
Resveratrol Treatment in Human Parkin-Mutant Fibroblasts Modulates cAMP and Calcium Homeostasis Regulating the Expression of Mitochondria-Associated Membranes Resident Proteins

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Cyclic Adenosine Monophosphate (cAMP) Assay
2.3. Quantitative Fluorimetric Measurement of Cytosolic and Mitochondrial Ca2+ Levels
2.4. Western Blot Analysis
2.5. Protein Measurement
2.6. Statistical Analysis
3. Results
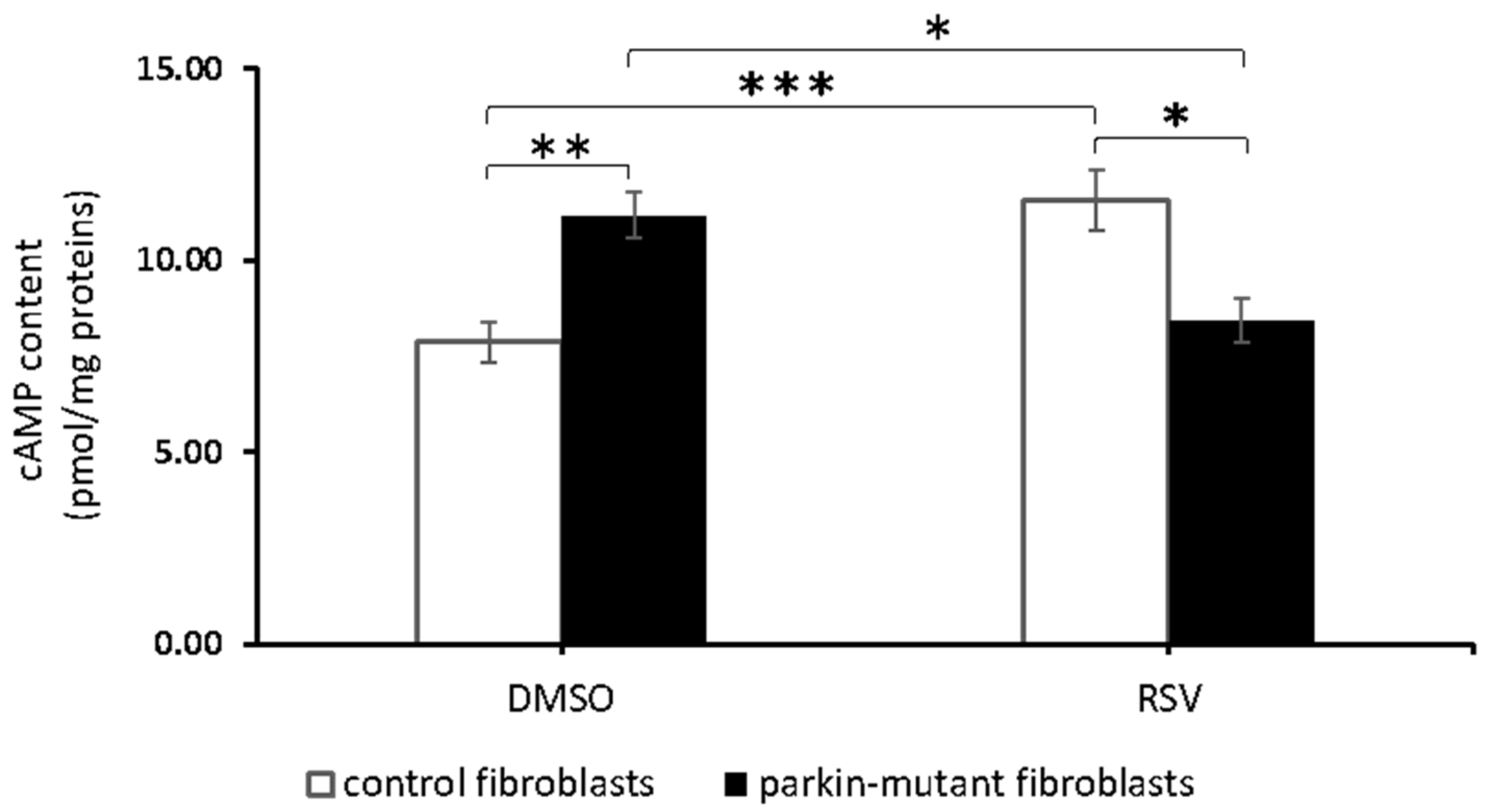
3.1. Resveratrol Decreases cAMP Level in Parkin-Mutant Fibroblasts
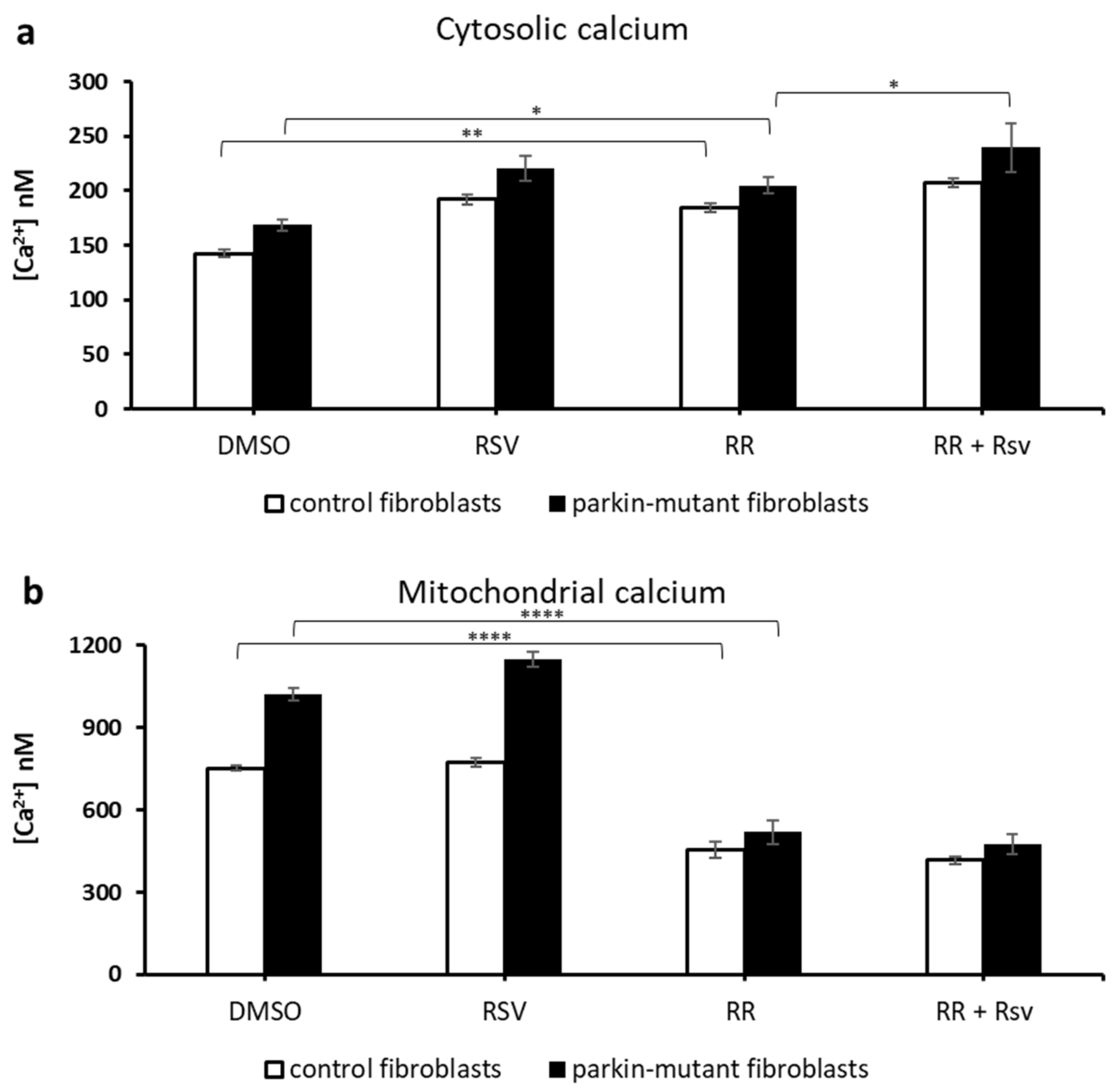
3.2. Resveratrol Further Increases Cytosolic and Mitochondrial Ca2+ Levels in Parkin-Mutant Fibroblasts
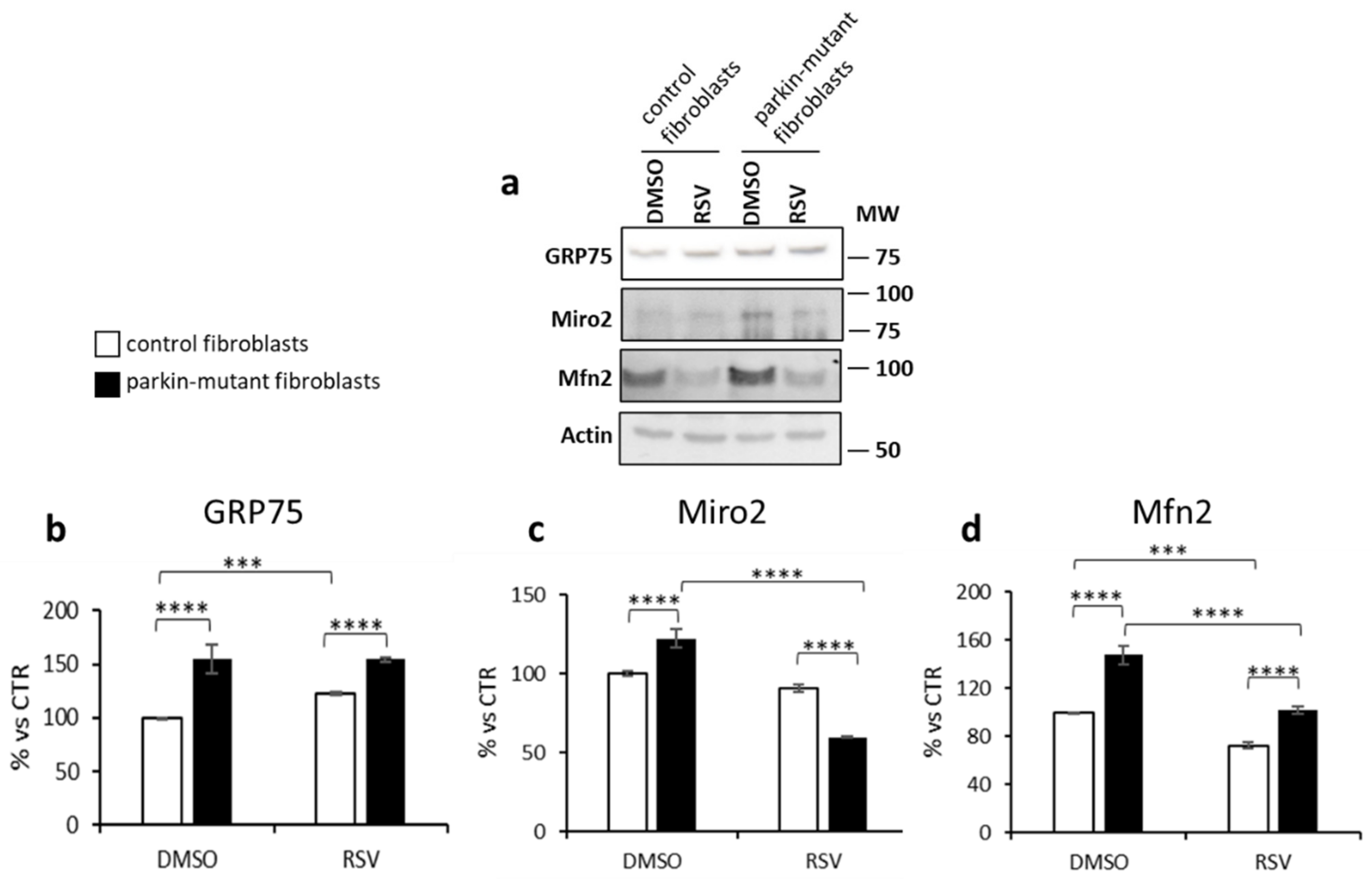
3.3. Parkin-Mutant Fibroblasts Show Higher Levels of GRP75, Miro2, and Mfn2 Proteins; Resveratrol Treatment Decreases Miro2 and Mfn2 Protein Levels
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cocco, T.; Pacelli, C.; Sgobbo, P.; Villani, G. Control of OXPHOS Efficiency by Complex I in Brain Mitochondria. Neurobiol. Aging 2009, 30, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Beal, M.F. Mitochondrial Dysfunction in Parkinson’s Disease. J. Neurochem. 2016, 139 (Suppl. S1), 216–231. [Google Scholar] [CrossRef] [PubMed]
- Zanellati, M.C.; Monti, V.; Barzaghi, C.; Reale, C.; Nardocci, N.; Albanese, A.; Valente, E.M.; Ghezzi, D.; Garavaglia, B. Mitochondrial Dysfunction in Parkinson Disease: Evidence in Mutant PARK2 Fibroblasts. Front. Genet. 2015, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Pacelli, C.; De Rasmo, D.; Signorile, A.; Grattagliano, I.; di Tullio, G.; D’Orazio, A.; Nico, B.; Comi, G.P.; Ronchi, D.; Ferranini, E.; et al. Mitochondrial Defect and PGC-1α Dysfunction in Parkin-Associated Familial Parkinson’s Disease. Biochim. Biophys. Acta 2011, 1812, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Ferretta, A.; Gaballo, A.; Tanzarella, P.; Piccoli, C.; Capitanio, N.; Nico, B.; Annese, T.; Di Paola, M.; Dell’aquila, C.; De Mari, M.; et al. Effect of Resveratrol on Mitochondrial Function: Implications in Parkin-Associated Familiar Parkinson’s Disease. Biochim. Biophys. Acta 2014, 1842, 902–915. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Guzman, J.N.; Sanchez-Padilla, J.; Schumacker, P.T. The Role of Calcium and Mitochondrial Oxidant Stress in the Loss of Substantia Nigra Pars Compacta Dopaminergic Neurons in Parkinson’s Disease. Neuroscience 2011, 198, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Calì, T.; Ottolini, D.; Brini, M. Mitochondria, Calcium, and Endoplasmic Reticulum Stress in Parkinson’s Disease. Biofactors 2011, 37, 228–240. [Google Scholar] [CrossRef]
- Tan, J.M.M.; Wong, E.S.P.; Lim, K.-L. Protein Misfolding and Aggregation in Parkinson’s Disease. Antioxid. Redox Signal. 2009, 11, 2119–2134. [Google Scholar] [CrossRef]
- Prasuhn, J.; Davis, R.L.; Kumar, K.R. Targeting Mitochondrial Impairment in Parkinson’s Disease: Challenges and Opportunities. Front. Cell Dev. Biol. 2020, 8, 615461. [Google Scholar] [CrossRef]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New Insights into the Complex Role of Mitochondria in Parkinson’s Disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Muqit, M.M. PINK1 and Parkin: Emerging Themes in Mitochondrial Homeostasis. Curr. Opin. Cell Biol. 2017, 45, 83–91. [Google Scholar] [CrossRef]
- Narendra, D.P.; Youle, R.J. Targeting Mitochondrial Dysfunction: Role for PINK1 and Parkin in Mitochondrial Quality Control. Antioxid. Redox Signal. 2011, 14, 1929–1938. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The Pathways of Mitophagy for Quality Control and Clearance of Mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial Dysfunction and Oxidative Damage in Parkin-Deficient Mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar] [CrossRef]
- Giguère, N.; Pacelli, C.; Saumure, C.; Bourque, M.-J.; Matheoud, D.; Levesque, D.; Slack, R.S.; Park, D.S.; Trudeau, L.-É. Comparative Analysis of Parkinson’s Disease-Associated Genes in Mice Reveals Altered Survival and Bioenergetics of Parkin-Deficient Dopamine Neurons. J. Biol. Chem. 2018, 293, 9580–9593. [Google Scholar] [CrossRef]
- Botella, J.A.; Bayersdorfer, F.; Gmeiner, F.; Schneuwly, S. Modelling Parkinson’s Disease in Drosophila. Neuromol. Med. 2009, 11, 268–280. [Google Scholar] [CrossRef]
- Greene, J.C.; Whitworth, A.J.; Kuo, I.; Andrews, L.A.; Feany, M.B.; Pallanck, L.J. Mitochondrial Pathology and Apoptotic Muscle Degeneration in Drosophila Parkin Mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 4078–4083. [Google Scholar] [CrossRef]
- Müftüoglu, M.; Elibol, B.; Dalmizrak, O.; Ercan, A.; Kulaksiz, G.; Ogüs, H.; Dalkara, T.; Ozer, N. Mitochondrial Complex I and IV Activities in Leukocytes from Patients with Parkin Mutations. Mov. Disord. 2004, 19, 544–548. [Google Scholar] [CrossRef]
- Mortiboys, H.; Thomas, K.J.; Koopman, W.J.H.; Klaffke, S.; Abou-Sleiman, P.; Olpin, S.; Wood, N.W.; Willems, P.H.G.M.; Smeitink, J.A.M.; Cookson, M.R.; et al. Mitochondrial Function and Morphology Are Impaired in Parkin-Mutant Fibroblasts. Ann. Neurol. 2008, 64, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Auburger, G.; Klinkenberg, M.; Drost, J.; Marcus, K.; Morales-Gordo, B.; Kunz, W.S.; Brandt, U.; Broccoli, V.; Reichmann, H.; Gispert, S.; et al. Primary Skin Fibroblasts as a Model of Parkinson’s Disease. Mol. Neurobiol. 2012, 46, 20–27. [Google Scholar] [CrossRef] [PubMed]
- González-Casacuberta, I.; Morén, C.; Juárez-Flores, D.-L.; Esteve-Codina, A.; Sierra, C.; Catalán-García, M.; Guitart-Mampel, M.; Tobías, E.; Milisenda, J.C.; Pont-Sunyer, C.; et al. Transcriptional Alterations in Skin Fibroblasts from Parkinson’s Disease Patients with Parkin Mutations. Neurobiol. Aging 2018, 65, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Vergara, D.; Ferraro, M.M.; Cascione, M.; del Mercato, L.L.; Leporatti, S.; Ferretta, A.; Tanzarella, P.; Pacelli, C.; Santino, A.; Maffia, M.; et al. Cytoskeletal Alterations and Biomechanical Properties of Parkin-Mutant Human Primary Fibroblasts. Cell Biochem. Biophys. 2015, 71, 1395–1404. [Google Scholar] [CrossRef]
- Vergara, D.; Gaballo, A.; Signorile, A.; Ferretta, A.; Tanzarella, P.; Pacelli, C.; Di Paola, M.; Cocco, T.; Maffia, M. Resveratrol Modulation of Protein Expression in Parkin-Mutant Human Skin Fibroblasts: A Proteomic Approach. Oxid. Med. Cell. Longev. 2017, 2017, 2198243. [Google Scholar] [CrossRef]
- Lippolis, R.; Siciliano, R.A.; Pacelli, C.; Ferretta, A.; Mazzeo, M.F.; Scacco, S.; Papa, F.; Gaballo, A.; Dell’Aquila, C.; De Mari, M.; et al. Altered Protein Expression Pattern in Skin Fibroblasts from Parkin-Mutant Early-Onset Parkinson’s Disease Patients. Biochim. Biophys. Acta 2015, 1852, 1960–1970. [Google Scholar] [CrossRef]
- Lobasso, S.; Tanzarella, P.; Vergara, D.; Maffia, M.; Cocco, T.; Corcelli, A. Lipid Profiling of Parkin-Mutant Human Skin Fibroblasts. J. Cell Physiol. 2017, 232, 3540–3551. [Google Scholar] [CrossRef]
- Guerra, F.; Girolimetti, G.; Beli, R.; Mitruccio, M.; Pacelli, C.; Ferretta, A.; Gasparre, G.; Cocco, T.; Bucci, C. Synergistic Effect of Mitochondrial and Lysosomal Dysfunction in Parkinson’s Disease. Cells 2019, 8, 452. [Google Scholar] [CrossRef]
- Pacelli, C.; Rotundo, G.; Lecce, L.; Menga, M.; Bidollari, E.; Scrima, R.; Cela, O.; Piccoli, C.; Cocco, T.; Vescovi, A.L.; et al. Parkin Mutation Affects Clock Gene-Dependent Energy Metabolism. Int. J. Mol. Sci. 2019, 20, 2772. [Google Scholar] [CrossRef]
- Tanzarella, P.; Ferretta, A.; Barile, S.N.; Ancona, M.; De Rasmo, D.; Signorile, A.; Papa, S.; Capitanio, N.; Pacelli, C.; Cocco, T. Increased Levels of CAMP by the Calcium-Dependent Activation of Soluble Adenylyl Cyclase in Parkin-Mutant Fibroblasts. Cells 2019, 8, 250. [Google Scholar] [CrossRef]
- Bhat, A.; Ray, B.; Mahalakshmi, A.M.; Tuladhar, S.; Nandakumar, D.N.; Srinivasan, M.; Essa, M.M.; Chidambaram, S.B.; Guillemin, G.J.; Sakharkar, M.K. Phosphodiesterase-4 Enzyme as a Therapeutic Target in Neurological Disorders. Pharm. Res. 2020, 160, 105078. [Google Scholar] [CrossRef]
- Papa, S.; Sardanelli, A.M.; Scacco, S.; Petruzzella, V.; Technikova-Dobrova, Z.; Vergari, R.; Signorile, A. The NADH: Ubiquinone Oxidoreductase (Complex I) of the Mammalian Respiratory Chain and the CAMP Cascade. J. Bioenerg. Biomembr. 2002, 34, 1–10. [Google Scholar] [CrossRef]
- Piccoli, C.; Scacco, S.; Bellomo, F.; Signorile, A.; Iuso, A.; Boffoli, D.; Scrima, R.; Capitanio, N.; Papa, S. CAMP Controls Oxygen Metabolism in Mammalian Cells. FEBS Lett. 2006, 580, 4539–4543. [Google Scholar] [CrossRef]
- Papa, S.; Scacco, S.; De Rasmo, D.; Signorile, A.; Papa, F.; Panelli, D.; Nicastro, A.; Scaringi, R.; Santeramo, A.; Roca, E.; et al. CAMP-Dependent Protein Kinase Regulates Post-Translational Processing and Expression of Complex I Subunits in Mammalian Cells. Biochim. Biophys. Acta 2010, 1797, 649–658. [Google Scholar] [CrossRef]
- Valsecchi, F.; Ramos-Espiritu, L.S.; Buck, J.; Levin, L.R.; Manfredi, G. CAMP and Mitochondria. Physiology 2013, 28, 199–209. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, L.; Qi, Y.; Xu, H. Mitochondrial CAMP Signaling. Cell. Mol. Life Sci. 2016, 73, 4577–4590. [Google Scholar] [CrossRef]
- De Rasmo, D.; Micelli, L.; Santeramo, A.; Signorile, A.; Lattanzio, P.; Papa, S. CAMP Regulates the Functional Activity, Coupling Efficiency and Structural Organization of Mammalian FOF1 ATP Synthase. Biochim. Biophys. Acta 2016, 1857, 350–358. [Google Scholar] [CrossRef]
- Signorile, A.; Santeramo, A.; Tamma, G.; Pellegrino, T.; D’Oria, S.; Lattanzio, P.; De Rasmo, D. Mitochondrial CAMP Prevents Apoptosis Modulating Sirt3 Protein Level and OPA1 Processing in Cardiac Myoblast Cells. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 355–366. [Google Scholar] [CrossRef]
- Valsecchi, F.; Konrad, C.; D’Aurelio, M.; Ramos-Espiritu, L.S.; Stepanova, A.; Burstein, S.R.; Galkin, A.; Magranè, J.; Starkov, A.; Buck, J.; et al. Distinct Intracellular SAC-CAMP Domains Regulate ER Ca2+ Signaling and OXPHOS Function. J. Cell Sci. 2017, 130, 3713–3727. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Gerbino, A.; Lefkimmiatis, K. Shaping Mitochondrial Dynamics: The Role of CAMP Signalling. Biochem. Biophys. Res. Commun. 2018, 500, 65–74. [Google Scholar] [CrossRef]
- Modesti, L.; Danese, A.; Angela Maria Vitto, V.; Ramaccini, D.; Aguiari, G.; Gafà, R.; Lanza, G.; Giorgi, C.; Pinton, P. Mitochondrial Ca2+ Signaling in Health, Disease and Therapy. Cells 2021, 10, 1317. [Google Scholar] [CrossRef]
- Del Rio, D.; Rodriguez-Mateos, A.; Spencer, J.P.E.; Tognolini, M.; Borges, G.; Crozier, A. Dietary (Poly)Phenolics in Human Health: Structures, Bioavailability, and Evidence of Protective Effects against Chronic Diseases. Antioxid. Redox Signal. 2013, 18, 1818–1892. [Google Scholar] [CrossRef]
- Sun, A.Y.; Wang, Q.; Simonyi, A.; Sun, G.Y. Resveratrol as a Therapeutic Agent for Neurodegenerative Diseases. Mol. Neurobiol. 2010, 41, 375–383. [Google Scholar] [CrossRef]
- Magalingam, K.B.; Radhakrishnan, A.K.; Haleagrahara, N. Protective Mechanisms of Flavonoids in Parkinson’s Disease. Oxid. Med. Cell. Longev. 2015, 2015, 314560. [Google Scholar] [CrossRef]
- Park, S.-J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol Ameliorates Aging-Related Metabolic Phenotypes by Inhibiting CAMP Phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef]
- Sareen, D.; Darjatmoko, S.R.; Albert, D.M.; Polans, A.S. Mitochondria, Calcium, and Calpain Are Key Mediators of Resveratrol-Induced Apoptosis in Breast Cancer. Mol. Pharm. 2007, 72, 1466–1475. [Google Scholar] [CrossRef]
- Campos-Toimil, M.; Elíes, J.; Orallo, F. Trans- and Cis-Resveratrol Increase Cytoplasmic Calcium Levels in A7r5 Vascular Smooth Muscle Cells. Mol. Nutr. Food Res. 2005, 49, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Elíes, J.; Cuíñas, A.; García-Morales, V.; Orallo, F.; Campos-Toimil, M. Trans-Resveratrol Simultaneously Increases Cytoplasmic Ca2+ Levels and Nitric Oxide Release in Human Endothelial Cells. Mol. Nutr. Food Res. 2011, 55, 1237–1248. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.A.; Oblad, R.V.; Mecham, J.C.; Kenealey, J.D. Resveratrol Inhibits Plasma Membrane Ca2+-ATPase Inducing an Increase in Cytoplasmic Calcium. Biochem. Biophys. Rep. 2016, 7, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, D.H.W.; Bezstarosti, K.; Gurusamy, N.; Luijk, K.; Verhoeven, A.J.M.; Rijkers, E.-J.; Demmers, J.A.; Lamers, J.M.J.; Maulik, N.; Das, D.K. Identification by a Differential Proteomic Approach of the Induced Stress and Redox Proteins by Resveratrol in the Normal and Diabetic Rat Heart. J. Cell. Mol. Med. 2008, 12, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Luo, B.; Gong, L. Resveratrol Reduces the Apoptosis Induced by Cigarette Smoke Extract by Upregulating MFN2. PLoS ONE 2017, 12, e0175009. [Google Scholar] [CrossRef]
- Dolgin, E. How Secret Conversations inside Cells Are Transforming Biology. Nature 2019, 567, 162–164. [Google Scholar] [CrossRef]
- Vance, J.E. MAM (Mitochondria-Associated Membranes) in Mammalian Cells: Lipids and Beyond. Biochim. Biophys. Acta 2014, 1841, 595–609. [Google Scholar] [CrossRef]
- Marchi, S.; Patergnani, S.; Pinton, P. The Endoplasmic Reticulum-Mitochondria Connection: One Touch, Multiple Functions. Biochim. Biophys. Acta 2014, 1837, 461–469. [Google Scholar] [CrossRef]
- Krols, M.; Bultynck, G.; Janssens, S. ER-Mitochondria Contact Sites: A New Regulator of Cellular Calcium Flux Comes into Play. J. Cell Biol. 2016, 214, 367–370. [Google Scholar] [CrossRef]
- Csordás, G.; Renken, C.; Várnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnóczky, G. Structural and Functional Features and Significance of the Physical Linkage between ER and Mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef]
- Csordás, G.; Várnai, P.; Golenár, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnóczky, G. Imaging Interorganelle Contacts and Local Calcium Dynamics at the ER-Mitochondrial Interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef]
- Filadi, R.; Theurey, P.; Pizzo, P. The Endoplasmic Reticulum-Mitochondria Coupling in Health and Disease: Molecules, Functions and Significance. Cell Calcium 2017, 62, 1–15. [Google Scholar] [CrossRef]
- Guardia-Laguarta, C.; Liu, Y.; Lauritzen, K.H.; Erdjument-Bromage, H.; Martin, B.; Swayne, T.C.; Jiang, X.; Przedborski, S. PINK1 Content in Mitochondria Is Regulated by ER-Associated Degradation. J. NeuroSci. 2019, 39, 7074–7085. [Google Scholar] [CrossRef]
- Gelmetti, V.; De Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; Di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. PINK1 and BECN1 Relocalize at Mitochondria-Associated Membranes during Mitophagy and Promote ER-Mitochondria Tethering and Autophagosome Formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef]
- Vance, J.E. Phospholipid Synthesis in a Membrane Fraction Associated with Mitochondria. J. Biol. Chem. 1990, 265, 7248–7256. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-Mediated Coupling of Endoplasmic Reticulum and Mitochondrial Ca2+ Channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Erpapazoglou, Z.; Mouton-Liger, F.; Corti, O. From Dysfunctional Endoplasmic Reticulum-Mitochondria Coupling to Neurodegeneration. Neurochem. Int. 2017, 109, 171–183. [Google Scholar] [CrossRef]
- Kornmann, B.; Currie, E.; Collins, S.R.; Schuldiner, M.; Nunnari, J.; Weissman, J.S.; Walter, P. An ER-Mitochondria Tethering Complex Revealed by a Synthetic Biology Screen. Science 2009, 325, 477–481. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Bononi, A.; Romagnoli, A.; Messina, A.; De Pinto, V.; Pinton, P.; Rizzuto, R. VDAC1 Selectively Transfers Apoptotic Ca2+ Signals to Mitochondria. Cell Death Differ. 2012, 19, 267–273. [Google Scholar] [CrossRef]
- Modi, S.; López-Doménech, G.; Halff, E.F.; Covill-Cooke, C.; Ivankovic, D.; Melandri, D.; Arancibia-Cárcamo, I.L.; Burden, J.J.; Lowe, A.R.; Kittler, J.T. Miro Clusters Regulate ER-Mitochondria Contact Sites and Link Cristae Organization to the Mitochondrial Transport Machinery. Nat. Commun. 2019, 10, 4399. [Google Scholar] [CrossRef]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 Tethers Endoplasmic Reticulum to Mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Lee, S.; Lee, K.-S.; Huh, S.; Liu, S.; Lee, D.-Y.; Hong, S.H.; Yu, K.; Lu, B. Polo Kinase Phosphorylates Miro to Control ER-Mitochondria Contact Sites and Mitochondrial Ca2+ Homeostasis in Neural Stem Cell Development. Dev. Cell 2016, 37, 174–189. [Google Scholar] [CrossRef]
- Hajnóczky, G.; Robb-Gaspers, L.D.; Seitz, M.B.; Thomas, A.P. Decoding of Cytosolic Calcium Oscillations in the Mitochondria. Cell 1995, 82, 415–424. [Google Scholar] [CrossRef]
- Mirabet, M.; Mallol, J.; Lluis, C.; Franco, R. Calcium Mobilization in Jurkat Cells via A2b Adenosine Receptors. Br. J. Pharm. 1997, 122, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A New Generation of Ca2+ Indicators with Greatly Improved Fluorescence Properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]
- Fabbri, E.; Brighenti, L.; Ottolenghi, C. Inhibition of Adenylate Cyclase of Catfish and Rat Hepatocyte Membranes by 9-(Tetrahydro-2-Furyl)Adenine (SQ 22536). J. Enzyme Inhib. 1991, 5, 87–98. [Google Scholar] [CrossRef]
- Sánchez-Melgar, A.; Albasanz, J.L.; Guixà-González, R.; Saleh, N.; Selent, J.; Martín, M. The Antioxidant Resveratrol Acts as a Non-Selective Adenosine Receptor Agonist. Free Radic. Biol. Med. 2019, 135, 261–273. [Google Scholar] [CrossRef]
- Halls, M.L.; Cooper, D.M.F. Regulation by Ca2+-Signaling Pathways of Adenylyl Cyclases. Cold Spring Harb. Perspect. Biol. 2011, 3, a004143. [Google Scholar] [CrossRef]
- Zhang, J.-Q.; Wu, P.-F.; Long, L.-H.; Chen, Y.; Hu, Z.-L.; Ni, L.; Wang, F.; Chen, J.-G. Resveratrol Promotes Cellular Glucose Utilization in Primary Cultured Cortical Neurons via Calcium-Dependent Signaling Pathway. J. Nutr. Biochem. 2013, 24, 629–637. [Google Scholar] [CrossRef]
- Rizzuto, R.; Simpson, A.W.; Brini, M.; Pozzan, T. Rapid Changes of Mitochondrial Ca2+ Revealed by Specifically Targeted Recombinant Aequorin. Nature 1992, 358, 325–327. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. The Machineries, Regulation and Cellular Functions of Mitochondrial Calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The Mitochondrial Calcium Uniporter Is a Highly Selective Ion Channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef]
- Liu, J.C.; Liu, J.; Holmström, K.M.; Menazza, S.; Parks, R.J.; Fergusson, M.M.; Yu, Z.-X.; Springer, D.A.; Halsey, C.; Liu, C.; et al. MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cell Rep. 2016, 16, 1561–1573. [Google Scholar] [CrossRef]
- Calì, T.; Ottolini, D.; Negro, A.; Brini, M. Enhanced Parkin Levels Favor ER-Mitochondria Crosstalk and Guarantee Ca2+ Transfer to Sustain Cell Bioenergetics. Biochim. Biophys. Acta 2013, 1832, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Basso, V.; Marchesan, E.; Peggion, C.; Chakraborty, J.; von Stockum, S.; Giacomello, M.; Ottolini, D.; Debattisti, V.; Caicci, F.; Tasca, E.; et al. Regulation of ER-Mitochondria Contacts by Parkin via Mfn2. Pharm. Res. 2018, 138, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Bernard-Marissal, N.; Moullan, N.; D’Amico, D.; Auwerx, J.; Moore, D.J.; Knott, G.; Aebischer, P.; Schneider, B.L. Parkin Functionally Interacts with PGC-1α to Preserve Mitochondria and Protect Dopaminergic Neurons. Hum. Mol. Genet. 2017, 26, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Gautier, C.A.; Erpapazoglou, Z.; Mouton-Liger, F.; Muriel, M.P.; Cormier, F.; Bigou, S.; Duffaure, S.; Girard, M.; Foret, B.; Iannielli, A.; et al. The Endoplasmic Reticulum-Mitochondria Interface Is Perturbed in PARK2 Knockout Mice and Patients with PARK2 Mutations. Hum. Mol. Genet. 2016, 25, 2972–2984. [Google Scholar] [CrossRef]
- Treiman, M.; Caspersen, C.; Christensen, S.B. A Tool Coming of Age: Thapsigargin as an Inhibitor of Sarco-Endoplasmic Reticulum Ca2+-ATPases. Trends Pharmacol. Sci. 1998, 19, 131–135. [Google Scholar] [CrossRef]
- Fruen, B.R.; Mickelson, J.R.; Louis, C.F. Dantrolene Inhibition of Sarcoplasmic Reticulum Ca2+ Release by Direct and Specific Action at Skeletal Muscle Ryanodine Receptors. J. Biol. Chem. 1997, 272, 26965–26971. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef]
- Csordás, G.; Hajnóczky, G. Sorting of Calcium Signals at the Junctions of Endoplasmic Reticulum and Mitochondria. Cell Calcium 2001, 29, 249–262. [Google Scholar] [CrossRef]
- Wu, H.; Carvalho, P.; Voeltz, G.K. Here, there, and everywhere: The importance of ER membrane contact sites. Science 2018, 361, eaan5835. [Google Scholar] [CrossRef]
- Tellone, E.; Galtieri, A.; Russo, A.; Giardina, B.; Ficarra, S. Resveratrol: A Focus on Several Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2015, 2015, 392169. [Google Scholar] [CrossRef]
- Xia, N.; Daiber, A.; Förstermann, U.; Li, H. Antioxidant Effects of Resveratrol in the Cardiovascular System. Br. J. Pharm. 2017, 174, 1633–1646. [Google Scholar] [CrossRef]
- De Oliveira, M.R.; Nabavi, S.F.; Manayi, A.; Daglia, M.; Hajheydari, Z.; Nabavi, S.M. Resveratrol and the Mitochondria: From Triggering the Intrinsic Apoptotic Pathway to Inducing Mitochondrial Biogenesis, a Mechanistic View. Biochim. Biophys. Acta 2016, 1860, 727–745. [Google Scholar] [CrossRef]
- Dasgupta, B.; Milbrandt, J. Resveratrol Stimulates AMP Kinase Activity in Neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 7217–7222. [Google Scholar] [CrossRef]
- Chiang, M.-C.; Nicol, C.J.; Cheng, Y.-C. Resveratrol Activation of AMPK-Dependent Pathways Is Neuroprotective in Human Neural Stem Cells against Amyloid-Beta-Induced Inflammation and Oxidative Stress. Neurochem. Int. 2018, 115, 1–10. [Google Scholar] [CrossRef]
- Price, N.L.; Gomes, A.P.; Ling, A.J.Y.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 Is Required for AMPK Activation and the Beneficial Effects of Resveratrol on Mitochondrial Function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E.; Janle, E.M.; Lobo, J.; Ferruzzi, M.G.; Davies, P.; et al. AMP-Activated Protein Kinase Signaling Activation by Resveratrol Modulates Amyloid-Beta Peptide Metabolism. J. Biol. Chem. 2010, 285, 9100–9113. [Google Scholar] [CrossRef]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol Improves Mitochondrial Function and Protects against Metabolic Disease by Activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-Dependent Protein Kinase Kinase-Beta Is an Alternative Upstream Kinase for AMP-Activated Protein Kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef]
- Hurley, R.L.; Anderson, K.A.; Franzone, J.M.; Kemp, B.E.; Means, A.R.; Witters, L.A. The Ca2+/Calmodulin-Dependent Protein Kinase Kinases Are AMP-Activated Protein Kinase Kinases. J. Biol. Chem. 2005, 280, 29060–29066. [Google Scholar] [CrossRef]
- Nakamura, K.; Zuppini, A.; Arnaudeau, S.; Lynch, J.; Ahsan, I.; Krause, R.; Papp, S.; De Smedt, H.; Parys, J.B.; Muller-Esterl, W.; et al. Functional Specialization of Calreticulin Domains. J. Cell Biol. 2001, 154, 961–972. [Google Scholar] [CrossRef]
- Gonzalez, L.L.; Garrie, K.; Turner, M.D. Role of S100 Proteins in Health and Disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118677. [Google Scholar] [CrossRef] [PubMed]
- Sandebring, A.; Dehvari, N.; Perez-Manso, M.; Thomas, K.J.; Karpilovski, E.; Cookson, M.R.; Cowburn, R.F.; Cedazo-Mínguez, A. Parkin Deficiency Disrupts Calcium Homeostasis by Modulating Phospholipase C Signalling. FEBS J. 2009, 276, 5041–5052. [Google Scholar] [CrossRef] [PubMed]
- Pozzan, T.; Rizzuto, R.; Volpe, P.; Meldolesi, J. Molecular and Cellular Physiology of Intracellular Calcium Stores. Physiol. Rev. 1994, 74, 595–636. [Google Scholar] [CrossRef] [PubMed]
- Matteucci, A.; Patron, M.; Vecellio Reane, D.; Gastaldello, S.; Amoroso, S.; Rizzuto, R.; Brini, M.; Raffaello, A.; Calì, T. Parkin-Dependent Regulation of the MCU Complex Component MICU1. Sci. Rep. 2018, 8, 14199. [Google Scholar] [CrossRef]
- García-Casas, P.; Arias-Del-Val, J.; Alvarez-Illera, P.; Fonteriz, R.I.; Montero, M.; Alvarez, J. Inhibition of Sarco-Endoplasmic Reticulum Ca2+ ATPase Extends the Lifespan in C. Elegans Worms. Front. Pharm. 2018, 9, 669. [Google Scholar] [CrossRef] [PubMed]
- Arnaudeau, S.; Kelley, W.L.; Walsh, J.V.; Demaurex, N. Mitochondria Recycle Ca2+ to the Endoplasmic Reticulum and Prevent the Depletion of Neighboring Endoplasmic Reticulum Regions. J. Biol. Chem. 2001, 276, 29430–29439. [Google Scholar] [CrossRef] [PubMed]
- Kopach, O.; Kruglikov, I.; Pivneva, T.; Voitenko, N.; Fedirko, N. Functional Coupling between Ryanodine Receptors, Mitochondria and Ca2+ ATPases in Rat Submandibular Acinar Cells. Cell Calcium 2008, 43, 469–481. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pozzan, T. Microdomains of Intracellular Ca2+: Molecular Determinants and Functional Consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef]
- Bravo-Sagua, R.; Rodriguez, A.E.; Kuzmicic, J.; Gutierrez, T.; Lopez-Crisosto, C.; Quiroga, C.; Díaz-Elizondo, J.; Chiong, M.; Gillette, T.G.; Rothermel, B.A.; et al. Cell Death and Survival through the Endoplasmic Reticulum-Mitochondrial Axis. Curr. Mol. Med. 2013, 13, 317–329. [Google Scholar] [CrossRef]
- Jakob, R.; Beutner, G.; Sharma, V.K.; Duan, Y.; Gross, R.A.; Hurst, S.; Jhun, B.S.; O-Uchi, J.; Sheu, S.-S. Molecular and Functional Identification of a Mitochondrial Ryanodine Receptor in Neurons. NeuroSci. Lett. 2014, 575, 7–12. [Google Scholar] [CrossRef]
- Green, D.R.; Wang, R. Calcium and Energy: Making the Cake and Eating It Too? Cell 2010, 142, 200–202. [Google Scholar] [CrossRef][Green Version]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; et al. PINK1-Associated Parkinson’s Disease Is Caused by Neuronal Vulnerability to Calcium-Induced Cell Death. Mol. Cell 2009, 33, 627–638. [Google Scholar] [CrossRef]
- Kwong, J.Q.; Molkentin, J.D. Physiological and Pathological Roles of the Mitochondrial Permeability Transition Pore in the Heart. Cell Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef]
- Guo, Y.-J.; Dong, S.-Y.; Cui, X.-X.; Feng, Y.; Liu, T.; Yin, M.; Kuo, S.-H.; Tan, E.-K.; Zhao, W.-J.; Wu, Y.-C. Resveratrol Alleviates MPTP-Induced Motor Impairments and Pathological Changes by Autophagic Degradation of α-Synuclein via SIRT1-Deacetylated LC3. Mol. Nutr. Food Res. 2016, 60, 2161–2175. [Google Scholar] [CrossRef]
- Liao, Z.; Liu, D.; Tang, L.; Yin, D.; Yin, S.; Lai, S.; Yao, J.; He, M. Long-Term Oral Resveratrol Intake Provides Nutritional Preconditioning against Myocardial Ischemia/Reperfusion Injury: Involvement of VDAC1 Downregulation. Mol. Nutr. Food Res. 2015, 59, 454–464. [Google Scholar] [CrossRef]
- Honrath, B.; Metz, I.; Bendridi, N.; Rieusset, J.; Culmsee, C.; Dolga, A.M. Glucose-Regulated Protein 75 Determines ER-Mitochondrial Coupling and Sensitivity to Oxidative Stress in Neuronal Cells. Cell Death Discov. 2017, 3, 17076. [Google Scholar] [CrossRef]
- Lee, S.; Wang, W.; Hwang, J.; Namgung, U.; Min, K.-T. Increased ER-Mitochondria Tethering Promotes Axon Regeneration. Proc. Natl. Acad. Sci. USA 2019, 116, 16074–16079. [Google Scholar] [CrossRef]
- Cosson, P.; Marchetti, A.; Ravazzola, M.; Orci, L. Mitofusin-2 Independent Juxtaposition of Endoplasmic Reticulum and Mitochondria: An Ultrastructural Study. PLoS ONE 2012, 7, e46293. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Mitofusin 2 Ablation Increases Endoplasmic Reticulum–Mitochondria Coupling. Proc. Natl. Acad. Sci. USA 2015, 112, E2174–E2181. [Google Scholar] [CrossRef]
- Detmer, S.A.; Chan, D.C. Functions and Dysfunctions of Mitochondrial Dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. On the Role of Mitofusin 2 in Endoplasmic Reticulum-Mitochondria Tethering. Proc. Natl. Acad. Sci. USA 2017, 114, E2266–E2267. [Google Scholar] [CrossRef]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernández-Alvarez, M.I.; et al. Critical Reappraisal Confirms That Mitofusin 2 Is an Endoplasmic Reticulum-Mitochondria Tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [Google Scholar] [CrossRef]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.-F.; Karbowski, M.; Youle, R.J. Proteasome and P97 Mediate Mitophagy and Degradation of Mitofusins Induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef]
- Gegg, M.E.; Cooper, J.M.; Chau, K.-Y.; Rojo, M.; Schapira, A.H.V.; Taanman, J.-W. Mitofusin 1 and Mitofusin 2 Are Ubiquitinated in a PINK1/Parkin-Dependent Manner upon Induction of Mitophagy. Hum. Mol. Genet. 2010, 19, 4861–4870. [Google Scholar] [CrossRef]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin Target Miro for Phosphorylation and Degradation to Arrest Mitochondrial Motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef]
- Liu, S.; Sawada, T.; Lee, S.; Yu, W.; Silverio, G.; Alapatt, P.; Millan, I.; Shen, A.; Saxton, W.; Kanao, T.; et al. Parkinson’s Disease-Associated Kinase PINK1 Regulates Miro Protein Level and Axonal Transport of Mitochondria. PLoS Genet. 2012, 8, e1002537. [Google Scholar] [CrossRef]
- Lee, K.-S.; Huh, S.; Lee, S.; Wu, Z.; Kim, A.-K.; Kang, H.-Y.; Lu, B. Altered ER-Mitochondria Contact Impacts Mitochondria Calcium Homeostasis and Contributes to Neurodegeneration in Vivo in Disease Models. Proc. Natl. Acad. Sci. USA 2018, 115, E8844–E8853. [Google Scholar] [CrossRef]
- Liang, T.; Hang, W.; Chen, J.; Wu, Y.; Wen, B.; Xu, K.; Ding, B.; Chen, J. ApoE4 (Δ272–299) Induces Mitochondrial-Associated Membrane Formation and Mitochondrial Impairment by Enhancing GRP75-Modulated Mitochondrial Calcium Overload in Neuron. Cell Biosci. 2021, 11, 50. [Google Scholar] [CrossRef]
- Yuan, M.; Gong, M.; Zhang, Z.; Meng, L.; Tse, G.; Zhao, Y.; Bao, Q.; Zhang, Y.; Yuan, M.; Liu, X.; et al. Hyperglycemia Induces Endoplasmic Reticulum Stress in Atrial Cardiomyocytes, and Mitofusin-2 Downregulation Prevents Mitochondrial Dysfunction and Subsequent Cell Death. Oxid. Med. Cell. Longev. 2020, 2020, 6569728. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Carreira, R.S. Autophagy in Health and Disease. 5. Mitophagy as a Way of Life. Am. J. Physiol. Cell Physiol. 2010, 299, C203–C210. [Google Scholar] [CrossRef]
- Chen, K.-G.; Kang, R.-R.; Sun, Q.; Liu, C.; Ma, Z.; Liu, K.; Deng, Y.; Liu, W.; Xu, B. Resveratrol Ameliorates Disorders of Mitochondrial Biogenesis and Mitophagy in Rats Continuously Exposed to Benzo(a)Pyrene from Embryonic Development through Adolescence. Toxicology 2020, 442, 152532. [Google Scholar] [CrossRef] [PubMed]
- Saunier, E.; Antonio, S.; Regazzetti, A.; Auzeil, N.; Laprévote, O.; Shay, J.W.; Coumoul, X.; Barouki, R.; Benelli, C.; Huc, L.; et al. Resveratrol Reverses the Warburg Effect by Targeting the Pyruvate Dehydrogenase Complex in Colon Cancer Cells. Sci. Rep. 2017, 7, 6945. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Signorile, A.; Ferretta, A.; Pacelli, C.; Capitanio, N.; Tanzarella, P.; Matrella, M.L.; Valletti, A.; De Rasmo, D.; Cocco, T. Resveratrol Treatment in Human Parkin-Mutant Fibroblasts Modulates cAMP and Calcium Homeostasis Regulating the Expression of Mitochondria-Associated Membranes Resident Proteins. Biomolecules 2021, 11, 1511. https://doi.org/10.3390/biom11101511
Signorile A, Ferretta A, Pacelli C, Capitanio N, Tanzarella P, Matrella ML, Valletti A, De Rasmo D, Cocco T. Resveratrol Treatment in Human Parkin-Mutant Fibroblasts Modulates cAMP and Calcium Homeostasis Regulating the Expression of Mitochondria-Associated Membranes Resident Proteins. Biomolecules. 2021; 11(10):1511. https://doi.org/10.3390/biom11101511
Chicago/Turabian StyleSignorile, Anna, Anna Ferretta, Consiglia Pacelli, Nazzareno Capitanio, Paola Tanzarella, Maria Laura Matrella, Alessio Valletti, Domenico De Rasmo, and Tiziana Cocco. 2021. "Resveratrol Treatment in Human Parkin-Mutant Fibroblasts Modulates cAMP and Calcium Homeostasis Regulating the Expression of Mitochondria-Associated Membranes Resident Proteins" Biomolecules 11, no. 10: 1511. https://doi.org/10.3390/biom11101511
APA StyleSignorile, A., Ferretta, A., Pacelli, C., Capitanio, N., Tanzarella, P., Matrella, M. L., Valletti, A., De Rasmo, D., & Cocco, T. (2021). Resveratrol Treatment in Human Parkin-Mutant Fibroblasts Modulates cAMP and Calcium Homeostasis Regulating the Expression of Mitochondria-Associated Membranes Resident Proteins. Biomolecules, 11(10), 1511. https://doi.org/10.3390/biom11101511