Yersinia pestis Plasminogen Activator




Abstract
1. Discovery, History, Genetic Control, Biosynthesis Conditions, Isolation, Purification, and Physicochemical Properties of Plasminogen Activator
1.1. The Discovery
1.2. Pla, Location, Processing, and Conformation
1.3. Regulation of Pla
1.4. Role of Temperature and Lipooligosaccharide (Los) on Pla Activities
2. Pla as a Broad-Spectrum Protease with Adhesion Properties
2.1. The Multiple Substrates Processed by Pla
2.1.1. The Substrates Related to Hemostasis
2.1.2. Immune-Related Targets
2.1.3. Ypestis Proteins Processed by Pla
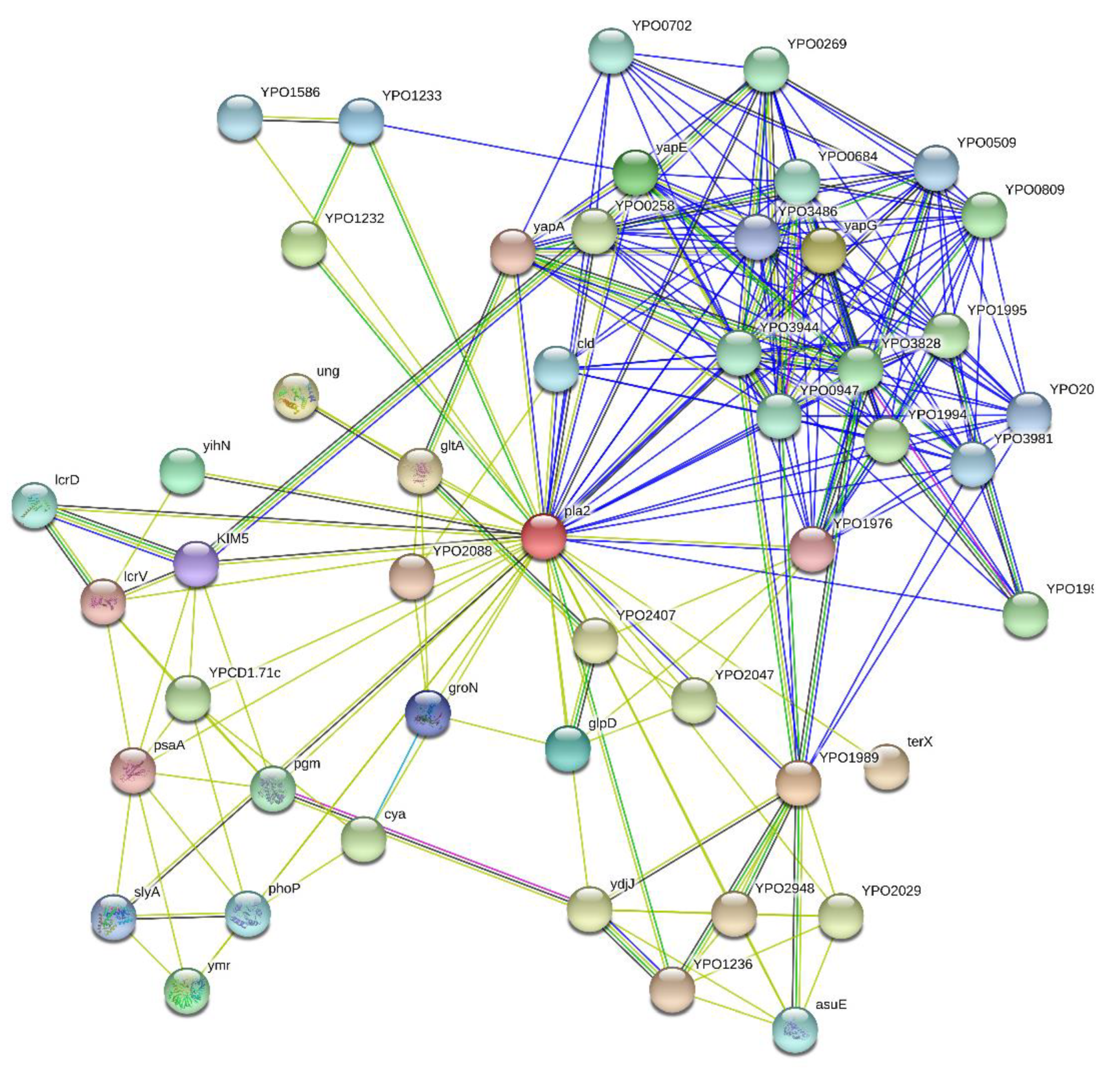
2.2. Overall Interactibility of Pla
2.3. Structure and Catalytic Mechanisms
2.4. Pla, an Adhesin with Multiple Binding Substrates
3. The Biological Role of Plasminogen Activator in Pathogenesis
3.1. The Proven or Unproven Role of Pla
3.2. The Role of Pla during Bubonic Plague
3.3. The Role of Pla during Primary Pneumonic Plague
4. The Evolution Aspect
5. Pla: Not a Vaccine Antigen, Maybe an Antibacterial Target, but a Diagnosis Tool
6. Concluding Remark
Author Contributions
Funding
Conflicts of Interest
References
- Yersin, A. Bubonic plague in Hong Kong. 1894. Rev. Med. Suisse Romande 1994, 114, 393–395. [Google Scholar] [PubMed]
- Butler, T. Plague and Other Yersinia Infections; Plenum Medical Book Company: New York, NY, USA, 1983. [Google Scholar]
- Madison, R.R. Fibrinolytic specificity of bacillus pestis. Proc. Soc. Exptl. Biol. Med. 1936, 34, 301–302. [Google Scholar] [CrossRef]
- Jawetz, E.; Meyer, K.F. Studies on plague immunity in experimental animals. II. Some factors of the immunity mechanism in bubonic plague. J. Immunol. 1944, 49, 15–30. [Google Scholar]
- Beesley, E.D.; Brubaker, R.R.; Janssen, W.A.; Surgalla, M.J. Pesticins. 3. Expression of coagulase and mechanism of fibrinolysis. J. Bacteriol. 1967, 94, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Sodeinde, O.A.; Subrahmanyam, Y.V.; Stark, K.; Quan, T.; Bao, Y.; Goguen, J.D. A surface protease and the invasive character of plague. Science 1992, 258, 1004–1007. [Google Scholar] [CrossRef]
- Domaradskiy, I.V. Plague; Meditsina Press: Moscow, Russia, 1998; p. 176. [Google Scholar]
- Domaradskiy, I.V. Outlines of Plague Pathogenesis; Meditsina Press: Moscow, Russia, 1966; p. 272. [Google Scholar]
- Brubaker, R.R.; Beesley, E.D.; Surgalla, M.J. Pasteurella pestis: Role of Pesticin I and Iron in Experimental Plague. Science 1965, 149, 422–424. [Google Scholar] [CrossRef]
- Brubaker, R.R.; Surgalla, M.J.; Beesley, E.D. Pesticinogeny and bacterial virulence. Zentr. Bakteriol. Parasitenk. Abt. I Orig. 1965, 196, 302–312. [Google Scholar]
- Ferber, D.M.; Brubaker, R.R. Plasmids in Yersinia pestis. Infect. Immun. 1981, 31, 839–841. [Google Scholar] [CrossRef]
- Protsenko, O.; Pi, A.; Mozharov, O.; Konnov, N.; Popov, I. Detection and characterization of the plasmids of the plague microbe which determine the synthesis of pesticin 1, fraction 1 antigen and “mouse” toxin exotoxin. Genetika 1983, 19, 1081–1090. [Google Scholar]
- Sodeinde, O.A.; Goguen, J.D. Nucleotide sequence of the plasminogen activator gene of Yersinia pestis: Relationship to ompT of Escherichia coli and gene E of Salmonella typhimurium. Infect. Immun. 1989, 57, 1517–1523. [Google Scholar] [CrossRef]
- Sodeinde, O.A.; Sample, A.K.; Brubaker, R.R.; Goguen, J.D. Plasminogen activator/coagulase gene of Yersinia pestis is responsible for degradation of plasmid-encoded outer membrane proteins. Infect. Immun. 1988, 56, 2749–2752. [Google Scholar] [CrossRef] [PubMed]
- Kienle, Z.; Emody, L.; Svanborg, C.; O′Toole, P.W. Adhesive properties conferred by the plasminogen activator of Yersinia pestis. J. Gen. Microbiol. 1992, 138 Pt 8, 1679–1687. [Google Scholar] [CrossRef][Green Version]
- Chu, M.C.; Dong, X.Q.; Zhou, X.; Garon, C.F. A cryptic 19-kilobase plasmid associated with U.S. isolates of Yersinia pestis: A dimer of the 9.5-kilobase plasmid. Am. J. Trop. Med. Hyg. 1998, 59, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Susat, J.; Bonczarowska, J.H.; Petersone-Gordina, E.; Immel, A.; Nebel, A.; Gerhards, G.; Krause-Kyora, B. Yersinia pestis strains from Latvia show depletion of the pla virulence gene at the end of the second plague pandemic. Sci. Rep. 2020, 10, 14628. [Google Scholar] [CrossRef] [PubMed]
- Eren, E.; Murphy, M.; Goguen, J.; van den Berg, B. An active site water network in the plasminogen activator pla from Yersinia pestis. Structure 2010, 18, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Eren, E.; van den Berg, B. Structural basis for activation of an integral membrane protease by lipopolysaccharide. J. Biol. Chem. 2012, 287, 23971–23976. [Google Scholar] [CrossRef]
- Kukkonen, M.; Korhonen, T.K. The omptin family of enterobacterial surface proteases/adhesins: From housekeeping in Escherichia coli to systemic spread of Yersinia pestis. Int. J. Med. Microbiol. 2004, 294, 7–14. [Google Scholar] [CrossRef]
- Mangel, W.F.; Toledo, D.L.; Brown, M.T.; Worzalla, K.; Lee, M.; Dunn, J.J. Omptin: An Escherichia coli outer membrane proteinase that activates plasminogen. Methods Enzymol. 1994, 244, 384–399. [Google Scholar] [CrossRef]
- Grodberg, J.; Dunn, J.J. ompT encodes the Escherichia coli outer membrane protease that cleaves T7 RNA polymerase during purification. J. Bacteriol. 1988, 170, 1245–1253. [Google Scholar] [CrossRef]
- Guina, T.; Yi, E.C.; Wang, H.; Hackett, M.; Miller, S.I. A PhoP-regulated outer membrane protease of Salmonella enterica serovar typhimurium promotes resistance to alpha-helical antimicrobial peptides. J. Bacteriol. 2000, 182, 4077–4086. [Google Scholar] [CrossRef]
- Egile, C.; d′Hauteville, H.; Parsot, C.; Sansonetti, P.J. SopA, the outer membrane protease responsible for polar localization of IcsA in Shigella flexneri. Mol. Microbiol. 1997, 23, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A.; Stierhof, Y.D.; Henning, U. New outer membrane-associated protease of Escherichia coli K-12. J. Bacteriol. 1994, 176, 359–367. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kutyrev, V.; Mehigh, R.J.; Motin, V.L.; Pokrovskaya, M.S.; Smirnov, G.B.; Brubaker, R.R. Expression of the plague plasminogen activator in Yersinia pseudotuberculosis and Escherichia coli. Infect. Immun. 1999, 67, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Sodeinde, O.A.; Goguen, J.D. Genetic analysis of the 9.5-kilobase virulence plasmid of Yersinia pestis. Infect. Immun. 1988, 56, 2743–2748. [Google Scholar] [CrossRef]
- Kukkonen, M.; Lahteenmaki, K.; Suomalainen, M.; Kalkkinen, N.; Emody, L.; Lang, H.; Korhonen, T.K. Protein regions important for plasminogen activation and inactivation of alpha2-antiplasmin in the surface protease Pla of Yersinia pestis. Mol. Microbiol. 2001, 40, 1097–1111. [Google Scholar] [CrossRef]
- Ruback, E.; Lobo, L.A.; Franca, T.C.; Pascutti, P.G. Structural analysis of Pla protein from the biological warfare agent Yersinia pestis: Docking and molecular dynamics of interactions with the mammalian plasminogen system. J. Biomol. Struct. Dyn. 2013, 31, 477–484. [Google Scholar] [CrossRef]
- Haiko, J.; Laakkonen, L.; Juuti, K.; Kalkkinen, N.; Korhonen, T.K. The omptins of Yersinia pestis and Salmonella enterica cleave the reactive center loop of plasminogen activator inhibitor 1. J. Bacteriol. 2010, 192, 4553–4561. [Google Scholar] [CrossRef]
- Iakoucheva, L.M.; Kimzey, A.L.; Masselon, C.D.; Bruce, J.E.; Garner, E.C.; Brown, C.J.; Dunker, A.K.; Smith, R.D.; Ackerman, E.J. Identification of intrinsic order and disorder in the DNA repair protein XPA. Protein Sci. 2001, 10, 560–571. [Google Scholar] [CrossRef]
- Receveur-Brechot, V.; Bourhis, J.M.; Uversky, V.N.; Canard, B.; Longhi, S. Assessing protein disorder and induced folding. Proteins 2006, 62, 24–45. [Google Scholar] [CrossRef]
- Uversky, V.N.; Dunker, A.K. Multiparametric analysis of intrinsically disordered proteins: Looking at intrinsic disorder through compound eyes. Anal. Chem. 2012, 84, 2096–2104. [Google Scholar] [CrossRef]
- Uversky, V.N. Biophysical Methods to Investigate Intrinsically Disordered Proteins: Avoiding an “Elephant and Blind Men” Situation. Adv. Exp. Med. Biol. 2015, 870, 215–260. [Google Scholar] [CrossRef] [PubMed]
- Haiko, J.; Kukkonen, M.; Ravantti, J.J.; Westerlund-Wikstrom, B.; Korhonen, T.K. The single substitution I259T, conserved in the plasminogen activator Pla of pandemic Yersinia pestis branches, enhances fibrinolytic activity. J. Bacteriol. 2009, 191, 4758–4766. [Google Scholar] [CrossRef] [PubMed]
- McDonough, K.A.; Falkow, S. A Yersinia pestis-specific DNA fragment encodes temperature-dependent coagulase and fibrinolysin-associated phenotypes. Mol. Microbiol. 1989, 3, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Motin, V.L.; Georgescu, A.M.; Fitch, J.P.; Gu, P.P.; Nelson, D.O.; Mabery, S.L.; Garnham, J.B.; Sokhansanj, B.A.; Ott, L.L.; Coleman, M.A.; et al. Temporal global changes in gene expression during temperature transition in Yersinia pestis. J. Bacteriol. 2004, 186, 6298–6305. [Google Scholar] [CrossRef] [PubMed]
- Felek, S.; Tsang, T.M.; Krukonis, E.S. Three Yersinia pestis adhesins facilitate Yop delivery to eukaryotic cells and contribute to plague virulence. Infect. Immun. 2010, 78, 4134–4150. [Google Scholar] [CrossRef] [PubMed]
- Chromy, B.A.; Choi, M.W.; Murphy, G.A.; Gonzales, A.D.; Corzett, C.H.; Chang, B.C.; Fitch, J.P.; McCutchen-Maloney, S.L. Proteomic characterization of Yersinia pestis virulence. J. Bacteriol. 2005, 187, 8172–8180. [Google Scholar] [CrossRef]
- Suomalainen, M.; Lobo, L.A.; Brandenburg, K.; Lindner, B.; Virkola, R.; Knirel, Y.A.; Anisimov, A.P.; Holst, O.; Korhonen, T.K. Temperature-induced changes in the lipopolysaccharide of Yersinia pestis affect plasminogen activation by the pla surface protease. Infect. Immun. 2010, 78, 2644–2652. [Google Scholar] [CrossRef]
- Chauvaux, S.; Rosso, M.L.; Frangeul, L.; Lacroix, C.; Labarre, L.; Schiavo, A.; Marceau, M.; Dillies, M.A.; Foulon, J.; Coppee, J.Y.; et al. Transcriptome analysis of Yersinia pestis in human plasma: An approach for discovering bacterial genes involved in septicaemic plague. Microbiology 2007, 153, 3112–3124. [Google Scholar] [CrossRef][Green Version]
- Smiley, S.T.; Szaba, F.M.; Kummer, L.W.; Duso, D.K.; Lin, J.S. Yersinia pestis Pla Protein Thwarts T Cell Defense against Plague. Infect. Immun. 2019, 87. [Google Scholar] [CrossRef]
- Sebbane, F.; Lemaitre, N.; Sturdevant, D.E.; Rebeil, R.; Virtaneva, K.; Porcella, S.F.; Hinnebusch, B.J. Adaptive response of Yersinia pestis to extracellular effectors of innate immunity during bubonic plague. Proc. Natl. Acad. Sci. USA 2006, 103, 11766–11771. [Google Scholar] [CrossRef]
- Lathem, W.W.; Crosby, S.D.; Miller, V.L.; Goldman, W.E. Progression of primary pneumonic plague: A mouse model of infection, pathology, and bacterial transcriptional activity. Proc. Natl. Acad. Sci. USA 2005, 102, 17786–17791. [Google Scholar] [CrossRef] [PubMed]
- Ritzert, J.T.; Lathem, W.W. Depletion of Glucose Activates Catabolite Repression during Pneumonic Plague. J. Bacteriol. 2018, 200. [Google Scholar] [CrossRef] [PubMed]
- Zhan, L.; Han, Y.; Yang, L.; Geng, J.; Li, Y.; Gao, H.; Guo, Z.; Fan, W.; Li, G.; Zhang, L.; et al. The cyclic AMP receptor protein, CRP, is required for both virulence and expression of the minimal CRP regulon in Yersinia pestis biovar microtus. Infect. Immun. 2008, 76, 5028–5037. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.J.; Chauhan, S.; Motin, V.L.; Goh, E.B.; Igo, M.M.; Young, G.M. Direct transcriptional control of the plasminogen activator gene of Yersinia pestis by the cyclic AMP receptor protein. J. Bacteriol. 2007, 189, 8890–8900. [Google Scholar] [CrossRef]
- Lathem, W.W.; Schroeder, J.A.; Bellows, L.E.; Ritzert, J.T.; Koo, J.T.; Price, P.A.; Caulfield, A.J.; Goldman, W.E. Posttranscriptional regulation of the Yersinia pestis cyclic AMP receptor protein Crp and impact on virulence. mBio 2014, 5, e01038-13. [Google Scholar] [CrossRef]
- Pouillot, F.; Derbise, A.; Kukkonen, M.; Foulon, J.; Korhonen, T.K.; Carniel, E. Evaluation of O-antigen inactivation on Pla activity and virulence of Yersinia pseudotuberculosis harbouring the pPla plasmid. Microbiology 2005, 151, 3759–3768. [Google Scholar] [CrossRef]
- Kukkonen, M.; Suomalainen, M.; Kyllonen, P.; Lahteenmaki, K.; Lang, H.; Virkola, R.; Helander, I.M.; Holst, O.; Korhonen, T.K. Lack of O-antigen is essential for plasminogen activation by Yersinia pestis and Salmonella enterica. Mol. Microbiol. 2004, 51, 215–225. [Google Scholar] [CrossRef]
- Rebeil, R.; Ernst, R.K.; Gowen, B.B.; Miller, S.I.; Hinnebusch, B.J. Variation in lipid A structure in the pathogenic yersiniae. Mol. Microbiol. 2004, 52, 1363–1373. [Google Scholar] [CrossRef]
- Dentovskaya, S.V.; Platonov, M.E.; Bakhteeva, I.V.; Anisimov, A.P. Presence of the full lipopolysaccharide core structure is necessary for activation of plasminogen by Yersinia pestis. Probl. Part. Danger. Infect. 2007, 93, 49–51. [Google Scholar]
- Montminy, S.W.; Khan, N.; McGrath, S.; Walkowicz, M.J.; Sharp, F.; Conlon, J.E.; Fukase, K.; Kusumoto, S.; Sweet, C.; Miyake, K.; et al. Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat. Immunol. 2006, 7, 1066–1073. [Google Scholar] [CrossRef]
- Knirel, Y.A.; Anisimov, A.P. Lipopolysaccharide of Yersinia pestis, the Cause of Plague: Structure, Genetics, Biological Properties. Acta Nat. 2012, 4, 46–58. [Google Scholar] [CrossRef]
- Dentovskaya, S.V.; Anisimov, A.P.; Kondakova, A.N.; Lindner, B.; Bystrova, O.V.; Svetoch, T.E.; Shaikhutdinova, R.Z.; Ivanov, S.A.; Bakhteeva, I.V.; Titareva, G.M.; et al. Functional characterization and biological significance of Yersinia pestis lipopolysaccharide biosynthesis genes. Biochem. (Mosc.) 2011, 76, 808–822. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.; Monroe, D.M., 3rd. A cell-based model of hemostasis. Thromb. Haemost. 2001, 85, 958–965. [Google Scholar] [PubMed]
- Swieringa, F.; Spronk, H.M.H.; Heemskerk, J.W.M.; van der Meijden, P.E.J. Integrating platelet and coagulation activation in fibrin clot formation. Res. Pract. Thromb. Haemost. 2018, 2, 450–460. [Google Scholar] [CrossRef]
- Bagoly, Z.; Szegedi, I.; Kalmandi, R.; Toth, N.K.; Csiba, L. Markers of Coagulation and Fibrinolysis Predicting the Outcome of Acute Ischemic Stroke Thrombolysis Treatment: A Review of the Literature. Front. Neurol. 2019, 10, 513. [Google Scholar] [CrossRef] [PubMed]
- Yun, T.H.; Cott, J.E.; Tapping, R.I.; Slauch, J.M.; Morrissey, J.H. Proteolytic inactivation of tissue factor pathway inhibitor by bacterial omptins. Blood 2009, 113, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Welkos, S.L.; Friedlander, A.M.; Davis, K.J. Studies on the role of plasminogen activator in systemic infection by virulent Yersinia pestis strain C092. Microb. Pathog. 1997, 23, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Jarvinen, H.M.; Laakkonen, L.; Haiko, J.; Johansson, T.; Juuti, K.; Suomalainen, M.; Buchrieser, C.; Kalkkinen, N.; Korhonen, T.K. Human single-chain urokinase is activated by the omptins PgtE of Salmonella enterica and Pla of Yersinia pestis despite mutations of active site residues. Mol. Microbiol. 2013, 89, 507–517. [Google Scholar] [CrossRef]
- Eddy, J.L.; Schroeder, J.A.; Zimbler, D.L.; Caulfield, A.J.; Lathem, W.W. Proteolysis of plasminogen activator inhibitor-1 by Yersinia pestis remodulates the host environment to promote virulence. J. Thromb. Haemost. 2016, 14, 1833–1843. [Google Scholar] [CrossRef]
- Bartra, S.S.; Ding, Y.; Miya Fujimoto, L.; Ring, J.G.; Jain, V.; Ram, S.; Marassi, F.M.; Plano, G.V. Yersinia pestis uses the Ail outer membrane protein to recruit vitronectin. Microbiology 2015, 161, 2174–2183. [Google Scholar] [CrossRef]
- Valls Seron, M.; Haiko, J.; PG, D.E.G.; Korhonen, T.K.; Meijers, J.C. Thrombin-activatable fibrinolysis inhibitor is degraded by Salmonella enterica and Yersinia pestis. J. Thromb. Haemost. 2010, 8, 2232–2240. [Google Scholar] [CrossRef] [PubMed]
- Eddy, J.L.; Schroeder, J.A.; Zimbler, D.L.; Bellows, L.E.; Lathem, W.W. Impact of the Pla protease substrate alpha2-antiplasmin on the progression of primary pneumonic plague. Infect. Immun. 2015, 83, 4837–4847. [Google Scholar] [CrossRef] [PubMed]
- Zimbler, D.L.; Eddy, J.L.; Schroeder, J.A.; Lathem, W.W. Inactivation of Peroxiredoxin 6 by the Pla Protease of Yersinia pestis. Infect. Immun. 2016, 84, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Caulfield, A.J.; Walker, M.E.; Gielda, L.M.; Lathem, W.W. The Pla protease of Yersinia pestis degrades fas ligand to manipulate host cell death and inflammation. Cell Host Microbe 2014, 15, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B. Peroxiredoxin 6: A bifunctional enzyme with glutathione peroxidase and phospholipase A(2) activities. Antioxid Redox Signal 2011, 15, 831–844. [Google Scholar] [CrossRef]
- Sharapov, M.G.; Novoselov, V.I.; Gudkov, S.V. Radioprotective Role of Peroxiredoxin 6. Antioxidants 2019, 8, 15. [Google Scholar] [CrossRef]
- Wang, X.; Phelan, S.A.; Forsman-Semb, K.; Taylor, E.F.; Petros, C.; Brown, A.; Lerner, C.P.; Paigen, B. Mice with targeted mutation of peroxiredoxin 6 develop normally but are susceptible to oxidative stress. J. Biol. Chem. 2003, 278, 25179–25190. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Phelan, S.A.; Manevich, Y.; Feinstein, S.I.; Fisher, A.B. Transgenic mice overexpressing peroxiredoxin 6 show increased resistance to lung injury in hyperoxia. Am. J. Respir. Cell Mol. Biol. 2006, 34, 481–486. [Google Scholar] [CrossRef]
- Nagata, S. Apoptosis by death factor. Cell 1997, 88, 355–365. [Google Scholar] [CrossRef]
- Caulfield, A.J.; Lathem, W.W. Disruption of fas-fas ligand signaling, apoptosis, and innate immunity by bacterial pathogens. PLoS Pathog. 2014, 10, e1004252. [Google Scholar] [CrossRef]
- Yamada, A.; Arakaki, R.; Saito, M.; Kudo, Y.; Ishimaru, N. Dual Role of Fas/FasL-Mediated Signal in Peripheral Immune Tolerance. Front. Immunol. 2017, 8, 403. [Google Scholar] [CrossRef] [PubMed]
- Galvan, E.M.; Lasaro, M.A.; Schifferli, D.M. Capsular antigen fraction 1 and Pla modulate the susceptibility of Yersinia pestis to pulmonary antimicrobial peptides such as cathelicidin. Infect. Immun. 2008, 76, 1456–1464. [Google Scholar] [CrossRef] [PubMed]
- Agarkov, A.; Chauhan, S.; Lory, P.J.; Gilbertson, S.R.; Motin, V.L. Substrate specificity and screening of the integral membrane protease Pla. Bioorg. Med. Chem. Lett. 2008, 18, 427–431. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Feodorova, V.A.; Devdariani, Z.L. Expression of acid-stable proteins and modified lipopolysaccharide of Yersinia pestis in acidic growth medium. J. Med. Microbiol. 2001, 50, 979–985. [Google Scholar] [CrossRef][Green Version]
- Lawrenz, M.B.; Lenz, J.D.; Miller, V.L. A novel autotransporter adhesin is required for efficient colonization during bubonic plague. Infect. Immun. 2009, 77, 317–326. [Google Scholar] [CrossRef][Green Version]
- Lenz, J.D.; Lawrenz, M.B.; Cotter, D.G.; Lane, M.C.; Gonzalez, R.J.; Palacios, M.; Miller, V.L. Expression during host infection and localization of Yersinia pestis autotransporter proteins. J. Bacteriol. 2011, 193, 5936–5949. [Google Scholar] [CrossRef]
- Henderson, I.R.; Navarro-Garcia, F.; Desvaux, M.; Fernandez, R.C.; Ala’Aldeen, D. Type V protein secretion pathway: The autotransporter story. Microbiol. Mol. Biol. Rev. 2004, 68, 692–744. [Google Scholar] [CrossRef]
- Lane, M.C.; Lenz, J.D.; Miller, V.L. Proteolytic processing of the Yersinia pestis YapG autotransporter by the omptin protease Pla and the contribution of YapG to murine plague pathogenesis. J. Med. Microbiol. 2013, 62, 1124–1134. [Google Scholar] [CrossRef]
- Caulfield, A.J.; Lathem, W.W. Substrates of the plasminogen activator protease of Yersinia pestis. Adv. Exp. Med. Biol. 2012, 954, 253–260. [Google Scholar] [CrossRef]
- Yen, Y.T.; Karkal, A.; Bhattacharya, M.; Fernandez, R.C.; Stathopoulos, C. Identification and characterization of autotransporter proteins of Yersinia pestis KIM. Mol. Membr. Biol. 2007, 24, 28–40. [Google Scholar] [CrossRef]
- Lawrenz, M.B.; Pennington, J.; Miller, V.L. Acquisition of omptin reveals cryptic virulence function of autotransporter YapE in Yersinia pestis. Mol. Microbiol. 2013, 89, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Garcia, E.; Nedialkov, Y.A.; Elliott, J.; Motin, V.L.; Brubaker, R.R. Molecular characterization of KatY (antigen 5), a thermoregulated chromosomally encoded catalase-peroxidase of Yersinia pestis. J. Bacteriol. 1999, 181, 3114–3122. [Google Scholar] [CrossRef] [PubMed]
- Mehigh, R.J.; Sample, A.K.; Brubaker, R.R. Expression of the low calcium response in Yersinia pestis. Microb. Pathog. 1989, 6, 203–217. [Google Scholar] [CrossRef]
- Plano, G.V.; Straley, S.C. Multiple effects of lcrD mutations in Yersinia pestis. J. Bacteriol. 1993, 175, 3536–3545. [Google Scholar] [CrossRef] [PubMed]
- Plano, G.V.; Schesser, K. The Yersinia pestis type III secretion system: Expression, assembly and role in the evasion of host defenses. Immunol. Res. 2013, 57, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Degen, J.L.; Bugge, T.H.; Goguen, J.D. Fibrin and fibrinolysis in infection and host defense. J. Thromb. Haemost. 2007, 5 (Suppl. S1), 24–31. [Google Scholar] [CrossRef]
- Sebbane, F.; Jarrett, C.O.; Gardner, D.; Long, D.; Hinnebusch, B.J. Role of the Yersinia pestis plasminogen activator in the incidence of distinct septicemic and bubonic forms of flea-borne plague. Proc. Natl. Acad. Sci. USA 2006, 103, 5526–5530. [Google Scholar] [CrossRef]
- Agar, S.L.; Sha, J.; Foltz, S.M.; Erova, T.E.; Walberg, K.G.; Baze, W.B.; Suarez, G.; Peterson, J.W.; Chopra, A.K. Characterization of the rat pneumonic plague model: Infection kinetics following aerosolization of Yersinia pestis CO92. Microbes Infect. 2009, 11, 205–214. [Google Scholar] [CrossRef]
- Lathem, W.W.; Price, P.A.; Miller, V.L.; Goldman, W.E. A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science 2007, 315, 509–513. [Google Scholar] [CrossRef]
- Yun, T.H.; Morrissey, J.H. Polyphosphate and omptins: Novel bacterial procoagulant agents. J. Cell Mol. Med. 2009, 13, 4146–4153. [Google Scholar] [CrossRef]
- Suomalainen, M.; Haiko, J.; Ramu, P.; Lobo, L.; Kukkonen, M.; Westerlund-Wikstrom, B.; Virkola, R.; Lahteenmaki, K.; Korhonen, T.K. Using every trick in the book: The Pla surface protease of Yersinia pestis. Adv. Exp. Med. Biol. 2007, 603, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Cowan, C.; Jones, H.A.; Kaya, Y.H.; Perry, R.D.; Straley, S.C. Invasion of epithelial cells by Yersinia pestis: Evidence for a Y. pestis-specific invasin. Infect. Immun. 2000, 68, 4523–4530. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef]
- Dunker, A.K.; Brown, C.J.; Obradovic, Z. Identification and functions of usefully disordered proteins. Adv. Protein Chem. 2002, 62, 25–49. [Google Scholar]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef]
- Uversky, V.N. What does it mean to be natively unfolded? Eur. J. Biochem. 2002, 269, 2–12. [Google Scholar] [CrossRef]
- Uversky, V.N. Protein folding revisited. A polypeptide chain at the folding-misfolding-nonfolding cross-roads: Which way to go? Cell. Mol. Life Sci. 2003, 60, 1852–1871. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta 2013, 1834, 932–951. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. A decade and a half of protein intrinsic disorder: Biology still waits for physics. Protein Sci. 2013, 22, 693–724. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Functional roles of transiently and intrinsically disordered regions within proteins. FEBS J. 2015, 282, 1182–1189. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef]
- Lahteenmaki, K.; Virkola, R.; Saren, A.; Emody, L.; Korhonen, T.K. Expression of plasminogen activator pla of Yersinia pestis enhances bacterial attachment to the mammalian extracellular matrix. Infect. Immun. 1998, 66, 5755–5762. [Google Scholar] [CrossRef]
- Lobo, L.A. Adhesive properties of the purified plasminogen activator Pla of Yersinia pestis. FEMS Microbiol. Lett. 2006, 262, 158–162. [Google Scholar] [CrossRef][Green Version]
- Lahteenmaki, K.; Kukkonen, M.; Korhonen, T.K. The Pla surface protease/adhesin of Yersinia pestis mediates bacterial invasion into human endothelial cells. FEBS Lett. 2001, 504, 69–72. [Google Scholar] [CrossRef]
- Banerjee, S.K.; Huckuntod, S.D.; Mills, S.D.; Kurten, R.C.; Pechous, R.D. Modeling Pneumonic Plague in Human Precision-Cut Lung Slices Highlights a Role for the Plasminogen Activator Protease in Facilitating Type 3 Secretion. Infect. Immun. 2019, 87. [Google Scholar] [CrossRef]
- Benedek, O.; Khan, A.S.; Schneider, G.; Nagy, G.; Autar, R.; Pieters, R.J.; Emody, L. Identification of laminin-binding motifs of Yersinia pestis plasminogen activator by phage display. Int. J. Med. Microbiol. 2005, 295, 87–98. [Google Scholar] [CrossRef]
- Zhang, S.S.; Park, C.G.; Zhang, P.; Bartra, S.S.; Plano, G.V.; Klena, J.D.; Skurnik, M.; Hinnebusch, B.J.; Chen, T. Plasminogen activator Pla of Yersinia pestis utilizes murine DEC-205 (CD205) as a receptor to promote dissemination. J. Biol. Chem. 2008, 283, 31511–31521. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, B.J.; Fischer, E.R.; Schwan, T.G. Evaluation of the role of the Yersinia pestis plasminogen activator and other plasmid-encoded factors in temperature-dependent blockage of the flea. J. Infect. Dis. 1998, 178, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.K.; Crane, S.D.; Pechous, R.D. A dual role for the Plasminogen activator protease during the pre-inflammatory phase of primary pneumonic plague. J. Infect. Dis. 2020, 222, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Cavanaugh, D.C. Specific effect of temperature upon transmission of the plague bacillus by the oriental rat flea, Xenopsylla cheopis. Am. J. Trop. Med. Hyg. 1971, 20, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Bacot, A.W.; Martin, C.J. LXVII. Observations on the mechanism of the transmission of plague by fleas. J. Hyg. 1914, 13, 423–439. [Google Scholar] [PubMed]
- Lorange, E.A.; Race, B.L.; Sebbane, F.; Joseph Hinnebusch, B. Poor vector competence of fleas and the evolution of hypervirulence in Yersinia pestis. J. Infect. Dis. 2005, 191, 1907–1912. [Google Scholar] [CrossRef] [PubMed]
- Filippov, A.A.; Solodovnikov, N.S.; Kookleva, L.M.; Protsenko, O.A. Plasmid content in Yersinia pestis strains of different origin. FEMS Microbiol. Lett. 1990, 55, 45–48. [Google Scholar] [CrossRef]
- Cavalcanti, Y.V.; Leal, N.C.; De Almeida, A.M. Typing of Yersinia pestis isolates from the state of Ceara, Brazil. Lett. Appl. Microbiol. 2002, 35, 543–547. [Google Scholar] [CrossRef]
- Sebbane, F.; Jarrett, C.; Gardner, D.; Long, D.; Hinnebusch, B.J. The Yersinia pestis caf1M1A1 fimbrial capsule operon promotes transmission by flea bite in a mouse model of bubonic plague. Infect. Immun. 2009, 77, 1222–1229. [Google Scholar] [CrossRef]
- Sebbane, F.; Jarrett, C.; Gardner, D.; Long, D.; Hinnebusch, B.J. Role of the Yersinia pestis yersiniabactin iron acquisition system in the incidence of flea-borne plague. PLoS ONE 2010, 5, e14379. [Google Scholar] [CrossRef]
- Guinet, F.; Ave, P.; Filali, S.; Huon, C.; Savin, C.; Huerre, M.; Fiette, L.; Carniel, E. Dissociation of Tissue Destruction and Bacterial Expansion during Bubonic Plague. PLoS Pathog. 2015, 11, e1005222. [Google Scholar] [CrossRef] [PubMed]
- Koster, F.; Perlin, D.S.; Park, S.; Brasel, T.; Gigliotti, A.; Barr, E.; Myers, L.; Layton, R.C.; Sherwood, R.; Lyons, C.R. Milestones in progression of primary pneumonic plague in cynomolgus macaques. Infect. Immun. 2010, 78, 2946–2955. [Google Scholar] [CrossRef] [PubMed]
- Agar, S.L.; Sha, J.; Foltz, S.M.; Erova, T.E.; Walberg, K.G.; Parham, T.E.; Baze, W.B.; Suarez, G.; Peterson, J.W.; Chopra, A.K. Characterization of a mouse model of plague after aerosolization of Yersinia pestis CO92. Microbiology 2008, 154, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- Bubeck, S.S.; Cantwell, A.M.; Dube, P.H. Delayed inflammatory response to primary pneumonic plague occurs in both outbred and inbred mice. Infect. Immun. 2007, 75, 697–705. [Google Scholar] [CrossRef]
- Derbise, A.; Pierre, F.; Merchez, M.; Pradel, E.; Laouami, S.; Ricard, I.; Sirard, J.C.; Fritz, J.; Lemaitre, N.; Akinbi, H.; et al. Inheritance of the lysozyme inhibitor Ivy was an important evolutionary step by Yersinia pestis to avoid the host innate immune response. J. Infect. Dis. 2013, 207, 1535–1543. [Google Scholar] [CrossRef]
- Chain, P.S.; Carniel, E.; Larimer, F.W.; Lamerdin, J.; Stoutland, P.O.; Regala, W.M.; Georgescu, A.M.; Vergez, L.M.; Land, M.L.; Motin, V.L.; et al. Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. USA 2004, 101, 13826–13831. [Google Scholar] [CrossRef]
- Cui, Y.; Yu, C.; Yan, Y.; Li, D.; Li, Y.; Jombart, T.; Weinert, L.A.; Wang, Z.; Guo, Z.; Xu, L.; et al. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis. Proc. Natl. Acad. Sci. USA 2013, 110, 577–582. [Google Scholar] [CrossRef]
- Platonov, M.E.; Evseeva, V.V.; Dentovskaya, S.V.; Anisimov, A.P. Molecular typing of Yersinia pestis. Mol. Gen. Mikrobiol. Virusol. 2013, 3–12. [Google Scholar] [CrossRef]
- Achtman, M. How old are bacterial pathogens? Proc. Biol. Sci. 2016, 283. [Google Scholar] [CrossRef]
- Rascovan, N.; Sjogren, K.G.; Kristiansen, K.; Nielsen, R.; Willerslev, E.; Desnues, C.; Rasmussen, S. Emergence and Spread of Basal Lineages of Yersinia pestis during the Neolithic Decline. Cell 2019, 176, 295–305.e10. [Google Scholar] [CrossRef]
- Kislichkina, A.A.; Platonov, M.E.; Vagaiskaya, A.S.; Bogun, A.G.; Dentovskaya, S.V.; Anisimov, A.P. Rational Taxonomy of Yersinia pestis. Mol. Genet. Microbiol. Virol. 2019, 34, 110–117. [Google Scholar] [CrossRef]
- Kutyrev, V.V.; Filippov, A.A.; Shavina, N.; Protsenko, O.A. [Genetic analysis and simulation of the virulence of Yersinia pestis]. Mol. Gen. Mikrobiol. Virusol. 1989, 42–47. [Google Scholar]
- Samoilova, S.V.; Samoilova, L.V.; Yezhov, I.N.; Drozdov, I.G.; Anisimov, A.P. Virulence of pPst+ and pPst- strains of Yersinia pestis for guinea-pigs. J. Med. Microbiol. 1996, 45, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Haiko, J.; Laakkonen, L.; Westerlund-Wikstrom, B.; Korhonen, T.K. Molecular adaptation of a plant-bacterium outer membrane protease towards plague virulence factor Pla. BMC Evol. Biol. 2011, 11, 43. [Google Scholar] [CrossRef]
- Spyrou, M.A.; Tukhbatova, R.I.; Wang, C.C.; Valtuena, A.A.; Lankapalli, A.K.; Kondrashin, V.V.; Tsybin, V.A.; Khokhlov, A.; Kuhnert, D.; Herbig, A.; et al. Analysis of 3800-year-old Yersinia pestis genomes suggests Bronze Age origin for bubonic plague. Nat. Commun. 2018, 9, 2234. [Google Scholar] [CrossRef]
- Zimbler, D.L.; Schroeder, J.A.; Eddy, J.L.; Lathem, W.W. Early emergence of Yersinia pestis as a severe respiratory pathogen. Nat. Commun. 2015, 6, 7487. [Google Scholar] [CrossRef]
- Dentovskaya, S.V.; Platonov, M.E.; Svetoch, T.E.; Kopylov, P.K.; Kombarova, T.I.; Ivanov, S.A.; Shaikhutdinova, R.Z.; Kolombet, L.V.; Chauhan, S.; Ablamunits, V.G.; et al. Two Isoforms of Yersinia pestis Plasminogen Activator Pla: Intraspecies Distribution, Intrinsic Disorder Propensity, and Contribution to Virulence. PLoS ONE 2016, 11, e0168089. [Google Scholar] [CrossRef]
- Anisimov, A.P.; Lindler, L.E.; Pier, G.B. Intraspecific diversity of Yersinia pestis. Clin. Microbiol. Rev. 2004, 17, 434–464. [Google Scholar] [CrossRef]
- Vernati, G.; Edwards, W.H.; Rocke, T.E.; Little, S.F.; Andrews, G.P. Antigenic profiling of yersinia pestis infection in the Wyoming coyote (Canis latrans). J. Wildl. Dis. 2011, 47, 21–29. [Google Scholar] [CrossRef]
- Benner, G.E.; Andrews, G.P.; Byrne, W.R.; Strachan, S.D.; Sample, A.K.; Heath, D.G.; Friedlander, A.M. Immune response to Yersinia outer proteins and other Yersinia pestis antigens after experimental plague infection in mice. Infect. Immun. 1999, 67, 1922–1928. [Google Scholar] [CrossRef]
- Erova, T.E.; Rosenzweig, J.A.; Sha, J.; Suarez, G.; Sierra, J.C.; Kirtley, M.L.; van Lier, C.J.; Telepnev, M.V.; Motin, V.L.; Chopra, A.K. Evaluation of protective potential of Yersinia pestis outer membrane protein antigens as possible candidates for a new-generation recombinant plague vaccine. Clin. Vaccine Immunol. 2013, 20, 227–238. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Easterbrook, T.J.; Reddin, K.; Robinson, A.; Modi, N. Studies on the immunogenicity of the Pla protein from Yersinia pestis. Contrib. Microbiol. Immunol. 1995, 13, 214–215. [Google Scholar]
- Wang, S.; Heilman, D.; Liu, F.; Giehl, T.; Joshi, S.; Huang, X.; Chou, T.H.; Goguen, J.; Lu, S. A DNA vaccine producing LcrV antigen in oligomers is effective in protecting mice from lethal mucosal challenge of plague. Vaccine 2004, 22, 3348–3357. [Google Scholar] [CrossRef] [PubMed]
- van Lier, C.J.; Tiner, B.L.; Chauhan, S.; Motin, V.L.; Fitts, E.C.; Huante, M.B.; Endsley, J.J.; Ponnusamy, D.; Sha, J.; Chopra, A.K. Further characterization of a highly attenuated Yersinia pestis CO92 mutant deleted for the genes encoding Braun lipoprotein and plasminogen activator protease in murine alveolar and primary human macrophages. Microb. Pathog. 2015, 80, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Agar, S.L.; Sha, J.; Baze, W.B.; Erova, T.E.; Foltz, S.M.; Suarez, G.; Wang, S.; Chopra, A.K. Deletion of Braun lipoprotein gene (lpp) and curing of plasmid pPCP1 dramatically alter the virulence of Yersinia pestis CO92 in a mouse model of pneumonic plague. Microbiology 2009, 155, 3247–3259. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tiner, B.L.; Sha, J.; Cong, Y.; Kirtley, M.L.; Andersson, J.A.; Chopra, A.K. Immunisation of two rodent species with new live-attenuated mutants of Yersinia pestis CO92 induces protective long-term humoral- and cell-mediated immunity against pneumonic plague. NPJ Vaccines 2016, 1, 16020. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Feng, K.; Zheng, X.; Wang, R.; Gao, N.; Fan, D.; Sheng, Z.; Zhou, H.; Chen, H.; An, J. Long-Term Protection Elicited by a DNA Vaccine Candidate Expressing the prM-E Antigen of Dengue Virus Serotype 3 in Mice. Front. Cell Infect. Microbiol. 2020, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Parkhill, J.; Wren, B.W.; Thomson, N.R.; Titball, R.W.; Holden, M.T.; Prentice, M.B.; Sebaihia, M.; James, K.D.; Churcher, C.; Mungall, K.L.; et al. Genome sequence of Yersinia pestis, the causative agent of plague. Nature 2001, 413, 523–527. [Google Scholar] [CrossRef]
- Thomas, R.E.; McDonough, K.A.; Schwan, T.G. Use of DNA hybridizations probes for detection of the plague bacillus (Yersinia pestis) in fleas (Siphonaptera: Pulicidae and Ceratophyllidae). J. Med. Entomol. 1989, 26, 342–348. [Google Scholar] [CrossRef]
- McDonough, K.A.; Schwan, T.G.; Thomas, R.E.; Falkow, S. Identification of a Yersinia pestis-specific DNA probe with potential for use in plague surveillance. J. Clin. Microbiol. 1988, 26, 2515–2519. [Google Scholar] [CrossRef]
- Campbell, J.; Lowe, J.; Walz, S.; Ezzell, J. Rapid and specific identification of Yersinia pestis by using a nested polymerase chain reaction procedure. J. Clin. Microbiol. 1993, 31, 758–759. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, J.; Schwan, T.G. New method for plague surveillance using polymerase chain reaction to detect Yersinia pestis in fleas. J. Clin. Microbiol. 1993, 31, 1511–1514. [Google Scholar] [CrossRef] [PubMed]
- Norkina, O.V.; Kulichenko, A.N.; Gintsburg, A.L.; Tuchkov, I.V.; Popov Yu, A.; Aksenov, M.U.; Drosdov, I.G. Development of a diagnostic test for Yersinia pestis by the polymerase chain reaction. J. Appl. Bacteriol. 1994, 76, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.A.; Ezzell, J.; Hinnebusch, B.J.; Shipley, M.; Henchal, E.A.; Ibrahim, M.S. 5′ nuclease PCR assay to detect Yersinia pestis. J. Clin. Microbiol. 1998, 36, 2284–2288. [Google Scholar] [CrossRef]
- Loiez, C.; Herwegh, S.; Wallet, F.; Armand, S.; Guinet, F.; Courcol, R.J. Detection of Yersinia pestis in sputum by real-time PCR. J. Clin. Microbiol. 2003, 41, 4873–4875. [Google Scholar] [CrossRef]
- Tomaso, H.; Reisinger, E.C.; Al Dahouk, S.; Frangoulidis, D.; Rakin, A.; Landt, O.; Neubauer, H. Rapid detection of Yersinia pestis with multiplex real-time PCR assays using fluorescent hybridisation probes. FEMS Immunol. Med. Microbiol. 2003, 38, 117–126. [Google Scholar] [CrossRef]
- Griffin, K.A.; Martin, D.J.; Rosen, L.E.; Sirochman, M.A.; Walsh, D.P.; Wolfe, L.L.; Miller, M.W. Detection of Yersinia pestis DNA in prairie dog-associated fleas by polymerase chain reaction assay of purified DNA. J. Wildl. Dis. 2010, 46, 636–643. [Google Scholar] [CrossRef]
- Janse, I.; Hamidjaja, R.A.; Bok, J.M.; van Rotterdam, B.J. Reliable detection of Bacillus anthracis, Francisella tularensis and Yersinia pestis by using multiplex qPCR including internal controls for nucleic acid extraction and amplification. BMC Microbiol. 2010, 10, 314. [Google Scholar] [CrossRef]
- Neubauer, H.; Meyer, H.; Prior, J.; Aleksic, S.; Hensel, A.; Splettstosser, W. A combination of different polymerase chain reaction (PCR) assays for the presumptive identification of Yersinia pestis. J. Vet. Med. B Infect. Dis. Vet. Public Health 2000, 47, 573–580. [Google Scholar] [CrossRef]
- Nyirenda, S.S.; Hang Ombe, B.M.; Simulundu, E.; Mulenga, E.; Moonga, L.; Machang, U.R.; Misinzo, G.; Kilonzo, B.S. Molecular epidemiological investigations of plague in Eastern Province of Zambia. BMC Microbiol. 2018, 18, 2. [Google Scholar] [CrossRef]
- Nyirenda, S.S.; Hang′ombe, B.M.; Kilonzo, B.S.; Kabeta, M.N.; Cornellius, M.; Sinkala, Y. Molecular, serological and epidemiological observations after a suspected outbreak of plague in Nyimba, eastern Zambia. Trop. Doct. 2017, 47, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Nyirenda, S.S.; Hang’ombe, B.M.; Mulenga, E.; Kilonzo, B.S. Serological and PCR investigation of Yersinia pestis in potential reservoir hosts from a plague outbreak focus in Zambia. BMC Res. Notes 2017, 10, 345. [Google Scholar] [CrossRef] [PubMed]
- Riehm, J.M.; Tserennorov, D.; Kiefer, D.; Stuermer, I.W.; Tomaso, H.; Zoller, L.; Otgonbaatar, D.; Scholz, H.C. Yersinia pestis in small rodents, Mongolia. Emerg. Infect. Dis. 2011, 17, 1320–1322. [Google Scholar] [CrossRef] [PubMed]
- Skottman, T.; Piiparinen, H.; Hyytiainen, H.; Myllys, V.; Skurnik, M.; Nikkari, S. Simultaneous real-time PCR detection of Bacillus anthracis, Francisella tularensis and Yersinia pestis. Eur. J. Clin. Microbiol. Infect. Dis. 2007, 26, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.; Satterfield, B.; Cohen, M.; O′Neill, K.; Robison, R. A quadruplex real-time PCR assay for the detection of Yersinia pestis and its plasmids. J. Med. Microbiol. 2008, 57, 324–331. [Google Scholar] [CrossRef]
- Tsukano, H.; Itoh, K.; Suzuki, S.; Watanabe, H. Detection and identification of Yersinia pestis by polymerase chain reaction (PCR) using multiplex primers. Microbiol. Immunol. 1996, 40, 773–775. [Google Scholar] [CrossRef]
- Ziwa, M.H.; Matee, M.I.; Kilonzo, B.S.; Hang′ombe, B.M. Evidence of Yersinia pestis DNA in rodents in plague outbreak foci in Mbulu and Karatu Districts, northern Tanzania. Tanzan J. Health Res. 2013, 15, 152–157. [Google Scholar] [CrossRef]
- Drancourt, M.; Aboudharam, G.; Signoli, M.; Dutour, O.; Raoult, D. Detection of 400-year-old Yersinia pestis DNA in human dental pulp: An approach to the diagnosis of ancient septicemia. Proc. Natl. Acad. Sci. USA 1998, 95, 12637–12640. [Google Scholar] [CrossRef]
- Seifert, L.; Harbeck, M.; Thomas, A.; Hoke, N.; Zoller, L.; Wiechmann, I.; Grupe, G.; Scholz, H.C.; Riehm, J.M. Strategy for sensitive and specific detection of Yersinia pestis in skeletons of the black death pandemic. PLoS ONE 2013, 8, e75742. [Google Scholar] [CrossRef]
- Bianucci, R.; Rahalison, L.; Ferroglio, E.; Massa, E.R.; Signoli, M. A rapid diagnostic test for plague detects Yersinia pestis F1 antigen in ancient human remains. C. R. Biol. 2007, 330, 747–754. [Google Scholar] [CrossRef]
- Collins, M.L.; Irvine, B.; Tyner, D.; Fine, E.; Zayati, C.; Chang, C.; Horn, T.; Ahle, D.; Detmer, J.; Shen, L.P.; et al. A branched DNA signal amplification assay for quantification of nucleic acid targets below 100 molecules/mL. Nucleic Acids Res. 1997, 25, 2979–2984. [Google Scholar] [CrossRef] [PubMed]
- Horn, T.; Chang, C.A.; Urdea, M.S. Chemical synthesis and characterization of branched oligodeoxyribonucleotides (bDNA) for use as signal amplifiers in nucleic acid quantification assays. Nucleic Acids Res. 1997, 25, 4842–4849. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Horn, T.; Urdea, M.S. Forks and combs and DNA: The synthesis of branched oligodeoxyribonucleotides. Nucleic Acids Res. 1989, 17, 6959–6967. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.S.; Chambers, J.P.; Brubaker, R.R.; Goode, M.T.; Valdes, J.J. Detection of Yersinia pestis using branched DNA. Mol. Cell. Probes 1999, 13, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Feodorova, V.A.; Devdariani, Z.L. Development, characterisation and diagnostic application of monoclonal antibodies against Yersinia pestis fibrinolysin and coagulase. J. Med. Microbiol. 2000, 49, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Mahesh, S.; Shukla, J.; Tuteja, U.; Batra, H.V. Molecular detection of Yersinia pestis isolates of Indian origin by using Pla specific monoclonal antibodies. Comp. Immunol. Microbiol. Infect. Dis. 2005, 28, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.; Demeure, C.; Lamourette, P.; Filali, S.; Plaisance, M.; Creminon, C.; Volland, H.; Carniel, E. Fast and simple detection of Yersinia pestis applicable to field investigation of plague foci. PLoS ONE 2013, 8, e54947. [Google Scholar] [CrossRef]
- Feodorova, V.A.; Sayapina, L.V.; Corbel, M.J.; Motin, V.L. Russian vaccines against especially dangerous bacterial pathogens. Emerg. Microbes. Infect. 2014, 3, e86. [Google Scholar] [CrossRef]
- Engelthaler, D.M.; Hinnebusch, B.J.; Rittner, C.M.; Gage, K.L. Quantitative competitive PCR as a technique for exploring flea-Yersina pestis dynamics. Am. J. Trop. Med. Hyg. 2000, 62, 552–560. [Google Scholar] [CrossRef]
- Feodorova, V.A.; Lyapina, A.M.; Khizhnyakova, M.A.; Zaitsev, S.S.; Sayapina, L.V.; Arseneva, T.E.; Trukhachev, A.L.; Lebedeva, S.A.; Telepnev, M.V.; Ulianova, O.V.; et al. Humoral and cellular immune responses to Yersinia pestis Pla antigen in humans immunized with live plague vaccine. PLoS Negl. Trop. Dis. 2018, 12, e0006511. [Google Scholar] [CrossRef]
- Janse, I.; Hamidjaja, R.A.; Reusken, C. Yersinia pestis plasminogen activator gene homolog in rat tissues. Emerg. Infect. Dis. 2013, 19, 342–344. [Google Scholar] [CrossRef] [PubMed]
- Hansch, S.; Cilli, E.; Catalano, G.; Gruppioni, G.; Bianucci, R.; Stenseth, N.C.; Bramanti, B.; Pallen, M.J. The pla gene, encoding plasminogen activator, is not specific to Yersinia pestis. BMC Res. Notes 2015, 8, 535. [Google Scholar] [CrossRef] [PubMed]
- Armougom, F.; Bitam, I.; Croce, O.; Merhej, V.; Barassi, L.; Nguyen, T.T.; La Scola, B.; Raoult, D. Genomic Insights into a New Citrobacter koseri Strain Revealed Gene Exchanges with the Virulence-Associated Yersinia pestis pPCP1 Plasmid. Front. Microbiol. 2016, 7, 340. [Google Scholar] [CrossRef] [PubMed]
- Woron, A.M.; Nazarian, E.J.; Egan, C.; McDonough, K.A.; Cirino, N.M.; Limberger, R.J.; Musser, K.A. Development and evaluation of a 4-target multiplex real-time polymerase chain reaction assay for the detection and characterization of Yersinia pestis. Diagn. Microbiol. Infect. Dis. 2006, 56, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Matero, P.; Pasanen, T.; Laukkanen, R.; Tissari, P.; Tarkka, E.; Vaara, M.; Skurnik, M. Real-time multiplex PCR assay for detection of Yersinia pestis and Yersinia pseudotuberculosis. APMIS 2009, 117, 34–44. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrates | Biological Function/Process | Hydrolyzable Amino Acids of Substrate | Consequence | Proven or Not Substrate In Vivo | Contribution to Virulence/Pathogenesis | References |
|---|---|---|---|---|---|---|
| Host proteins processed by Pla | ||||||
| Peroxiredoxin 6 (Prdx6) | Immune system process; ROS metabolic process | Cleaves at sites Lis173/Arg174, Lys201/Leu202, and the undefined site located in the C-terminal region | Disrupt peroxidase and phospholipase A2 activities | yes | The cleavage of Prdx6 has a little detectable impact on the progression or outcome of pneumonic plague | [66] |
| Alpha2-antiplasmin (A2AP) | proteolysis; contributes to control of the pulmonary inflammatory response to infection by reducing neutrophil recruitment and cytokine production | ND | uncontrolled production of active plasmin and resulting clearance of fibrin depositions | no | A2AP is not significantly affected by the Pla protease during pneumonic plague; A2AP participating in immune modulation in the lungs has a limited impact on the course or ultimate outcome of the infection | [65,66,82] |
| Plasminogen activator inhibitor-1 (PAI-1) | inhibition of activation of plasminogen. | cleaves between residues R346 and M347 | prevent inhibition of tPA and uPA | yes | PAI-1 deficiency results in a decreased level of neutrophil influx to the pulmonary compartment during pneumonia. This leads to increased bacterial out-growth, enhanced dissemination, and decreased survival of infected mice | [66,82] |
| Urokinase plasminogen activator (uPA) | activation of plasminogen | cleaves the single-chain uPA (scuPA) between residues Lys158 and Ile159 | cleavage led to the activation of scuPA | no | activates fibrinolysis, cell migration, and tissue remodeling | [61,66] |
| Complement component C3 | cytokine activity; complement activation | ND 1 | ND | no | cleavage of C3 disrupts chemotaxis of inflammatory cells to foci of infection, leads to disturbances in their phagocytic activity and the inability of the complement system to form the cytolytic end product of the complement system activation, membrane attack complex | [66,82] |
| Apoptotic molecule Fas ligand (FasL) | immune system process; its binding with its receptor induces apoptosis | cleaves at multiple sites located within the extracellular domain of FasL | ND | no | contribute to the progression of pneumonic plague | [66] |
| Glutathione S-transferase A3 | immune system process; ROS metabolic process | ND | ND | no | ND | [66] |
| Glutathione peroxidase 3 | immune system process; response to toxin | ND | ND | no | ND | [66] |
| Tubulin polymerization-promoting protein family | cell component; structure | ND | ND | no | ND | [66] |
| Pigment epithelium-derived factor | protein binding; proteolysis | ND | ND | no | ND | [66] |
| Alpha-2-HS-glycoprotein | protein binding; immune system process; proteolysis | ND | ND | no | ND | [66] |
| Glutathione S-transferase Mu 1 | immune system process; transferase activity | ND | ND | no | ND | [66] |
| BPI fold-containing family A member 1 (sPlunc) | immune system response | ND | ND | no | ND | [66] |
| Carboxypeptidase N subunit 2 | immune system process; cytokine-mediated signaling | ND | ND | no | ND | [66] |
| Sulfated glycoprotein 1 | protein binding; lipid transport | ND | ND | no | ND | [66] |
| BPI fold-containing family b member 1 (Lplunc1) | MAC activation, response to stress | ND | ND | no | ND | [66] |
| Vinculin | actin binding; cell adhesion | ND | ND | no | ND | [66] |
| Plasminogen | serine-type peptidase activity; proteolysis | cleavage at a single site between residues Arg561 and Val562 of the proenzyme | activates plasminogen through cleavage this zymogen at a single site | no | activates fibrinolysis | [66,82] |
| Actin gamma | cell component; structure | ND | ND | no | ND | [66] |
| Plastin-2 | structure; actin binding | ND | ND | no | ND | [66] |
| Lipoprotein lipase | lipase activity; lipid transport | ND | ND | no | ND | [66] |
| Phosphoglycerate mutase 1 | glycolysis | ND | ND | no | ND | [66] |
| Complement C4-B | complement activation; signal transduction | ND | ND | no | ND | [66] |
| Hypoxanthine-guanine phosphoribosyltransferase | monosaccharide metabolic process | ND | ND | no | ND | [66] |
| Calmodulin | Ca2+ binding | ND | ND | no | ND | [66] |
| Apolipoprotein A-IV | lipid transporter activity; blood circulation | ND | ND | no | ND | [66] |
| Thrombin-activatable fibrinolysis inhibitor (TAFI) | antifibrinolytic plasma protein | ND | Pla can cleave TAFI near its C-terminus, preventing activation to TAFIa during subsequent incubation with thrombin–thrombomodulin; in addition to the direct inactivation of TAFI by Pla, TAFIa can also be inactivated through proteolysis by plasmin | yes | ND | [82] |
| Tissue factor pathway inhibitor (TFPI) | TFPI is an anticoagulant protein that reversibly binds to coagulation factor Xa (FXa). This bimolecular TFPI–FXa complex is a potent inhibitor of the procoagulant complex TF:FVIIa (the primary initiator of coagulation in vivo), which acts to block further coagulation at this point in the cascade | Cleavage of TFPI by Pla occurs between residues K249 and G250 | cleavage by Pla is predicted to have procoagulant consequences; Pla disrupts the TFPI-mediated inhibition of clot formation | no | TFPI inactivation enhances coagulation | [82] |
| Cathelicidins | cationic antimicrobial peptides (CAMPs) | CAMPs permeabilize bacterial lipid bilayers, resulting in the lysis of affected cells; Pla inhibit CAMPs chemoattractant properties that recruit neutrophils, monocytes, and T cells in response to infection | no | ND | [82] | |
| α-2-macroglobuline | impede the plasmin activity | ND | ND | no | activates proteolysis | [28] |
| Y. pestis proteins processed by Pla | ||||||
| Type-III secretion system effectors | inhibit phagocytosis, induce apoptosis of macrophages, destroy actin cytoskeleton and signaling pathway of activation of inflammatory cells, suppress production of cytokines and chemokines | ND | degrades most Yops in vitro including the Yops B, C, D, E, F, H, J, and M, but is unable to degrade LcrV | no | it is supposed that Pla coordinates the degradation of extracellular Yops that may otherwise compromise innate immunity evasion | [14,82] |
| YapA | autotransporter | processes at multiple sites (Lys512, Lys548/Lys549, Lys594/Lys595, Lys558, and Lys604) | cleavage at the C terminus released the protein from the cell surface | no | it is supposed that YapA might be an adhesin | [83] |
| YapG | autotransporter | processes at multiple sites | ND | no | does not contribute to Y. pestis virulence in established mouse models of bubonic and pneumonic infection | [81] |
| YapE | autotransporter | processes at two sites (Lys232 and Lys338 but preferentially at Lys232) | cleavage is required to proteolytical activation of the protein | no | contributes to disease in the mouse model of bubonic plague by mediating bacterial aggregation and adherence to eukaryotic cells | [84] |
| KatY | catalase-peroxidase | Cleavage of α-KatY (78.8 kDa) by Pla resulted in its smaller forms, β-KatY (∼50 kDa), γ-KatY (∼36 kDa) and δ-KatY (∼34 kDa) | ND | no | ND | [85] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sebbane, F.; Uversky, V.N.; Anisimov, A.P. Yersinia pestis Plasminogen Activator. Biomolecules 2020, 10, 1554. https://doi.org/10.3390/biom10111554
Sebbane F, Uversky VN, Anisimov AP. Yersinia pestis Plasminogen Activator. Biomolecules. 2020; 10(11):1554. https://doi.org/10.3390/biom10111554
Chicago/Turabian StyleSebbane, Florent, Vladimir N. Uversky, and Andrey P. Anisimov. 2020. "Yersinia pestis Plasminogen Activator" Biomolecules 10, no. 11: 1554. https://doi.org/10.3390/biom10111554
APA StyleSebbane, F., Uversky, V. N., & Anisimov, A. P. (2020). Yersinia pestis Plasminogen Activator. Biomolecules, 10(11), 1554. https://doi.org/10.3390/biom10111554