-Apoptosis Induced by P2X7-Mediated Ca(2+) Influx in Osteoarthritis. Front. Cell Dev. Biol. 2021, 9, 695041. [Google Scholar] [CrossRef]
- Morrison, M.S.; Turin, L.; King, B.F.; Burnstock, G.; Arnett, T.R. ATP is a potent stimulator of the activation and formation of rodent osteoclasts. J. Physiol. 1998, 511 Pt 2, 495–500. [Google Scholar] [CrossRef]
- Jorgensen, N.R. Role of the purinergic P2X receptors in osteoclast pathophysiology. Curr. Opin. Pharmacol. 2019, 47, 97–101. [Google Scholar] [CrossRef]
- Wang, N.; Agrawal, A.; Jorgensen, N.R.; Gartland, A. P2X7 receptor regulates osteoclast function and bone loss in a mouse model of osteoporosis. Sci. Rep. 2018, 8, 3507. [Google Scholar] [CrossRef]
- Jorgensen, N.R.; Husted, L.B.; Skarratt, K.K.; Stokes, L.; Tofteng, C.L.; Kvist, T.; Jensen, J.E.; Eiken, P.; Brixen, K.; Fuller, S.; et al. Single-nucleotide polymorphisms in the P2X7 receptor gene are associated with post-menopausal bone loss and vertebral fractures. Eur. J. Hum. Genet. 2012, 20, 675–681. [Google Scholar] [CrossRef]
- Syberg, S.; Schwarz, P.; Petersen, S.; Steinberg, T.H.; Jensen, J.E.; Teilmann, J.; Jorgensen, N.R. Association between P2X7 Receptor Polymorphisms and Bone Status in Mice. J. Osteoporos. 2012, 2012, 637986. [Google Scholar] [CrossRef]
- Naemsch, L.N.; Dixon, S.J.; Sims, S.M. Activity-dependent development of P2X7 current and Ca2+ entry in rabbit osteoclasts. J. Biol. Chem. 2001, 276, 39107–39114. [Google Scholar] [CrossRef]
- Gartland, A.; Buckley, K.A.; Hipskind, R.A.; Perry, M.J.; Tobias, J.H.; Buell, G.; Chessell, I.; Bowler, W.B.; Gallagher, J.A. Multinucleated osteoclast formation in vivo and in vitro by P2X7 receptor-deficient mice. Crit. Rev. Eukaryot. Gene Expr. 2003, 13, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Buckley, K.A.; Bowers, K.; Furber, M.; Gallagher, J.A.; Gartland, A. The effects of P2X7 receptor antagonists on the formation and function of human osteoclasts in vitro. Purinergic. Signal. 2010, 6, 307–315. [Google Scholar] [CrossRef]
- Schilling, W.P.; Wasylyna, T.; Dubyak, G.R.; Humphreys, B.D.; Sinkins, W.G. Maitotoxin and P2Z/P2X(7) purinergic receptor stimulation activate a common cytolytic pore. Am. J. Physiol. 1999, 277, C766–C776. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, T.H.; Hiken, J.F. P2 receptors in macrophage fusion and osteoclast formation. Purinergic. Signal. 2007, 3, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Hanaka, M.; Iba, K.; Dohke, T.; Kanaya, K.; Okazaki, S.; Yamashita, T. Antagonists to TRPV1, ASICs and P2X have a potential role to prevent the triggering of regional bone metabolic disorder and pain-like behavior in tail-suspended mice. Bone 2018, 110, 284–294. [Google Scholar] [CrossRef]
- Tanaka, M.; Hosoya, A.; Mori, H.; Kayasuga, R.; Nakamura, H.; Ozawa, H. Minodronic acid induces morphological changes in osteoclasts at bone resorption sites and reaches a level required for antagonism of purinergic P2X2/3 receptors. J. Bone Miner. Metab. 2018, 36, 54–63. [Google Scholar] [CrossRef]
- Wewers, M.D.; Sarkar, A. P2X(7) receptor and macrophage function. Purinergic. Signal. 2009, 5, 189–195. [Google Scholar] [CrossRef]
- Fan, Z.D.; Zhang, Y.Y.; Guo, Y.H.; Huang, N.; Ma, H.H.; Huang, H.; Yu, H.G. Involvement of P2X7 receptor signaling on regulating the differentiation of Th17 cells and type II collagen-induced arthritis in mice. Sci. Rep. 2016, 6, 35804. [Google Scholar] [CrossRef]
- Lopez-Castejon, G.; Theaker, J.; Pelegrin, P.; Clifton, A.D.; Braddock, M.; Surprenant, A. P2X(7) receptor-mediated release of cathepsins from macrophages is a cytokine-independent mechanism potentially involved in joint diseases. J. Immunol. 2010, 185, 2611–2619. [Google Scholar] [CrossRef]
- Putney, J.W., Jr. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef]
- Bhuvaneshwari, S.; Sankaranarayanan, K. Structural and Mechanistic Insights of CRAC Channel as a Drug Target in Autoimmune Disorder. Curr. Drug Targets 2020, 21, 55–75. [Google Scholar] [CrossRef] [PubMed]
- Lunz, V.; Romanin, C.; Frischauf, I. STIM1 activation of Orai1. Cell Calcium 2019, 77, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Hori, K.; Tsujikawa, S.; Novakovic, M.M.; Yamashita, M.; Prakriya, M. Regulation of chemoconvulsant-induced seizures by store-operated Orai1 channels. J. Physiol. 2020, 598, 5391–5409. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Diez-Bello, R.; Lopez, J.J.; Redondo, P.C.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC6 Channels Are Required for Proliferation, Migration and Invasion of Breast Cancer Cell Lines by Modulation of Orai1 and Orai3 Surface Exposure. Cancers 2018, 10, 331. [Google Scholar] [CrossRef]
- Yen, J.H.; Chang, C.M.; Hsu, Y.W.; Lee, C.H.; Wu, M.S.; Hwang, D.Y.; Chen, B.K.; Liao, H.T.; Wu, M.T.; Chang, W.C. A polymorphism of ORAI1 rs7135617, is associated with susceptibility to rheumatoid arthritis. Mediators Inflamm. 2014, 2014, 834831. [Google Scholar] [CrossRef]
- Lu, J.; Yang, J.; Zheng, Y.; Fang, S.; Chen, X. Resveratrol reduces store-operated Ca(2+) entry and enhances the apoptosis of fibroblast-like synoviocytes in adjuvant arthritis rats model via targeting ORAI1-STIM1 complex. Biol. Res. 2019, 52, 45. [Google Scholar] [CrossRef]
- Liu, S.; Kiyoi, T.; Takemasa, E.; Maeyama, K. Systemic lentivirus-mediated delivery of short hairpin RNA targeting calcium release-activated calcium channel 3 as gene therapy for collagen-induced arthritis. J. Immunol. 2015, 194, 76–83. [Google Scholar] [CrossRef]
- Janczi, T.; Bohm, B.B.; Fehrl, Y.; DeGiacomo, P.; Kinne, R.W.; Burkhardt, H. ADAM15 in Apoptosis Resistance of Synovial Fibroblasts: Converting Fas/CD95 Death Signals Into the Activation of Prosurvival Pathways by Calmodulin Recruitment. Arthritis Rheumatol. 2019, 71, 63–72. [Google Scholar] [CrossRef]
- Robinson, L.J.; Soboloff, J.; Tourkova, I.L.; Larrouture, Q.C.; Witt, M.R.; Gross, S.; Hooper, R.; Samakai, E.; Worley, P.F.; Barnett, J.B.; et al. The function of the calcium channel Orai1 in osteoclast development. FASEB J. 2021, 35, e21653. [Google Scholar] [CrossRef]
- Li, P.; Bian, X.; Liu, C.; Wang, S.; Guo, M.; Tao, Y.; Huo, B. STIM1 and TRPV4 regulate fluid flow-induced calcium oscillation at early and late stages of osteoclast differentiation. Cell Calcium 2018, 71, 45–52. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Putney, J.W. Orai1-mediated calcium entry plays a critical role in osteoclast differentiation and function by regulating activation of the transcription factor NFATc1. FASEB J. 2012, 26, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Robinson, L.J.; Mancarella, S.; Songsawad, D.; Tourkova, I.L.; Barnett, J.B.; Gill, D.L.; Soboloff, J.; Blair, H.C. Gene disruption of the calcium channel Orai1 results in inhibition of osteoclast and osteoblast differentiation and impairs skeletal development. Lab. Invest. 2012, 92, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Coste, B.; Mathur, J.; Schmidt, M.; Earley, T.J.; Ranade, S.; Petrus, M.J.; Dubin, A.E.; Patapoutian, A. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 2010, 330, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Coste, B.; Xiao, B.; Santos, J.S.; Syeda, R.; Grandl, J.; Spencer, K.S.; Kim, S.E.; Schmidt, M.; Mathur, J.; Dubin, A.E.; et al. Piezo proteins are pore-forming subunits of mechanically activated channels. Nature 2012, 483, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Leddy, H.A.; Chen, Y.; Lee, S.H.; Zelenski, N.A.; McNulty, A.L.; Wu, J.; Beicker, K.N.; Coles, J.; Zauscher, S.; et al. Synergy between Piezo1 and Piezo2 channels confers high-strain mechanosensitivity to articular cartilage. Proc. Natl. Acad. Sci. USA 2014, 111, E5114–E5122. [Google Scholar] [CrossRef]
- Li, X.F.; Zhang, Z.; Li, X.D.; Wang, T.B.; Zhang, H.N. [Mechanism of the Piezo1 protein-induced apoptosis of the chondrocytes through the MAPK/ERK1/2 signal pathway]. Zhonghua Yi Xue Za Zhi 2016, 96, 2472–2477. [Google Scholar] [CrossRef]
- Sun, Y.; Leng, P.; Guo, P.; Gao, H.; Liu, Y.; Li, C.; Li, Z.; Zhang, H. G protein coupled estrogen receptor attenuates mechanical stress-mediated apoptosis of chondrocyte in osteoarthritis via suppression of Piezo1. Mol. Med. 2021, 27, 96. [Google Scholar] [CrossRef]
- Hendrickx, G.; Fischer, V.; Liedert, A.; von Kroge, S.; Haffner-Luntzer, M.; Brylka, L.; Pawlus, E.; Schweizer, M.; Yorgan, T.; Baranowsky, A.; et al. Piezo1 Inactivation in Chondrocytes Impairs Trabecular Bone Formation. J. Bone Miner. Res. 2021, 36, 369–384. [Google Scholar] [CrossRef]
- Morris, J.A.; Kemp, J.P.; Youlten, S.E.; Laurent, L.; Logan, J.G.; Chai, R.C.; Vulpescu, N.A.; Forgetta, V.; Kleinman, A.; Mohanty, S.T.; et al. An atlas of genetic influences on osteoporosis in humans and mice. Nat. Genet. 2019, 51, 258–266. [Google Scholar] [CrossRef]
- Jin, Y.; Li, J.; Wang, Y.; Ye, R.; Feng, X.; Jing, Z.; Zhao, Z. Functional role of mechanosensitive ion channel Piezo1 in human periodontal ligament cells. Angle Orthod. 2015, 85, 87–94. [Google Scholar] [CrossRef]
- Wang, L.; You, X.; Lotinun, S.; Zhang, L.; Wu, N.; Zou, W. Mechanical sensing protein PIEZO1 regulates bone homeostasis via osteoblast-osteoclast crosstalk. Nat. Commun. 2020, 11, 282. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
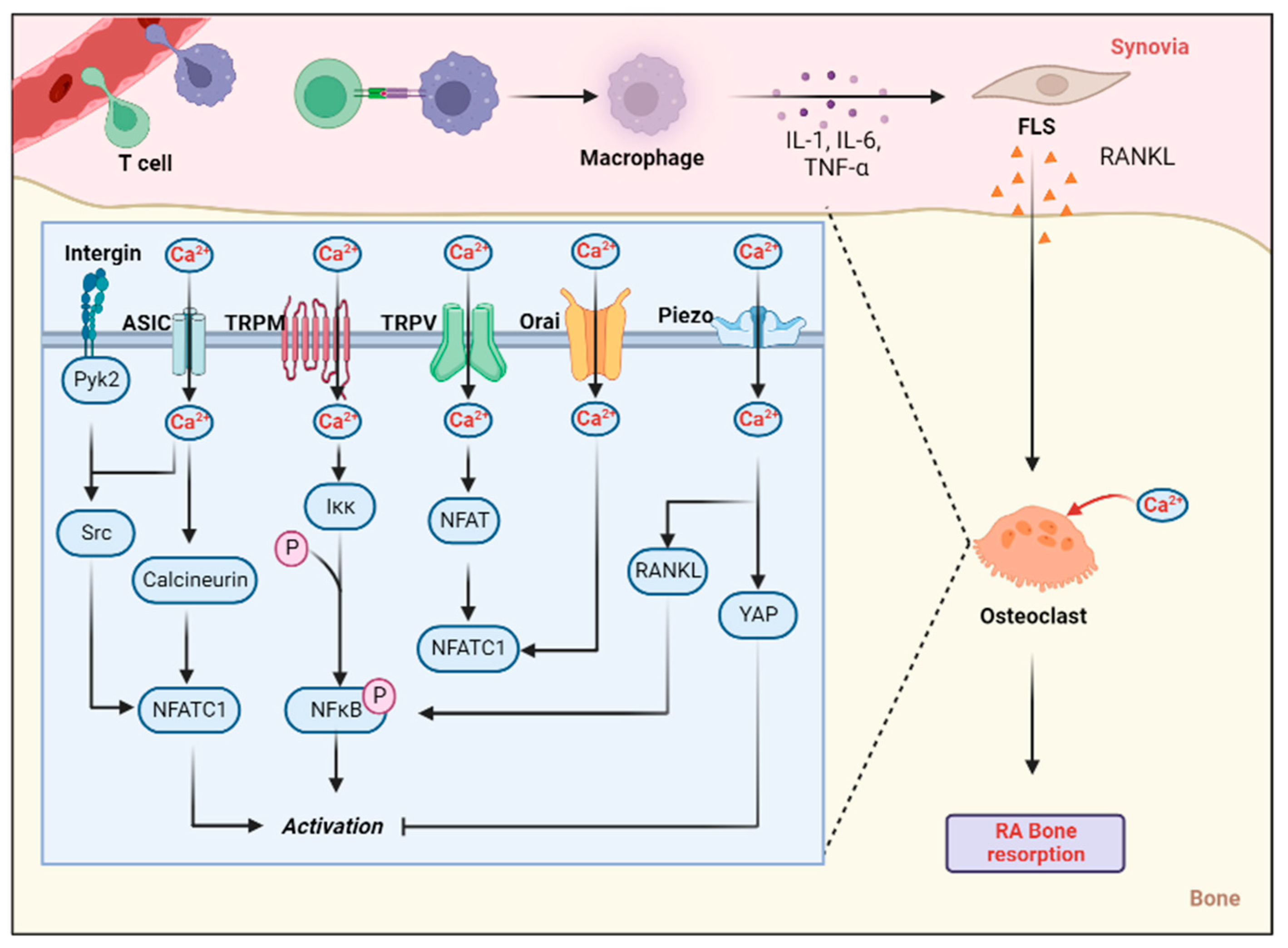{kind=link}
| Synovial Hyperplasia | Cartilage Destruction | Bone Damage | Inflammation | ||
|---|---|---|---|---|---|
| ASIC | ASIC1a | RA synovial invasion, Ras-associated C3 botulinum toxin substrate1 [48]; RA FLSs proliferation, ERK/MAPK [49,50] | ECM expression [53]; chondrocyte pyroptosis, calpain-2/calcineurin [56,57]; chondrocyte apoptosis, NF-κB [23] | osteoclast migration, integrin/Pyk2/Src [61]; osteoclast genesis, NFATc1 [63] | MIP-1a, IL-8, NFATc3/RANTES [49] |
| ASIC2a | chondroprotective effects of PcTx1 and APETx2, ERK/MAPK [64] | ||||
| ASIC3 | FLS apoptosis, [Ca2+]i [55]; Sensitivity of FLS to inflammatory mediators [69] | hyaluronic acid [68] | IL-6, MMP [69]; Pain behavior [70] | ||
| TRPM | TRPM7 | FLS apoptosis [80] | Ihh, chondrocyte damage [82]; collagen X, PI3K/Akt [81]; ferroptosis, PKCα-NOX4 [83] | osteoclast genesis [85]; osteoblast proliferation [84] | neutrophil chemotaxis, adhesion, invasiveness, Ca2+ influx [89]; Akt/mTOR [90] |
| TRPM8 | FLS apoptosis [79] | ||||
| TRPC | TRPC1 | osteoclast genesis [92] | |||
| TRPC5 | joint pain and swelling [94] | ||||
| TRPC6 | osteoclast activity [93] | ||||
| TRPV | TRPV1 | FLS apoptosis, [Ca2+]i, ROS mitochondrial membrane depolarization [100] | osteoblast differentiation [103]; bone resorption [101] | painful behavior [109] | |
| TRPV2 | aggressiveness of FLS [97] | osteoclast differentiation, Ca2+-NFAT signaling [111] | MMP-2, MMP-3 [97] | ||
| TRPV4 | proliferation of FLSs [113] | chondrocyte apoptosis [114] | osteoclast differentiation, RANKL [117]; NFATc1 transcription, cellular autophagy [118] | ||
| TRPV5 | chondrocyte apoptosis, MAPK and Akt/mTOR pathways [120] | osteoclast genesis [130]; osteoclast [Ca2+]i, bone resorption [129] | |||
| P2XR | P2X7R | chondrocyte apoptosis, IRE1-mTOR-PERK [144] | osteoclasts activation [150]; number of osteoclasts [147] | histone proteases [159]; IL-1β, IL-6 [158] | |
| Other channel | Orai1 | Fas signaling [168] | osteoclast synthesis, NFATc1 transduction [171]; osteoblast function, bone trabeculae [172] | ||
| Piezo1 | chondrocyte apoptosis, MAPK/ERK [177] | RANKL, RUNX2, osteoclasts differentiation [180]; osteoclast activity, bone resorption [181] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, H.-Y.; Yin, H.-X.; Li, S.-F.; Chen, Y.; Zhao, Y.-J.; Hu, W.; Zhou, R.-P. Calcium-Permeable Channels Cooperation for Rheumatoid Arthritis: Therapeutic Opportunities. Biomolecules 2022, 12, 1383. https://doi.org/10.3390/biom12101383
Liang H-Y, Yin H-X, Li S-F, Chen Y, Zhao Y-J, Hu W, Zhou R-P. Calcium-Permeable Channels Cooperation for Rheumatoid Arthritis: Therapeutic Opportunities. Biomolecules. 2022; 12(10):1383. https://doi.org/10.3390/biom12101383
Chicago/Turabian StyleLiang, Hong-Yu, Huan-Xin Yin, Shu-Fang Li, Yong Chen, Ying-Jie Zhao, Wei Hu, and Ren-Peng Zhou. 2022. "Calcium-Permeable Channels Cooperation for Rheumatoid Arthritis: Therapeutic Opportunities" Biomolecules 12, no. 10: 1383. https://doi.org/10.3390/biom12101383
APA StyleLiang, H.-Y., Yin, H.-X., Li, S.-F., Chen, Y., Zhao, Y.-J., Hu, W., & Zhou, R.-P. (2022). Calcium-Permeable Channels Cooperation for Rheumatoid Arthritis: Therapeutic Opportunities. Biomolecules, 12(10), 1383. https://doi.org/10.3390/biom12101383