The Ubiquitin–Proteasome System in Immune Cells



Abstract
1. The Ubiquitin–Proteasome System (UPS): Structure and Function
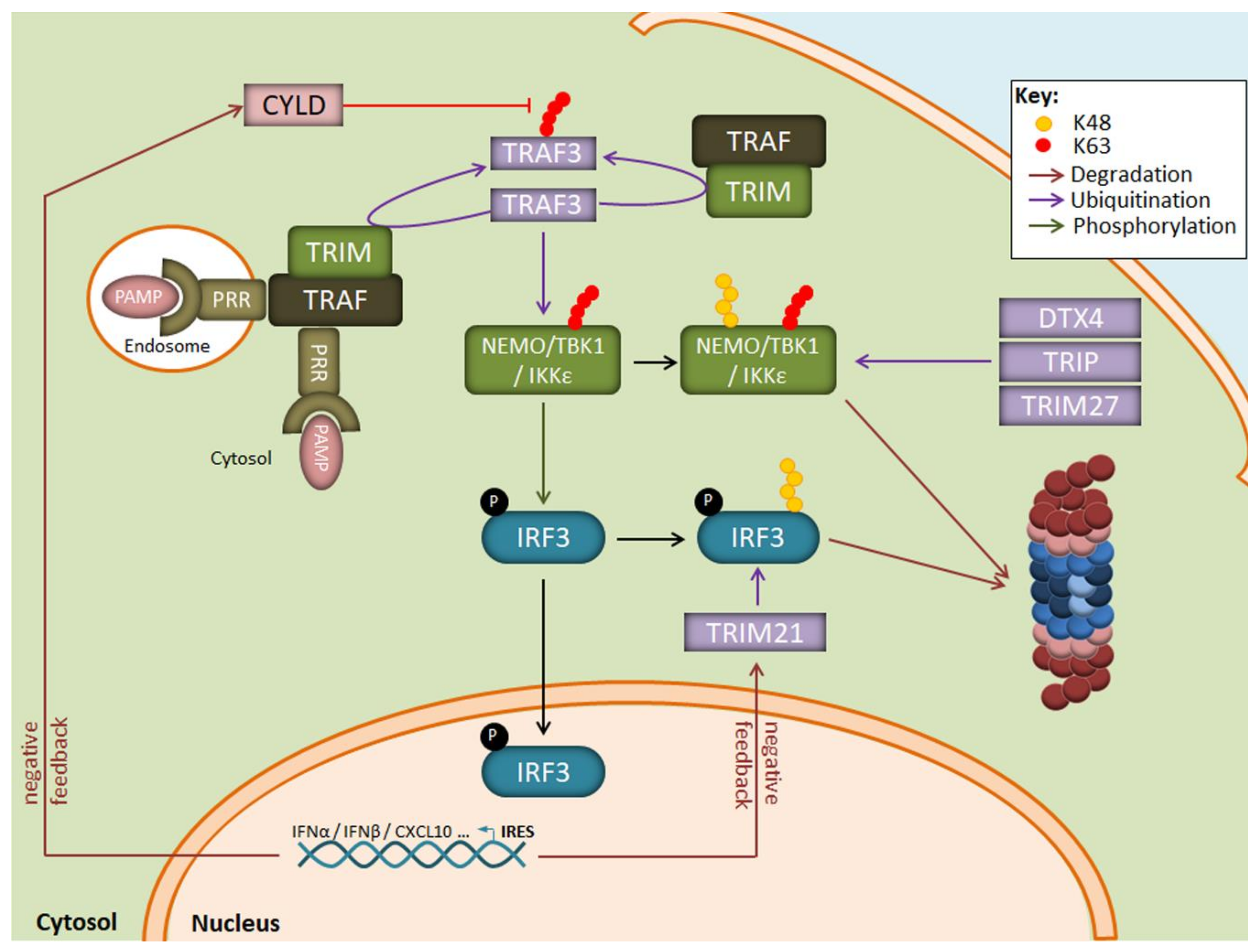
2. The UPS Is a Major Regulator of Cell Signalling in Response to Pathogens
3. The UPS as a Major Guardian of Proteostasis during Activation of the Immune System
4. Role of the UPS in MHC Class I Antigen Processing and Presentation
5. The UPS in Disease
5.1. Autoinflammatory Diseases
5.2. Manipulation of the UPS by Pathogens
5.3. Tumour Diseases
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ciehanover, A.; Hod, Y.; Hershko, A. A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem. Biophys. Res. Commun. 1978, 81, 1100–1105. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Elias, S.; Heller, H.; Ferber, S.; Hershko, A. Characterization of the heat-stable polypeptide of the ATP-dependent proteolytic system from reticulocytes. J. Biol. Chem. 1980, 255, 7525–7528. [Google Scholar] [PubMed]
- Hershko, A.; Ciechanover, A.; Rose, I.A. Resolution of the ATP-dependent proteolytic system from reticulocytes: A component that interacts with ATP. Proc. Natl. Acad. Sci. USA 1979, 76, 3107–3110. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, K.D.; Urban, M.K.; Haas, A.L. Ubiquitin is the ATP-dependent proteolysis factor I of rabbit reticulocytes. J. Biol. Chem. 1980, 255, 7529–7532. [Google Scholar] [PubMed]
- Pickart, C.M. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2004, 1695, 55–72. [Google Scholar] [CrossRef]
- Li, W.; Bengtson, M.H.; Ulbrich, A.; Matsuda, A.; Reddy, V.A.; Orth, A.; Chanda, S.K.; Batalov, S.; Joazeiro, C.A.P. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle’s dynamics and signaling. PLoS ONE 2008, 3, e1487. [Google Scholar] [CrossRef]
- Berndsen, C.E.; Wolberger, C. New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 2014, 21, 301–307. [Google Scholar] [CrossRef]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef]
- McDowell, G.; Kucerova, R.; Philpott, A. Non-canonical ubiquitylation of the proneural protein Ngn2 occurs in both Xenopus embryos and mammalian cells. Biochem. Biophys. Res. Commun. 2010, 400, 655–660. [Google Scholar] [CrossRef]
- Golnik, R.; Lehmann, A.; Kloetzel, P.M.; Ebstein, F. Major histocompatibility complex (MHC) class I processing of the NY-ESO-1 antigen is regulated by Rpn10 and Rpn13 proteins and immunoproteasomes following non-lysine ubiquitination. J. Biol. Chem. 2016, 291, 8805–8815. [Google Scholar] [CrossRef]
- Tait, S.W.; De Vries, E.; Maas, C.; Keller, A.M.; D’Santos, C.S.; Borst, J. Apoptosis induction by Bid requires unconventional ubiquitination and degradation of its N-terminal fragment. J. Cell Biol. 2007, 179, 1453–1466. [Google Scholar] [CrossRef] [PubMed]
- Vosper, J.M.D.; McDowell, G.S.; Hindley, C.J.; Fiore-Heriche, C.S.; Kucerova, R.; Horan, I.; Philpott, A. Ubiquitylation on canonical and non-canonical sites targets the transcription factor neurogenin for ubiquitin-mediated proteolysis. J. Biol. Chem. 2009, 284, 15458–15468. [Google Scholar] [CrossRef]
- Oh, E.; Akopian, D.; Rape, M. Principles of ubiquitin-dependent signaling. Annu. Rev. Cell Dev. Biol. 2018, 34, 137–162. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, M.; Suryadinata, R.; Tan, A.R.; Roesley, S.N.A.; Sarcevic, B. Protein monoubiquitination and polyubiquitination generate structural diversity to control distinct biological processes. IUBMB Life 2012, 64, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Kravtsova-Ivantsiv, Y.; Ciechanover, A. Non-canonical ubiquitin-based signals for proteasomal degradation. J. Cell Sci. 2012, 125, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Grumati, P.; Dikic, I. Ubiquitin signaling and autophagy. J. Biol. Chem. 2018, 293, 5404–5413. [Google Scholar] [CrossRef] [PubMed]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef]
- Dahlmann, B. Proteasomes. Essays Biochem. 2005, 41, 31–48. [Google Scholar] [CrossRef]
- Tanaka, K.; Mizushima, T.; Saeki, Y. The proteasome: Molecular machinery and pathophysiological roles. Biol. Chem. 2012, 393, 217–234. [Google Scholar] [CrossRef]
- Smith, D.M.; Chang, S.-C.; Park, S.; Finley, D.; Cheng, Y.; Goldberg, A.L. Docking of the proteasomal ATPases’ Carboxyl termini in the 20S proteasome’s α ring opens the gate for substrate entry. Mol. Cell 2007, 27, 731–744. [Google Scholar] [CrossRef] [PubMed]
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef]
- Bard, J.A.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and function of the 26S proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef] [PubMed]
- Finley, D.; Chen, X.; Walters, K.J. Gates, channels, and switches: Elements of the proteasome machine. Trends Biochem. Sci. 2016, 41, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Takahama, Y.; Kasahara, M.; Tanaka, K. The immunoproteasome and thymoproteasome: Functions, evolution and human disease. Nat. Immunol. 2018, 19, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Ebstein, F.; Kloetzel, P.-M.; Krüger, E.; Seifert, U.; Krüger, E. Emerging roles of immunoproteasomes beyond MHC class I antigen processing. Cell. Mol. Life Sci. 2012, 69, 2543–2558. [Google Scholar] [CrossRef]
- Seifert, U.; Bialy, L.P.; Ebstein, F.; Bech-Otschir, D.; Voigt, A.; Schröter, F.; Prozorovski, T.; Lange, N.; Steffen, J.; Rieger, M.; et al. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell 2010, 142, 613–624. [Google Scholar] [CrossRef]
- Yun, Y.S.; Kim, K.H.; Tschida, B.; Sachs, Z.; Noble-Orcutt, K.E.; Moriarity, B.S.; Ai, T.; Ding, R.; Williams, J.; Chen, L.; et al. mTORC1 coordinates protein synthesis and immunoproteasome formation via PRAS40 to prevent accumulation of protein stress. Mol. Cell 2016, 61, 625–639. [Google Scholar] [CrossRef]
- Niewerth, D.; Kaspers, G.J.L.; Assaraf, Y.G.; Van Meerloo, J.; Kirk, C.J.; Anderl, J.; Blank, J.L.; Van de Ven, P.M.; Zweegman, S.; Jansen, G.; et al. Interferon-γ-induced upregulation of immunoproteasome subunit assembly overcomes bortezomib resistance in human hematological cell lines. J. Hematol. Oncol. 2014, 7, 7. [Google Scholar] [CrossRef]
- Opitz, E.; Koch, A.; Klingel, K.; Schmidt, F.; Prokop, S.; Rahnefeld, A.; Sauter, M.; Heppner, F.L.; Völker, U.; Kandolf, R.; et al. Impairment of immunoproteasome function by β5i/LMP7 subunit deficiency results in severe enterovirus myocarditis. PLoS Pathog. 2011, 7, e1002233. [Google Scholar] [CrossRef]
- Krüger, E.; Kloetzel, P.-M. Immunoproteasomes at the interface of innate and adaptive immune responses: Two faces of one enzyme. Curr. Opin. Immunol. 2012, 24, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Guillaume, B.; Chapiro, J.; Stroobant, V.; Colau, D.; Van Holle, B.; Parvizi, G.; Bousquet-Dubouch, M.-P.; Théate, I.; Parmentier, N.; Eynde, B.J.V.D. Two abundant proteasome subtypes that uniquely process some antigens presented by HLA class I molecules. Proc. Natl. Acad. Sci. USA 2010, 107, 18599–18604. [Google Scholar] [CrossRef] [PubMed]
- Guillaume, B.; Stroobant, V.; Bousquet-Dubouch, M.-P.; Colau, D.; Chapiro, J.; Parmentier, N.; Dalet, A.; Eynde, B.J.V.D. Analysis of the processing of seven human tumor antigens by intermediate proteasomes. J. Immunol. 2012, 189, 3538–3547. [Google Scholar] [CrossRef] [PubMed]
- Dahlmann, B. Mammalian proteasome subtypes: Their diversity in structure and function. Arch. Biochem. Biophys. 2016, 591, 132–140. [Google Scholar] [CrossRef]
- Gohlke, S.; Kloss, A.; Tsokos, M.; Textoris-Taube, K.; Keller, C.; Kloetzel, P.M.; Dahlmann, B. Adult human liver con-tains intermediate-type proteasomes with different enzymatic properties. Ann. Hepatol. 2014, 13, 429–438. [Google Scholar] [CrossRef]
- Ma, C.P.; Slaughter, C.A.; DeMartino, G.N. Identification, purification, and characterization of a protein activator (PA28) of the 20 S proteasome (macropain). J. Biol. Chem. 1992, 267, 10515–10523. [Google Scholar]
- Wójcik, C.; Tanaka, K.; Paweletz, N.; Naab, U.; Wilk, S. Proteasome activator (PA28) subunits, α, β and γ (Ki antigen) in NT2 neuronal precursor cells and HeLa S3 cells. Eur. J. Cell Biol. 1998, 77, 151–160. [Google Scholar] [CrossRef]
- Ustrell, V.; Hoffman, L.; Pratt, G.; Rechsteiner, M. PA200, a nuclear proteasome activator involved in DNA repair. EMBO J. 2002, 21, 3516–3525. [Google Scholar] [CrossRef]
- McCutchen-Maloney, S.L.; Matsuda, K.; Shimbara, N.; Binns, D.D.; Tanaka, K.; Slaughter, C.A.; DeMartino, G.N. cDNA cloning, expression, and functional characterization of PI31, a proline-rich inhibitor of the proteasome. J. Biol. Chem. 2000, 275, 18557–18565. [Google Scholar] [CrossRef]
- Zaiss, D.M.; Standera, S.; Kloetzel, P.-M.; Sijts, A.J.A.M. PI31 is a modulator of proteasome formation and antigen processing. Proc. Natl. Acad. Sci. USA 2002, 99, 14344–14349. [Google Scholar] [CrossRef]
- Cohen-Kaplan, V.; Livneh, I.; Avni, N.; Fabre, B.; Ziv, T.; Kwon, Y.T.; Ciechanover, A. p62- and ubiquitin-dependent stress-induced autophagy of the mammalian 26S proteasome. Proc. Natl. Acad. Sci. USA 2016, 113, E7490–E7499. [Google Scholar] [CrossRef] [PubMed]
- Bochmann, I.; Ebstein, F.; Lehmann, A.; Wohlschlaeger, J.; Sixt, S.U.; Kloetzel, P.; Dahlmann, B. T lymphocytes export proteasomes by way of microparticles: A possible mechanism for generation of extracellular proteasomes. J. Cell. Mol. Med. 2013, 18, 59–68. [Google Scholar] [CrossRef]
- Sengupta, D.; Graham, M.; Liu, X.; Cresswell, P. Proteasomal degradation within endocytic organelles mediates antigen cross-presentation. EMBO J. 2019, 38, e99266. [Google Scholar] [CrossRef] [PubMed]
- DeMartino, G.N.; Gillette, T.G. Proteasomes: Machines for all reasons. Cell 2007, 129, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Laiosa, C.V.; Stadtfeld, M.; Graf, T. Determinants of lymphoid-myeloid lineage diversification. Annu. Rev. Immunol. 2006, 24, 705–738. [Google Scholar] [CrossRef]
- Owens, T.; Benmamar-Badel, A.; Wlodarczyk, A.; Marczynska, J.; Mørch, M.T.; Dubik, M.; Arengoth, D.S.; Asgari, N.; Webster, G.; Khorooshi, R. Protective roles for myeloid cells in neuroinflammation. Scand. J. Immunol. 2020, 92, e12963. [Google Scholar] [CrossRef] [PubMed]
- Hiscott, J. Convergence of the NF-κB and IRF pathways in the regulation of the innate antiviral response. Cytokine Growth Factor Rev. 2007, 18, 483–490. [Google Scholar] [CrossRef]
- Hiscott, J.; Grandvaux, N.; Sharma, S.; Tenoever, B.R.; Servant, M.J.; Lin, R. Convergence of the NF-κB and interferon signaling pathways in the regulation of antiviral defense and apoptosis. Ann. N. Y. Acad. Sci. 2003, 1010, 237–248. [Google Scholar] [CrossRef]
- Heaton, S.M.; Borg, N.A.; Dixit, V.M. Ubiquitin in the activation and attenuation of innate antiviral immunity. J. Exp. Med. 2016, 213, 1–13. [Google Scholar] [CrossRef]
- Wu, X.-F.; Karin, M. Emerging roles of Lys63-linked polyubiquitylation in immune responses. Immunol. Rev. 2015, 266, 161–174. [Google Scholar] [CrossRef]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-κB activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; Dixit, V.M. Ubiquitin-mediated regulation of TNFR1 signaling. Cytokine Growth Factor Rev. 2008, 19, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Arron, J.R.; Lamothe, B.; Cirilli, M.; Kobayashi, T.; Shevde, N.K.; Segal, D.; Dzivenu, O.K.; Vologodskaia, M.; Yim, M.; et al. Distinct molecular mechanism for initiating TRAF6 signalling. Nature 2002, 418, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Lin, S.-C.; Lamothe, B.; Lu, M.; Lo, Y.-C.; Hura, G.; Zheng, L.; Rich, R.L.; Campos, A.D.; Myszka, D.G.; et al. E2 interaction and dimerization in the crystal structure of TRAF6. Nat. Struct. Mol. Biol. 2009, 16, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Rothwarf, D.M.; Zandi, E.; Natoli, G.; Karin, M. IKK-γ is an essential regulatory subunit of the IκB kinase complex. Nature 1998, 395, 297–300. [Google Scholar] [CrossRef]
- Yamaoka, S.; Courtois, G.; Bessia, C.; Whiteside, S.T.; Weil, R.; Agou, F.; Kirk, H.E.; Kay, R.J.; Israël, A. Complementation cloning of NEMO, a component of the IκB kinase complex essential for NF-κB activation. Cell 1998, 93, 1231–1240. [Google Scholar] [CrossRef]
- Cohen, P.; Strickson, S. The role of hybrid ubiquitin chains in the MyD88 and other innate immune signalling pathways. Cell Death Differ. 2017, 24, 1153–1159. [Google Scholar] [CrossRef]
- Emmerich, C.H.; Ordureau, A.; Strickson, S.; Arthur, J.S.C.; Pedrioli, P.G.A.; Komander, D.; Cohen, P. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc. Natl. Acad. Sci. USA 2013, 110, 15247–15252. [Google Scholar] [CrossRef]
- Lo, Y.-C.; Lin, S.-C.; Rospigliosi, C.C.; Conze, D.B.; Wu, C.-J.; Ashwell, J.D.; Eliezer, D.; Wu, H. Structural basis for recognition of diubiquitins by NEMO. Mol. Cell 2009, 33, 602–615. [Google Scholar] [CrossRef]
- Rahighi, S.; Ikeda, F.; Kawasaki, M.; Akutsu, M.; Suzuki, N.; Kato, R.; Kensche, T.; Uejima, T.; Bloor, S.; Komander, D.; et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-κB activation. Cell 2009, 136, 1098–1109. [Google Scholar] [CrossRef]
- Häcker, H.; Karin, M. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, 2006, re13. [Google Scholar] [CrossRef] [PubMed]
- Winston, J.T.; Strack, P.; Beer-Romero, P.; Chu, C.Y.; Elledge, S.J.; Harper, J.W. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev. 1999, 13, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Chiba, T.; Kobayashi, M.; Takeuchi, M.; Suzuki, T.; Ichiyama, A.; Ikenoue, T.; Omata, M.; Furuichi, K.; Tanaka, K. IκBα ubiquitination is catalyzed by an SCF-like complex containing Skp1, cullin-1, and two F-box/WD40-repeat proteins, βTrCP1 and βTrCP2. Biochem. Biophys. Res. Commun. 1999, 256, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Visekruna, A.; Joeris, T.; Seidel, D.; Kroesen, A.; Loddenkemper, C.; Zeitz, M.; Kaufmann, S.H.; Schmidt-Ullrich, R.; Steinhoff, U. Proteasome-mediated degradation of IκBα and processing of p105 in Crohn disease and ulcerative colitis. J. Clin. Investig. 2006, 116, 3195–3203. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.; Gonzalez, E.; Visekruna, A.; Kühl, A.A.; Loddenkemper, C.; Mollenkopf, H.; Kaufmann, S.H.E.; Steinhoff, U.; Joeris, T. Targeting the proteasome: Partial inhibition of the proteasome by bortezomib or deletion of the immunosubunit LMP7 attenuates experimental colitis. Gut 2010, 59, 896–906. [Google Scholar] [CrossRef]
- Mitchell, S.; Mercado, E.L.; Adelaja, A.; Ho, J.Q.; Cheng, Q.J.; Ghosh, G.; Hoffmann, A. An NFκB activity calculator to delineate signaling crosstalk: Type I and II interferons enhance NFκB via distinct mechanisms. Front. Immunol. 2019, 10, 1425. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, L.; Ji, L.; Chen, F.; Fortmann, K.; Zhang, K.; Liu, Q.; Li, K.; Wang, W.; Wang, H.; et al. The REGγ-proteasome forms a regulatory circuit with IκBɛ and NFκB in experimental colitis. Nat. Commun. 2016, 7, 10761. [Google Scholar] [CrossRef]
- Ebstein, F.; Lange, N.; Urban, S.; Seifert, U.; Krüger, E.; Kloetzel, P.-M. Maturation of human dendritic cells is accompanied by functional remodelling of the ubiquitin-proteasome system. Int. J. Biochem. Cell Biol. 2009, 41, 1205–1215. [Google Scholar] [CrossRef]
- Muchamuel, T.; Basler, M.; Aujay, M.A.; Suzuki, E.; Kalim, K.W.; Lauer, C.; Sylvain, C.; Ring, E.R.; Shields, J.; Jiang, J.; et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 15, 781–787. [Google Scholar] [CrossRef]
- Basler, M.; Dajee, M.; Moll, C.; Groettrup, M.; Kirk, C.J. Prevention of experimental colitis by a selective inhibitor of the immunoproteasome. J. Immunol. 2010, 185, 634–641. [Google Scholar] [CrossRef]
- Basler, M.; Mundt, S.; Muchamuel, T.; Moll, C.; Jiang, J.; Groettrup, M.; Kirk, C.J. Inhibition of the immunoproteasome ameliorates experimental autoimmune encephalomyelitis. EMBO Mol. Med. 2014, 6, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wan, C.; Ding, Y.; Han, R.; He, Y.; Xiao, J.; Hao, J. PR-957, a selective inhibitor of immunoproteasome subunit low-MW polypeptide 7, attenuates experimental autoimmune neuritis by suppressing Th17-cell differentiation and regulating cytokine production. FASEB J. 2017, 31, 1756–1766. [Google Scholar] [CrossRef]
- Vachharajani, N.; Joeris, T.; Luu, M.; Hartmann, S.; Pautz, S.; Jenike, E.; Pantazis, G.; Prinz, I.; Hofer, M.J.; Steinhoff, U.; et al. Prevention of colitis-associated cancer by selective targeting of immunoproteasome subunit LMP7. Oncotarget 2017, 8, 50447–50459. [Google Scholar] [CrossRef] [PubMed]
- Althof, N.; Goetzke, C.C.; Kespohl, M.; Voss, K.; Heuser, A.; Pinkert, S.; Kaya, Z.; Klingel, K.; Beling, A. The immunoproteasome-specific inhibitor ONX 0914 reverses susceptibility to acute viral myocarditis. EMBO Mol. Med. 2018, 10, 200–218. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Chen, X.; Li, D.; Liu, H.; Ding, Y.; Han, R.; Shi, Y.; Ma, X. PR-957 mediates neuroprotection by inhibiting Th17 differentiation and modulating cytokine production in a mouse model of ischaemic stroke. Clin. Exp. Immunol. 2018, 193, 194–206. [Google Scholar] [CrossRef]
- Volkov, A.; Hagner, S.; Löser, S.; Alnahas, S.; Raifer, H.; Hellhund, A.; Garn, H.; Steinhoff, U. β5i subunit deficiency of the immunoproteasome leads to reduced Th2 response in OVA induced acute asthma. PLoS ONE 2013, 8, e60565. [Google Scholar] [CrossRef]
- Kimura, H.; Usui, F.; Karasawa, T.; Kawashima, A.; Shirasuna, K.; Inoue, Y.; Komada, T.; Kobayashi, M.; Mizushina, Y.; Kasahara, T.; et al. Immunoproteasome subunit LMP7 deficiency improves obesity and metabolic disorders. Sci. Rep. 2015, 5, 15883. [Google Scholar] [CrossRef]
- Bitzer, A.; Basler, M.; Krappmann, D.; Groettrup, M. Immunoproteasome subunit deficiency has no influence on the canonical pathway of NF-κB activation. Mol. Immunol. 2017, 83, 147–153. [Google Scholar] [CrossRef]
- Heger, K.; Wickliffe, K.E.; Ndoja, A.; Zhang, J.; Murthy, A.; Dugger, D.L.; Maltzman, A.; Melo, F.D.S.E.; Hung, J.; Zeng, Y.; et al. OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature 2018, 559, 120–124. [Google Scholar] [CrossRef]
- Harhaj, E.W.; Dixit, V.M. Deubiquitinases in the regulation of NF-κB signaling. Cell Res. 2010, 21, 22–39. [Google Scholar] [CrossRef]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.T.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; Newton, K.; Seshasayee, D.; Kusam, S.; Lam, C.; Zhang, J.; Popovych, N.; Helgason, E.; Schoeffler, A.; Jeet, S.; et al. Phosphorylation and linear ubiquitin direct A20 inhibition of inflammation. Nature 2015, 528, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Dixit, V.M. A20 edits ubiquitin and autoimmune paradigms. Nat. Genet. 2011, 43, 822–823. [Google Scholar] [CrossRef] [PubMed]
- Häcker, H.; Tseng, P.-H.; Karin, M. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat. Rev. Immunol. 2011, 11, 457–468. [Google Scholar] [CrossRef]
- Häcker, H.; Redecke, V.; Blagoev, B.; Kratchmarova, I.; Hsu, L.-C.; Wang, G.G.; Kamps, M.P.; Raz, E.; Wagner, H.; Häcker, G.; et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 2005, 439, 204–207. [Google Scholar] [CrossRef]
- Tseng, P.-H.; Matsuzawa, A.; Zhang, W.; Mino, T.; Vignali, D.A.A.; Karin, M. Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat. Immunol. 2009, 11, 70–75. [Google Scholar] [CrossRef]
- Lei, C.-Q.; Zhang, Y.; Xia, T.; Jiang, L.-Q.; Zhong, B.; Shu, H.-B. FoxO1 negatively regulates cellular antiviral response by promoting degradation of IRF3. J. Biol. Chem. 2013, 288, 12596–12604. [Google Scholar] [CrossRef]
- Yu, Y.; Hayward, G.S. The ubiquitin E3 ligase RAUL negatively regulates type i interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 2010, 33, 863–877. [Google Scholar] [CrossRef]
- Cui, J.; Li, Y.; Zhu, L.; Liu, D.; Songyang, Z.; Wang, H.Y.; Wang, R.-F. NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat. Immunol. 2012, 13, 387–395. [Google Scholar] [CrossRef]
- Zheng, Q.; Hou, J.; Zhou, Y.; Yang, Y.; Xie, B.; Cao, X. Siglec1 suppresses antiviral innate immune response by inducing TBK1 degradation via the ubiquitin ligase TRIM27. Cell Res. 2015, 25, 1121–1136. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, L.; Zhao, X.; Zhao, K.; Meng, H.; Zhao, W.; Gao, C. TRAF-interacting protein (TRIP) negatively regulates IFN-β production and antiviral response by promoting proteasomal degradation of TANK-binding kinase 1. J. Exp. Med. 2012, 209, 1703–1711. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Mao, R.; Yu, Y.; Liu, S.; Shi, Z.; Cheng, J.; Zhang, H.; An, L.; Zhao, Y.; Xu, X.; et al. USP21 negatively regulates antiviral response by acting as a RIG-I deubiquitinase. J. Exp. Med. 2014, 211, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Friedman, C.S.; O’Donnell, M.A.; Legarda-Addison, D.; Ng, A.; Cárdenas, W.B.; Yount, J.S.; Moran, T.M.; Basler, C.F.; Komuro, A.; Horvath, C.M.; et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 2008, 9, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Song, Y.; Li, Y.; Zhu, Q.; Tan, P.; Qin, Y.; Wang, H.Y.; Wang, R.-F. USP3 inhibits type I interferon signaling by deubiquitinating RIG-I-like receptors. Cell Res. 2014, 24, 400–416. [Google Scholar] [CrossRef]
- Chen, M.; Meng, Q.; Qin, Y.; Liang, P.; Tan, P.; He, L.; Zhou, Y.; Chen, Y.; Huang, J.; Wang, R.F.; et al. TRIM14 Inhibits cGAS degradation mediated by selective autophagy receptor p62 to promote innate immune responses. Mol. Cell 2016, 64, 105–119. [Google Scholar] [CrossRef]
- Reis, J.; Hassan, F.; Guan, X.Q.; Shen, J.; Monaco, J.J.; Papasian, C.J.; Qureshi, A.A.; Van Way, C.W., III; Vogel, S.N.; Morrison, D.C.; et al. The immunoproteasomes regulate LPS-induced TRIF/TRAM signaling pathway in murine macrophages. Cell Biophys. 2011, 60, 119–126. [Google Scholar] [CrossRef]
- Morimoto, R.I.; Cuervo, A.M. Proteostasis and the aging proteome in health and disease. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, S33–S38. [Google Scholar] [CrossRef]
- Bett, J.S. Proteostasis regulation by the ubiquitin system. Essays Biochem. 2016, 60, 143–151. [Google Scholar] [CrossRef]
- Gomez-Navarro, N.; Miller, E.A. Protein sorting at the ER–Golgi interface. J. Cell Biol. 2016, 215, 769–778. [Google Scholar] [CrossRef]
- Maseda, D.; Meister, S.; Neubert, K.; Herrmann, M.; Voll, R.E. Proteasome inhibition drastically but reversibly impairs murine lymphocyte development. Cell Death Differ. 2008, 15, 600–612. [Google Scholar] [CrossRef]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef]
- Haigis, M.C.; Yankner, B.A. The aging stress response. Mol. Cell 2010, 40, 333–344. [Google Scholar] [CrossRef]
- Olzscha, H. Posttranslational modifications and proteinopathies: How guardians of the proteome are defeated. Biol. Chem. 2019, 400, 895–915. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Miles, J.; Scherz-Shouval, R.; Van Oosten-Hawle, P. Expanding the organismal proteostasis network: Linking systemic stress signaling with the innate immune response. Trends Biochem. Sci. 2019, 44, 927–942. [Google Scholar] [CrossRef] [PubMed]
- Limanaqi, F.; Biagioni, F.; Gambardella, S.; Familiari, P.; Frati, A.; Fornai, F. Promiscuous roles of autophagy and proteasome in neurodegenerative proteinopathies. Int. J. Mol. Sci. 2020, 21, 3028. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E.; Borchelt, D.R.; Giasson, B.I.; Lewis, J. Thinking laterally about neurodegenerative proteinopathies. J. Clin. Investig. 2013, 123, 1847–1855. [Google Scholar] [CrossRef] [PubMed]
- Dever, T.E.; Feng, L.; Wek, R.C.; Cigan, A.; Donahue, T.F.; Hinnebusch, A.G. Phosphorylation of initiation factor 2α by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 1992, 68, 585–596. [Google Scholar] [CrossRef]
- Kostura, M.; Mathews, M.B. Purification and activation of the double-stranded RNA-dependent eIF-2 kinase DAI. Mol. Cell. Biol. 1989, 9, 1576–1586. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Jiang, H.-Y.; Wek, R.C.; Anthony, T. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 2006, 34, 7–11. [Google Scholar] [CrossRef]
- Rousseau, A.; Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 697–712. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kunkeaw, N.; Lee, Y. Protein kinase R and its cellular regulators in cancer: An active player or a surveillant? Wiley Interdiscip. Rev. RNA 2020, 11, e1558. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef]
- Ma, Y.; Hendershot, L.M. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J. Biol. Chem. 2003, 278, 34864–34873. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Sommer, T.; Jarosch, E. BiP binding keeps ATF6 at bay. Dev. Cell 2002, 3, 1–2. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef]
- Fabre, B.; Lambour, T.; Garrigues, L.; Amalric, F.; Vigneron, N.; Menneteau, T.; Stella, A.; Monsarrat, B.; Eynde, B.V.D.; Burlet-Schiltz, O.; et al. Deciphering preferential interactions within supramolecular protein complexes: The proteasome case. Mol. Syst. Biol. 2015, 11, 771. [Google Scholar] [CrossRef]
- Tanahashi, N.; Murakami, Y.; Minami, Y.; Shimbara, N.; Hendil, K.B.; Tanaka, K. Hybrid proteasomes. J. Biol. Chem. 2000, 275, 14336–14345. [Google Scholar] [CrossRef]
- Jung, T.; Grune, T. The proteasome and the degradation of oxidized proteins: Part I—Structure of proteasomes. Redox Biol. 2013, 1, 178–182. [Google Scholar] [CrossRef]
- Kriegenburg, F.; Poulsen, E.G.; Koch, A.; Krüger, E.; Hartmann-Petersen, R. Redox control of the ubiquitin-proteasome system: From molecular mechanisms to functional significance. Antioxid. Redox Signal. 2011, 15, 2265–2299. [Google Scholar] [CrossRef]
- Respondek, D.; Voss, M.; Kühlewindt, I.; Klingel, K.; Krüger, E.; Beling, A. PA28 modulates antigen processing and viral replication during coxsackievirus B3 infection. PLoS ONE 2017, 12, e0173259. [Google Scholar] [CrossRef] [PubMed]
- Lelouard, H.; Gatti, E.; Cappello, F.; Gresser, O.; Camosseto, V.; Pierre, P. Transient aggregation of ubiquitinated proteins during dendritic cell maturation. Nature 2002, 417, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Szeto, J.; Kaniuk, N.A.; Canadien, V.; Nisman, R.; Mizushima, N.; Yoshimori, T.; Bazett-Jones, D.P.; Brumell, J.H. ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy 2006, 2, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Berger, T.; Groettrup, M.; Basler, M. Immunoproteasome inhibition impairs T and B cell activation by restraining ERK signaling and proteostasis. Front. Immunol. 2018, 9, 2386. [Google Scholar] [CrossRef]
- Studencka-Turski, M.; Çetin, G.; Junker, H.; Ebstein, F.; Krüger, E. Molecular insight into the IRE1α-mediated type I interferon response induced by proteasome impairment in myeloid cells of the brain. Front. Immunol. 2019, 10, 2900. [Google Scholar] [CrossRef]
- Jansen, A.H.P.; Reits, E.A.J.; Hol, E.M. The ubiquitin proteasome system in glia and its role in neurodegenerative diseases. Front. Mol. Neurosci. 2014, 7, 73. [Google Scholar] [CrossRef] [PubMed]
- Cianciulli, A.; Porro, C.; Calvello, R.; Trotta, T.; Lofrumento, N.E.; Panaro, M.A. Microglia mediated neuroinflammation: Focus on PI3K modulation. Biomolecules 2020, 10, 137. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Roche, P.A.; Furuta, K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat. Rev. Immunol. 2015, 15, 203–216. [Google Scholar] [CrossRef]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major histocompatibility complex (MHC) class I and MHC class II proteins: Conformational plasticity in antigen presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef]
- Van Kaert, L.; Ashton-Rickardt, P.G.; Eichelberger, M.; Gaczynska, M.; Nagashima, K.; Rock, K.L.; Goldberg, A.L.; Doherty, P.C.; Tonegawa, S. Altered peptidase and viral-specific T cell response in LMP2 mutant mice. Immunity 1994, 1, 533–541. [Google Scholar] [CrossRef]
- Kincaid, E.Z.; Che, J.W.; York, I.; Escobar, H.; Reyes-Vargas, E.; Delgado, J.C.; Welsh, R.M.; Karow, M.L.; Murphy, A.J.; Valenzuela, D.M.; et al. Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat. Immunol. 2011, 13, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, S.; Sims, S.; O’Hara, G.; Silk, J.; Gileadi, U.; Cerundolo, V.; Klenerman, P. A dominant role for the immunoproteasome in CD8+ T cell responses to murine cytomegalovirus. PLoS ONE 2011, 6, e14646. [Google Scholar] [CrossRef] [PubMed]
- Sijts, A.J.A.M.; Standera, S.; Toes, R.E.M.; Ruppert, T.; Beekman, N.J.C.M.; van Veelen, P.A.; Ossendorp, F.A.; Melief, C.J.M.; Kloetzel, P.M. MHC class I antigen processing of an adenovirus CTL epitope is linked to the levels of immunoproteasomes in infected cells. J. Immunol. 2000, 164, 4500–4506. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, N.; Van Helden, M.J.G.; Textoris-Taube, K.; Chiba, T.; Topham, D.J.; Kloetzel, P.-M.; Zaiss, D.; Sijts, A.J.A.M. PA28 and the proteasome immunosubunits play a central and independent role in the production of MHC class I-binding peptides in vivo. Eur. J. Immunol. 2011, 41, 926–935. [Google Scholar] [CrossRef]
- Guimarães, G.; Gomes, M.T.R.; Campos, P.C.; Marinho, F.V.; De Assis, N.R.G.; Silveira, T.N.; Oliveira, S.C. Immunoproteasome subunits are required for CD8+T Cell function and host resistance to Brucella abortus infection in mice. Infect. Immun. 2017, 86, 86. [Google Scholar] [CrossRef]
- Morel, S.; Lévy, F.; Burlet-Schiltz, O.; Brasseur, F.; Probst-Kepper, M.; Peitrequin, A.-L.; Monsarrat, B.; Van Velthoven, R.; Cerottini, J.-C.; Boon, T.; et al. Processing of some antigens by the standard proteasome but not by the immunoproteasome results in poor presentation by dendritic cells. Immunity 2000, 12, 107–117. [Google Scholar] [CrossRef]
- Chapatte, L.; Ayyoub, M.; Morel, S.; Peitrequin, A.-L.; Lévy, N.; Servis, C.; Eynde, B.J.V.D.; Valmori, D.; Lévy, F. Processing of tumor-associated antigen by the proteasomes of dendritic cells controls in vivo T-Cell responses. Cancer Res. 2006, 66, 5461–5468. [Google Scholar] [CrossRef]
- Chapiro, J.; Claverol, S.; Piette, F.; Ma, W.; Stroobant, V.; Guillaume, B.; Gairin, J.-E.; Morel, S.; Burlet-Schiltz, O.; Monsarrat, B.; et al. Destructive cleavage of antigenic peptides either by the immunoproteasome or by the standard proteasome results in differential antigen presentation. J. Immunol. 2006, 176, 1053–1061. [Google Scholar] [CrossRef]
- Keller, M.; Ebstein, F.; Bürger, E.; Textoris-Taube, K.; Gorny, X.; Urban, S.; Zhao, F.; Dannenberg, T.; Sucker, A.; Keller, C.; et al. The proteasome immunosubunits, PA28 and ER-aminopeptidase 1 protect melanoma cells from efficient MART-126-35-specific T-cell recognition. Eur. J. Immunol. 2015, 45, 3257–3268. [Google Scholar] [CrossRef] [PubMed]
- Mishto, M.; Liepe, J.; Textoris-Taube, K.; Keller, C.; Henklein, P.; Weberruß, M.; Dahlmann, B.; Enenkel, C.; Voigt, A.; Kuckelkorn, U.; et al. Proteasome isoforms exhibit only quantitative differences in cleavage and epitope generation. Eur. J. Immunol. 2014, 44, 3508–3521. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.-I.; Yewdell, J.W.; Yang, J.C. Immune recognition of a human renal cancer antigen through post-translational protein splicing. Nature 2004, 427, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Vigneron, N.; Stroobant, V.; Chapiro, J.; Ooms, A.; DeGiovanni, G.; Morel, S.; Van der Bruggen, P.; Boon, T.; Eynde, B.J.V.D. An antigenic peptide produced by peptide splicing in the proteasome. Science 2004, 304, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Warren, E.H.; Vigneron, N.; Gavin, M.A.; Coulie, P.G.; Stroobant, V.; Dalet, A.; Tykodi, S.S.; Xuereb, S.M.; Mito, J.K.; Riddell, S.R.; et al. An antigen produced by splicing of noncontiguous peptides in the reverse order. Science 2006, 313, 1444–1447. [Google Scholar] [CrossRef] [PubMed]
- Dalet, A.; Vigneron, N.; Stroobant, V.; Hanada, K.-I.; Eynde, B.J.V.D. Splicing of distant peptide fragments occurs in the proteasome by transpeptidation and produces the spliced antigenic peptide derived from fibroblast growth factor-5. J. Immunol. 2010, 184, 3016–3024. [Google Scholar] [CrossRef] [PubMed]
- Ebstein, F.; Textoris-Taube, K.; Keller, C.; Golnik, R.; Vigneron, N.; Eynde, B.J.V.D.; Schuler-Thurner, B.; Schadendorf, D.; Lorenz, F.K.M.; Uckert, W.; et al. Proteasomes generate spliced epitopes by two different mechanisms and as efficiently as non-spliced epitopes. Sci. Rep. 2016, 6, 24032. [Google Scholar] [CrossRef]
- Michaux, A.; Larrieu, P.; Stroobant, V.; Fonteneau, J.-F.; Jotereau, F.; Eynde, B.J.V.D.; Moreau-Aubry, A.; Vigneron, N. A spliced antigenic peptide comprising a single spliced amino acid is produced in the proteasome by reverse splicing of a longer peptide fragment followed by trimming. J. Immunol. 2014, 192, 1962–1971. [Google Scholar] [CrossRef]
- Platteel, A.C.M.; Mishto, M.; Textoris-Taube, K.; Keller, C.; Liepe, J.; Busch, D.H.; Kloetzel, P.M.; Sijts, A.J.A.M. CD8+T cells of Listeria monocytogenes-infected mice recognize both linear and spliced proteasome products. Eur. J. Immunol. 2016, 46, 1109–1118. [Google Scholar] [CrossRef]
- Dalet, A.; Robbins, P.F.; Stroobant, V.; Vigneron, N.; Li, Y.F.; El-Gamil, M.; Hanada, K.-I.; Yang, J.C.; Rosenberg, S.A.; Eynde, B.J.V.D. An antigenic peptide produced by reverse splicing and double asparagine deamidation. Proc. Natl. Acad. Sci. USA 2011, 108, E323–E331. [Google Scholar] [CrossRef]
- Mishto, M.; Liepe, J. Post-translational peptide splicing and T cell responses. Trends Immunol. 2017, 38, 904–915. [Google Scholar] [CrossRef]
- Platteel, A.C.M.; Liepe, J.; Van Eden, W.; Mishto, M.; Sijts, A.J.A.M. An unexpected major role for proteasome-catalyzed peptide splicing in generation of T Cell epitopes: Is there relevance for vaccine development? Front. Immunol. 2017, 8, 1441. [Google Scholar] [CrossRef] [PubMed]
- Mishto, M. What we see, what we do not see, and what we do not want to see in HLA Class I immunopeptidomes. Proteomics 2020, 20, 2000112. [Google Scholar] [CrossRef] [PubMed]
- Liepe, J.; Marino, F.; Sidney, J.; Jeko, A.; Bunting, D.E.; Sette, A.; Kloetzel, P.M.; Stumpf, M.P.H.; Heck, A.J.R.; Mishto, M. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science 2016, 354, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Parham, P. Transporters of delight. Nature 1990, 348, 674–675. [Google Scholar] [CrossRef] [PubMed]
- Spies, T.; Bresnahan, M.; Bahrain, S.; Arnold, D.; Blanck, G.; Mellins, E.; Pious, D.; Demars, R. A gene in the human major histocompatibility complex class II region controlling the class I antigen presentation pathway. Nature 1990, 348, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Trowsdale, J.; Hanson, I.; Mockridge, I.; Beck, S.; Townsend, A.R.; Kelly, A. Sequences encoded in the class II region of the MHC related to the ‘ABC’ superfamily of transporters. Nature 1990, 348, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Deverson, E.V.; Gow, I.R.; Coadwell, W.J.; Monaco, J.J.; Butcher, G.W.; Howard, J.C. MHC class II region encoding proteins related to the muKidrug resistance family of transmembrane transporters. Nature 1990, 348, 738–741. [Google Scholar] [CrossRef] [PubMed]
- Monaco, J.J.; Cho, S.; Attaya, M. Transport protein genes in the murine MHC: Possible implications for antigen processing. Science 1990, 250, 1723–1726. [Google Scholar] [CrossRef] [PubMed]
- Van De Weijer, M.L.; Luteijn, R.D.; Wiertz, E.J.H.J. Viral immune evasion: Lessons in MHC class I antigen presentation. Semin. Immunol. 2015, 27, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Kovacsovics-Bankowski, M.; Rock, K.L. A phagosome-to-cytosol pathway for exogenous antigens presented on MHC class I molecules. Science 1995, 267, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, A.L.; Kyritsis, C.; Tampé, R.; Cresswell, P. Early phagosomes in dendritic cells form a cellular compartment sufficient for cross presentation of exogenous antigens. Proc. Natl. Acad. Sci. USA 2003, 100, 12889–12894. [Google Scholar] [CrossRef] [PubMed]
- Palmowski, M.J.; Gileadi, U.; Salio, M.; Gallimore, A.; Millrain, M.; James, E.; Addey, C.; Scott, D.; Dyson, J.; Simpson, E.; et al. Role of immunoproteasomes in cross-presentation. J. Immunol. 2006, 177, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Guermonprez, P.; Saveanu, L.; Kleijmeer, M.J.; Davoust, J.; Van Endert, P.; Amigorena, S. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature 2003, 425, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Houde, M.; Bertholet, S.; Gagnon, E.; Brunet, S.; Goyette, G.; Laplante, A.; Princiotta, M.F.; Thibault, P.; Sacks, D.; Desjardins, M. Phagosomes are competent organelles for antigen cross-presentation. Nature 2003, 425, 402–406. [Google Scholar] [CrossRef]
- Ma, W.; Stroobant, V.; Heirman, C.; Sun, Z.; Thielemans, K.; Mulder, A.; Van der Bruggen, P.; Eynde, B.J.V.D. The vacuolar pathway of long peptide cross-presentation can be TAP dependent. J. Immunol. 2018, 202, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Sigal, L.J.; Boes, M.; Rock, K.L. Important role of cathepsin S in generating peptides for TAP-independent MHC class I crosspresentation in vivo. Immunity 2004, 21, 155–165. [Google Scholar] [CrossRef]
- Pfeifer, J.D.; Wick, M.J.; Roberts, R.L.; Findlay, K.; Normark, S.J.; Harding, C.V. Phagocytic processing of bacterial antigens for class I MHC presentation to T cells. Nature 1993, 361, 359–362. [Google Scholar] [CrossRef]
- Liu, T.; Chambers, B.; Diehl, A.D.; Van Kaer, L.; Jondal, M.; Ljunggren, H.G. TAP peptide transporter-independent presentation of heat-killed Sendai virus antigen on MHC class I molecules by splenic antigen-presenting cells. J. Immunol. 1997, 159, 5364–5371. [Google Scholar]
- Campbell, D.J.; Serwold, T.; Shastri, N. Bacterial proteins can be processed by macrophages in a transporter associated with antigen processing-independent, cysteine protease-dependent manner for presentation by MHC class I molecules. J. Immunol. 2000, 164, 168–175. [Google Scholar] [CrossRef]
- Bertholet, S.; Goldszmid, R.; Morrot, A.; Debrabant, A.; Afrin, F.; Collazo-Custodio, C.; Houde, M.; Desjardins, M.; Sher, A.; Sacks, D. Leishmania antigens are presented to CD8+T cells by a transporter associated with antigen processing-independent pathway in vitro and in vivo. J. Immunol. 2006, 177, 3525–3533. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Antón, L.C.; Bennink, J.R. Defective ribosomal products (DRiPs): A major source of antigenic peptides for MHC class I molecules? J. Immunol. 1996, 157, 1823–1826. [Google Scholar] [PubMed]
- Schubert, U.S.; Antón, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Reits, E.A.J.; Vos, J.C.; Grommé, M.; Neefjes, J. The major substrates for TAP in vivo are derived from newly synthesized proteins. Nature 2000, 404, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Yewdell, J.W. DRiPs solidify: Progress in understanding endogenous MHC class I antigen processing. Trends Immunol. 2011, 32, 548–558. [Google Scholar] [CrossRef]
- Antón, L.C.; Yewdell, J.W. Translating DRiPs: MHC class I immunosurveillance of pathogens and tumors. J. Leukoc. Biol. 2014, 95, 551–562. [Google Scholar] [CrossRef]
- Wei, J.; Yewdell, J.W. Immunoribosomes: Where’s there’s fire, there’s fire. Mol. Immunol. 2019, 113, 38–42. [Google Scholar] [CrossRef]
- Wei, J.; Yewdell, J.W. Flu DRiPs in MHC class I immunosurveillance. Virol. Sin. 2018, 34, 162–167. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Hollý, J. DRiPs get molecular. Curr. Opin. Immunol. 2020, 64, 130–136. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Reits, E.A.J.; Neefjes, J. Making sense of mass destruction: Quantitating MHC class I antigen presentation. Nat. Rev. Immunol. 2003, 3, 952–961. [Google Scholar] [CrossRef]
- Rock, K.L.; Reits, E.; Neefjes, J. Present Yourself! By MHC class I and MHC class II molecules. Trends Immunol. 2016, 37, 724–737. [Google Scholar] [CrossRef]
- Wei, J.; Kishton, R.J.; Angel, M.; Conn, C.S.; Dalla-Venezia, N.; Marcel, V.; Vincent, A.; Catez, F.; Ferré, S.; Ayadi, L.; et al. Ribosomal proteins regulate MHC class I peptide generation for immunosurveillance. Mol. Cell 2019, 73, 1162–1173.e5. [Google Scholar] [CrossRef] [PubMed]
- Lelouard, H.; Ferrand, V.; Marguet, D.; Bania, J.; Camosseto, V.; David, A.; Gatti, E.; Pierre, P. Dendritic cell aggresome-like induced structures are dedicated areas for ubiquitination and storage of newly synthesized defective proteins. J. Cell Biol. 2004, 164, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Fehling, H.J.; Swat, W.; LaPlace, C.; Kuhn, R.; Rajewsky, K.; Muller, U.; Von Boehmer, H. MHC class I expression in mice lacking the proteasome subunit LMP-7. Science 1994, 265, 1234–1237. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Sanchez, G.A.M.; Jesus, A.A. Insights from mendelian interferonopathies: Comparison of CANDLE, SAVI with AGS, monogenic lupus. J. Mol. Med. 2016, 94, 1111–1127. [Google Scholar] [CrossRef]
- Torrelo, A.; Patel, S.; Colmenero, I.; Gurbindo, D.; Lendínez, F.; Hernández, A.; López-Robledillo, J.C.; Dadban, A.; Requena, L.; Paller, A.S. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. J. Am. Acad. Dermatol. 2010, 62, 489–495. [Google Scholar] [CrossRef]
- Torrelo, A. CANDLE syndrome as a paradigm of proteasome-related autoinflammation. Front. Immunol. 2017, 8, 927. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Xing, C.; DeMartino, G.N.; Mizrachi, D.; Hernandez, M.D.; Sousa, A.B.; De Villarreal, L.M.; Dos Santos, H.G.; Garg, A. PSMB8 encoding the β5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am. J. Hum. Genet. 2010, 87, 866–872. [Google Scholar] [CrossRef]
- Arima, K.; Kinoshita, A.; Mishima, H.; Kanazawa, N.; Kaneko, T.; Mizushima, T.; Ichinose, K.; Nakamura, H.; Tsujino, A.; Kawakami, A.; et al. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 14914–14919. [Google Scholar] [CrossRef]
- Kitamura, A.; Maekawa, Y.; Uehara, H.; Izumi, K.; Kawachi, I.; Nishizawa, M.; Toyoshima, Y.; Takahashi, H.; Standley, D.M.; Tanaka, K.; et al. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J. Clin. Investig. 2011, 121, 4150–4160. [Google Scholar] [CrossRef]
- Liu, Y.; Ramot, Y.; Torrelo, A.; Paller, A.S.; Si, N.; Babay, S.; Kim, P.W.; Sheikh, A.; Lee, C.-C.R.; Chen, Y.; et al. Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 2011, 64, 895–907. [Google Scholar] [CrossRef]
- Brehm, A.; Liu, Y.; Sheikh, A.; Marrero, B.; Omoyinmi, E.; Zhou, Q.; Montealegre, G.; Biancotto, A.; Reinhardt, A.; De Jesus, A.A.; et al. Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J. Clin. Investig. 2015, 125, 4196–4211. [Google Scholar] [CrossRef] [PubMed]
- Poli, M.C.; Ebstein, F.; Nicholas, S.K.; De Guzman, M.M.; Forbes, L.R.; Chinn, I.K.; Mace, E.M.; Vogel, T.P.; Carisey, A.F.; Benavides, F.; et al. Heterozygous truncating variants in POMP escape nonsense-mediated decay and cause a unique immune dysregulatory syndrome. Am. J. Hum. Genet. 2018, 102, 1126–1142. [Google Scholar] [CrossRef] [PubMed]
- De Jesus, A.A.; Brehm, A.; VanTries, R.; Pillet, P.; Parentelli, A.-S.; Sanchez, G.A.M.; Deng, Z.; Paut, I.K.; Krüger, E. Novel proteasome assembly chaperone mutations in PSMG2/PAC2 cause the autoinflammatory interferonopathy CANDLE/PRAAS4. J. Allergy Clin. Immunol. 2019, 143, 1939–1943.e8. [Google Scholar] [CrossRef] [PubMed]
- Sarrabay, G.; Méchin, D.; Salhi, A.; Boursier, G.; Rittore, C.; Crow, Y.; Rice, G.; Tran, T.-A.; Cezar, R.; Duffy, D.; et al. PSMB10, the last immunoproteasome gene missing for PRAAS. J. Allergy Clin. Immunol. 2020, 145, 1015–1017.e6. [Google Scholar] [CrossRef]
- Wahl, C.; Kautzmann, S.; Krebiehl, G.; Strauss, K.; Woitalla, D.; Müller, T.; Bauer, P.; Riess, O.; Krüger, R. A comprehensive genetic study of the proteasomal subunit S6 ATPase in German Parkinson’s disease patients. J. Neural Transm. 2008, 115, 1141–1148. [Google Scholar] [CrossRef]
- Küry, S.; Besnard, T.; Ebstein, F.; Khan, T.N.; Gambin, T.; Douglas, J.; Bacino, C.A.; Sanders, S.J.; Lehmann, A.; Latypova, X.; et al. De Novo disruption of the proteasome regulatory subunit PSMD12 causes a syndromic neurodevelopmental disorder. Am. J. Hum. Genet. 2017, 100, 352–363. [Google Scholar] [CrossRef]
- Ansar, M.; Ebstein, F.; Özkoç, H.; Paracha, S.A.; Iwaszkiewicz, J.; Gesemann, M.; Zoete, V.; Ranza, E.; Santoni, F.A.; Sarwar, M.T.; et al. Biallelic variants in PSMB1 encoding the proteasome subunit β6 cause impairment of proteasome function, microcephaly, intellectual disability, developmental delay and short stature. Hum. Mol. Genet. 2020, 29, 1132–1143. [Google Scholar] [CrossRef]
- Liepe, J.; Ovaa, H.; Mishto, M. Why do proteases mess up with antigen presentation by re-shuffling antigen sequences? Curr. Opin. Immunol. 2018, 52, 81–86. [Google Scholar] [CrossRef]
- Bosanac, I.; Wertz, I.E.; Pan, B.; Yu, C.; Kusam, S.; Lam, C.; Phu, L.; Phung, Q.; Maurer, B.; Arnott, D.; et al. Ubiquitin binding to A20 ZnF4 is required for modulation of NF-κB signaling. Mol. Cell 2010, 40, 548–557. [Google Scholar] [CrossRef]
- Rigante, D. Phenotype variability of autoinflammatory disorders in the pediatric patient: A pictorial overview. J. Evid. Based Med. 2020, 13, 227–245. [Google Scholar] [CrossRef]
- Beck, D.B.; Aksentijevich, I. Biochemistry of autoinflammatory diseases: Catalyzing monogenic disease. Front. Immunol. 2019, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Lohr, N.J.; Molleston, J.P.; Strauss, K.A.; Torres-Martinez, W.; Sherman, E.A.; Squires, R.H.; Rider, N.L.; Chikwava, K.R.; Cummings, O.W.; Morton, D.H.; et al. Human ITCH E3 ubiquitin ligase deficiency causes syndromic multisystem autoimmune disease. Am. J. Hum. Genet. 2010, 86, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Aki, D.; Li, H.; Zhang, W.; Zheng, M.; Elly, C.; Lee, J.H.; Zou, W.; Liu, Y.-C. The E3 ligases Itch and WWP2 cooperate to limit TH2 differentiation by enhancing signaling through the TCR. Nat. Immunol. 2018, 19, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Bachmaier, K.; Krawczyk, C.M.; Kozieradzki, I.; Kong, Y.-Y.; Sasaki, T.; Oliveira-Dos-Santos, A.; Mariathasan, S.; Bouchard, D.; Wakeham, A.; Itie, A.; et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature 2000, 403, 211–216. [Google Scholar] [CrossRef]
- Ahmed, N.; Zeng, M.; Sinha, I.; Polin, L.; Wei, W.-Z.; Rathinam, C.; Flavell, R.; Massoumi, R.; Venuprasad, K. The E3 ligase Itch and deubiquitinase Cyld act together to regulate Tak1 and inflammation. Nat. Immunol. 2011, 12, 1176–1183. [Google Scholar] [CrossRef]
- Theivanthiran, B.; Kathania, M.; Zeng, M.; Anguiano, E.; Basrur, V.; Vandergriff, T.; Pascual, V.; Wei, W.-Z.; Massoumi, R.; Venuprasad, K. The E3 ubiquitin ligase Itch inhibits p38α signaling and skin inflammation through the ubiquitylation of Tab1. Sci. Signal. 2015, 8, ra22. [Google Scholar] [CrossRef]
- Layman, A.A.K.; Sprout, S.L.; Phillips, D.; Oliver, P.M. Ndfip1 restricts Th17 cell potency by limiting lineage stability and proinflammatory cytokine production. Sci. Rep. 2017, 7, 39649. [Google Scholar] [CrossRef]
- Kathania, M.; Khare, P.; Zeng, M.; Cantarel, B.; Zhang, H.; Ueno, H.; Venuprasad, K. Itch inhibits IL-17-mediated colon inflammation and tumorigenesis by ROR-γt ubiquitination. Nat. Immunol. 2016, 17, 997–1004. [Google Scholar] [CrossRef]
- Meuwissen, M.E.; Schot, R.; Buta, S.; Oudesluijs, G.; Tinschert, S.; Speer, S.D.; Li, Z.; Van Unen, L.; Heijsman, D.; Goldmann, T.; et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J. Exp. Med. 2016, 213, 1163–1174. [Google Scholar] [CrossRef]
- Zhang, X.; Bogunovic, D.; Payelle-Brogard, B.; Francois-Newton, V.; Speer, S.D.; Yuan, C.; Volpi, S.; Li, Z.; Sanal, O.; Mansouri, D.; et al. Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature 2015, 517, 89–93. [Google Scholar] [CrossRef]
- Kang, J.A.; Jeon, Y.J. Emerging roles of USP18: From biology to pathophysiology. Int. J. Mol. Sci. 2020, 21, 6825. [Google Scholar] [CrossRef] [PubMed]
- Kubori, T.; Hyakutake, A.; Nagai, H. Legionella translocates an E3 ubiquitin ligase that has multiple U-boxes with distinct functions. Mol. Microbiol. 2008, 67, 1307–1319. [Google Scholar] [CrossRef] [PubMed]
- Kubori, T.; Shinzawa, N.; Kanuka, H.; Nagai, H. Legionella metaeffector exploits host proteasome to temporally regulate cognate effector. PLoS Pathog. 2010, 6, e1001216. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Luo, Z.-Q. Hijacking of the host ubiquitin network by Legionella pneumophila. Front. Cell. Infect. Microbiol. 2017, 7, 487. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Sheedlo, M.J.; Yu, K.; Tan, Y.; Nakayasu, E.S.; Das, C.; Liu, X.; Luo, Z.-Q. Ubiquitination independent of E1 and E2 enzymes by bacterial effectors. Nature 2016, 533, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Kotewicz, K.M.; Ramabhadran, V.; Sjoblom, N.; Vogel, J.P.; Haenssler, E.; Zhang, M.; Behringer, J.; Scheck, R.A.; Isberg, R.R. A single Legionella effector catalyzes a multistep ubiquitination pathway to rearrange tubular endoplasmic reticulum for replication. Cell Host Microbe 2017, 21, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Bhogaraju, S.; Dikic, I. Ubiquitination without E1 and E2 enzymes. Nature 2016, 533, 43–44. [Google Scholar] [CrossRef] [PubMed]
- Gan, N.; Nakayasu, E.; Hollenbeck, P.J.; Luo, Z.-Q. Legionella pneumophila inhibits immune signalling via MavC-mediated transglutaminase-induced ubiquitination of UBE2N. Nat. Microbiol. 2018, 4, 134–143. [Google Scholar] [CrossRef]
- Kubori, T.; Kitao, T.; Nagai, H. Emerging insights into bacterial deubiquitinases. Curr. Opin. Microbiol. 2019, 47, 14–19. [Google Scholar] [CrossRef]
- Sheedlo, M.J.; Qiu, J.; Tan, Y.; Paul, L.N.; Luo, Z.-Q.; Das, C. Structural basis of substrate recognition by a bacterial deubiquitinase important for dynamics of phagosome ubiquitination. Proc. Natl. Acad. Sci. USA 2015, 112, 15090–15095. [Google Scholar] [CrossRef]
- Lindner, H.A.; Lytvyn, V.; Qi, H.; Lachance, P.; Ziomek, E.; Ménard, R. Selectivity in ISG15 and ubiquitin recognition by the SARS coronavirus papain-like protease. Arch. Biochem. Biophys. 2007, 466, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020, 39, e106275. [Google Scholar] [CrossRef] [PubMed]
- Kilianski, A.; Mielech, A.M.; Deng, X.; Baker, S.C. Assessing activity and inhibition of Middle East respiratory syndrome coronavirus papain-like and 3C-like proteases using luciferase-based biosensors. J. Virol. 2013, 87, 11955–11962. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, X.; Bian, G.; Tu, J.; Xing, Y.; Wang, Y.; Chen, Z. Proteolytic processing, deubiquitinase and interferon antagonist activities of Middle East respiratory syndrome coronavirus papain-like protease. J. Gen. Virol. 2014, 95, 614–626. [Google Scholar] [CrossRef] [PubMed]
- Frieman, M.; Ratia, K.; Johnston, R.E.; Mesecar, A.D.; Baric, R.S. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-κB signaling. J. Virol. 2009, 83, 6689–6705. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Hamilton, A.M.; Zito, K. Breaking it down: The ubiquitin proteasome system in neuronal morphogenesis. Neural Plast. 2013, 2013, 196848. [Google Scholar] [CrossRef]
- Boukhalfa, A.; Miceli, C.; Ávalos, Y.; Morel, E.; Dupont, N. Interplay between primary cilia, ubiquitin-proteasome system and autophagy. Biochimie 2019, 166, 286–292. [Google Scholar] [CrossRef]
- Gerhardt, C.; Wiegering, A.; Leu, T.; Rüther, U. Control of Hedgehog signalling by the cilia-regulated proteasome. J. Dev. Biol. 2016, 4, 27. [Google Scholar] [CrossRef]
- Baloghova, N.; Lidak, T.; Cermak, L. Ubiquitin ligases involved in the regulation of Wnt, TGF-β, and notch signaling pathways and their roles in mouse development and homeostasis. Genes 2019, 10, 815. [Google Scholar] [CrossRef]
- Micel, L.N.; Tentler, J.J.; Smith, P.G.; Eckhardt, G.S. Role of ubiquitin ligases and the proteasome in oncogenesis: Novel targets for anticancer therapies. J. Clin. Oncol. 2013, 31, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Cloos, J.; Roeten, M.S.; Franke, N.E.; Van Meerloo, J.; Zweegman, S.; Kaspers, G.J.; Jansen, G. (Immuno)proteasomes as therapeutic target in acute leukemia. Cancer Metastasis Rev. 2017, 36, 599–615. [Google Scholar] [CrossRef] [PubMed]
- Raedler, L. Velcade (Bortezomib) receives 2 new FDA indications: For retreatment of patients with multiple myeloma and for first-line treatment of patients with mantle-cell lymphoma. Am. Health Drug Benefits 2015, 8, 135–140. [Google Scholar] [PubMed]
- Horton, T.M.; Perentesis, J.P.; Gamis, A.S.; Alonzo, T.A.; Gerbing, R.B.; Ballard, J.; Adlard, K.; Howard, D.S.; Smith, F.O.; Jenkins, G.; et al. A phase 2 study of bortezomib combined with either idarubicin/cytarabine or cytarabine/etoposide in children with relapsed, refractory or secondary acute myeloid leukemia: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2014, 61, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Kraus, M.; Müller-Ide, H.; Rückrich, T.; Bader, J.; Overkleeft, H.; Driessen, C. Ritonavir, nelfinavir, saquinavir and lopinavir induce proteotoxic stress in acute myeloid leukemia cells and sensitize them for proteasome inhibitor treatment at low micromolar drug concentrations. Leuk. Res. 2014, 38, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Wartman, L.D.; Fiala, M.A.; Fletcher, T.; Hawkins, E.R.; Cashen, A.; DiPersio, J.F.; Jacoby, M.A.; Stockerl-Goldstein, K.E.; Pusic, I.; Uy, G.L.; et al. A phase I study of carfilzomib for relapsed or refractory acute myeloid and acute lymphoblastic leukemia. Leuk. Lymphoma 2015, 57, 728–730. [Google Scholar] [CrossRef] [PubMed]
- Dou, Q.P.; Zonder, J.A. Overview of proteasome inhibitor-based anti-cancer therapies: Perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin-proteasome system. Curr. Cancer Drug Targets 2014, 14, 517–536. [Google Scholar] [CrossRef]
- Zhang, X.-D.; Baladandayuthapani, V.; Lin, H.; Mulligan, G.; Li, B.; Esseltine, D.-L.W.; Qing, Z.; Xu, J.; Hunziker, W.; Barlogie, B.; et al. Tight junction protein 1 modulates proteasome capacity and proteasome inhibitor sensitivity in multiple myeloma via EGFR/JAK1/STAT3 signaling. Cancer Cell 2016, 29, 639–652. [Google Scholar] [CrossRef]
- Hamazaki, J.; Murata, S. ER-resident transcription factor Nrf1 regulates proteasome expression and beyond. Int. J. Mol. Sci. 2020, 21, 3683. [Google Scholar] [CrossRef]
- Steffen, J.; Seeger, M.; Koch, A.; Krüger, E. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol. Cell 2010, 40, 147–158. [Google Scholar] [CrossRef]
- Meiners, S.; Heyken, D.; Weller, A.; Ludwig, A.; Stangl, K.; Kloetzel, P.-M.; Krüger, E. Inhibition of proteasome activity induces concerted expression of proteasome genes and de novo formation of mammalian proteasomes. J. Biol. Chem. 2003, 278, 21517–21525. [Google Scholar] [CrossRef] [PubMed]
- Vangala, J.R.; Sotzny, F.; Krüger, E.; Deshaies, R.J.; Radhakrishnan, S.K. Nrf1 can be processed and activated in a proteasome-independent manner. Curr. Biol. 2016, 26, R834–R835. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Northrop, A.; Byers, H.A.; Radhakrishnan, S.K. Regulation of NRF1, a master transcription factor of proteasome genes: Implications for cancer and neurodegeneration. Mol. Biol. Cell 2020, 31, 2158–2163. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Wang, X.; Wang, Y.; Wang, Y.; Li, J.; Yu, F.-X. Nelfinavir inhibits human DDI2 and potentiates cytotoxicity of proteasome inhibitors. Cell. Signal. 2020, 75, 109775. [Google Scholar] [CrossRef] [PubMed]
- Tomlin, F.M.; Gerling-Driessen, U.I.M.; Liu, Y.-C.; Flynn, R.A.; Vangala, J.R.; Lentz, C.S.; Clauder-Muenster, S.; Jakob, P.; Mueller, W.F.; Ordoñez-Rueda, D.; et al. Inhibition of NGLY1 inactivates the transcription factor Nrf1 and potentiates proteasome inhibitor cytotoxicity. ACS Cent. Sci. 2017, 3, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef]
- Stewart, A.K. How thalidomide works against cancer. Science 2014, 343, 256–257. [Google Scholar] [CrossRef]
- Angers, S.; Li, T.; Yi, X.; MacCoss, M.J.; Moon, R.T.; Zheng, N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Naure 2006, 443, 590–593. [Google Scholar] [CrossRef]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Altered UPS Gene | Component |
|---|---|---|
| CANDLE/PRAAS | PSMB8 (β5i/LMP7) | 20S |
| PSMA3 (α7) | 20S | |
| PSMB4 (β7) | 20S | |
| PSMB9 (β1i/LMP2) | 20S | |
| POMP (POMP) | Proteasome assembly | |
| PSMG2 (PAC2) | Proteasome assembly | |
| PSMB10 (β2i/MECL-1) | 20S | |
| Neurodevelopmental Disorders | PSMC3 (Rpt5) | 19S |
| PSMD12 (Rpn5) | 19S | |
| PSMB1 (β6) | 20S | |
| A20 Haploinsufficiency | TNFAIP3 (A20) | DUB |
| Syndromic Multisystem Autoimmune Disease | ITCH (ITCH) | E3 ligase |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Çetin, G.; Klafack, S.; Studencka-Turski, M.; Krüger, E.; Ebstein, F. The Ubiquitin–Proteasome System in Immune Cells. Biomolecules 2021, 11, 60. https://doi.org/10.3390/biom11010060
Çetin G, Klafack S, Studencka-Turski M, Krüger E, Ebstein F. The Ubiquitin–Proteasome System in Immune Cells. Biomolecules. 2021; 11(1):60. https://doi.org/10.3390/biom11010060
Chicago/Turabian StyleÇetin, Gonca, Sandro Klafack, Maja Studencka-Turski, Elke Krüger, and Frédéric Ebstein. 2021. "The Ubiquitin–Proteasome System in Immune Cells" Biomolecules 11, no. 1: 60. https://doi.org/10.3390/biom11010060
APA StyleÇetin, G., Klafack, S., Studencka-Turski, M., Krüger, E., & Ebstein, F. (2021). The Ubiquitin–Proteasome System in Immune Cells. Biomolecules, 11(1), 60. https://doi.org/10.3390/biom11010060