Abstract
Dissociative electron attachment has shown site selectivity in aliphatic molecules based on the functional groups present in them. This selectivity arises from the core excited resonances that have excited parent states localized to a specific site of the functional group. Here, we show that such site selectivity is also observed in the amine group when present in aromatic molecules. However, the proximity of the aromatic ring to the functional group under investigation has a substantial effect on the dissociation dynamics. This effect is evident in the momentum distribution of the hydride ions generated from the amine group. Our results unravel the hitherto unknown facets of the site selectivity in aromatic organic molecules.
1. Introduction
Low-energy electron-induced chemistry plays a vital role in many phenomena, ranging from radiation biology to astrochemistry. Dissociative electron attachment (DEA) is a dominant channel in these processes where the electron energy is intrinsically translated to the nuclear motion via the dynamics of the negative ion resonance state (NIRS). The most fascinating aspect of DEA is the site selectivity shown in the dissociation process, as manifested in the hydride ion formation [1]. This site selectivity directly correlates with the functional group present at the site. Hence, it has been seen even in larger molecules such as DNA bases [2]. This site selectivity is beyond the conventional threshold energy-based selectivity shown in the dissociation processes [3]. The NIRSs that participate in the electron attachment and give site selectivity are core excited resonances. The underlying parent excited neutral states for these NIRSs show the localization of the excited molecular orbital [4]. The excitation of non-bonding electrons from a specific site to the anti-bonding molecular orbital with the capture of the incoming electron to the same or other orbital results in the localization of the dissociation process. For example, hydride ion formation in ammonia peaks at 5.5 eV electron energy. This peak arises from a core excited Feshbach resonance. The underlying parent neutral excited state is the first triplet excited state of ammonia known to dissociate along with the bond [5]. This excited state is formed by the excitation of the lone pair of electrons from the N atom to the Rydberg type orbital . This localized excited state provides the site selectivity to the DEA process. The localization due to the excitation of the lone pair of electrons in ammonia remains almost unaffected in simple saturated aliphatic amines such as n-propyl amine [4,6]. However, as in the unsaturated amines and particularly in the aromatic amines, the lone pair of electrons from the N-site may get easily influenced by the delocalized π electron cloud of the phenyl group, it would be interesting to see its effect on the site selectivity in the DEA process.
In DEA to various amines, the systematic difference between the saturated and unsaturated amines has been seen in terms of the ion yields and their peaks [7], which indicates the presence of low-lying shape resonance at lower electron energy in the channel around 2 eV. The 5.5 eV peak in the channel, which has been seen in ammonia and n-propyl amine (at 5.7 eV), is also consistently seen in the channel in the saturated amines with varying energy positions [7]. However, this peak loses its intensity substantially in the aromatic or unsaturated amines, and the low-energy shape resonance dominates the DEA channel. The presence of this resonance is understood to be due to π and π* orbitals in the unsaturated amines, which influence the Rydberg-type orbitals. With this observation in mind, we have investigated the DEA dynamics of the channel in the aromatic molecules aniline and benzylamine using the velocity slice imaging technique.
In aniline, the amine group is directly attached to the phenyl group, making it an unsaturated amine, whereas, in benzylamine, the amine group is attached to the benzyl group, making it another saturated amine. However, the benzylamine also contains the aromatic ring, giving a test bed to explore its influence on the site selectivity of DEA and its underlying dynamics.
The aromatic amines are also important from a biological point of view. Aromatic amines are present in many biological molecules, including DNA bases adenine, guanine, and cytosine, and, as mentioned earlier, it is well-recognized that the reactions induced by low-energy electrons in living cells represent an important step toward radiation damage [8,9]. Understanding the DEA dynamics in these molecules will also serve as a stepping stone to studying the DEA in higher aromatic compounds and complex biomolecules. We also present the absolute cross-sections for the DEA process in these two molecules as it is closely related to the dynamics of the process and is of importance in theoretical modeling as well as for possible use in practical applications.
2. Experimental Setup
We measured the kinetic energy and angular distribution of hydride anions formed in DEA to aniline and benzylamine by the velocity slice imaging (VSI) technique. Although the experimental setup for these measurements is described earlier, here we give its brief description [10]. We performed the experiments by crossing the magnetically collimated pulsed electron beam at right angles with the effusive molecular beam generated using a capillary array. The molecular beam coincided with the axis of the VSI spectrometer. The electron current was measured using a Faraday cup placed against the electron gun across the interaction region of the spectrometer. The pulsed electrostatic field on the pusher electrode extracted the ions. A four-element electrostatic lens with the appropriate voltage condition velocity focused the ions onto the position-sensitive detector made of a pair of microchannel plates (MCPs) of 75 mm in diameter in the chevron geometry, followed by a phosphor screen. We used a CCD camera to capture the illuminated spots on the phosphor screen due to the ion hit on the detector. While determining the ion yield as a function of the incoming electron energy, we used the spectrometer in the mass-spectrometer mode. In this mode, we applied DC electric potentials to the MCPs and the phosphor screen. We counted the ions by detecting the electrical signal pulses on the MCP back electrode. The voltage pulses were amplified and fed to a multiple-event time digitizer to obtain the time-of-flight mass spectra. Using these mass spectra as a function of electron energy, we determined the relative cross-sections for different DEA channels. We used the relative flow technique to put these cross-sections on the absolute scale using the mass spectra obtained for from molecular oxygen. To obtain accurate absolute cross-sections, we ensured the complete collection of all the ions onto the detector (as confirmed by the VSI data) and the elimination of the bias voltage dependence on the detection efficiency as a function of mass-to-charge ratios. We used the arrival time of ions of given m/e to determine the detector pulsing delay in the imaging mode. In this mode, we pulsed the MCP back voltage at an appropriate time delay to capture the signal from the ions of interest. We recorded the images on the CCD camera and analyzed the ion hit distribution in the offline mode using a homebuilt Matlab-based data analysis program. We used 80 ns pulses on the detector to obtain the slice images.
For creating the molecular beam, we used aniline (purity > 99.5%) and benzylamine (purity > 99.5%) from Sigma-Aldrich. The liquid sample was stored in an evacuated glass bulb connected to the vacuum chamber via a glass-to-metal seal using a ¼-inch stainless steel tube through a leak valve (make: Granville Phillips). To eliminate any dissolved gases, including water vapor, the sample liquid in the bulb was pumped by a rotary pump until its volume was reduced to 1/3rd before using it as the target source. The sample vapor beam was introduced in the chamber in two ways: (i) through the capillary array (we term this as a crossed-beam measurement) and (ii) directly into the chamber (we term this as a static gas background measurement). The overall chamber pressure rises when the target vapor is flown through the capillary array. As the electron beam passes through this raised background gas, it contributes to the measured mass spectrum. The static gas background measurements are carried out to subtract these background counts from the crossed beam measurements. The two spectra are subtracted on proper current and pressure normalization. The details of this technique to measure the absolute cross-section are given in ref. [4].
3. Results and Discussions
3.1. Absolute Cross-Sections
The earlier measurements of the negative ion formation in aniline by resonant electron capture by Pikhtovnikov et al. show ion signal peaking at 2.6, 5.3, 6.3, and 8.3 eV; ion signal peaking at 5.3, 7, 9.2, and 10.6 eV; ion signal peaking at 6.7 eV; peaking at 6 eV; peaking at 9.7 eV; peaking at 7.1 eV; and ion signal peaking at 9.2 eV [11]. They found the at 8.3 eV, , and as the most dominant channels. However, DEA to benzylamine has not been reported in the literature so far.
We found and as the most dominant channels in our measurements of DEA to aniline and benzylamine. The absolute cross-sections as a function of electron energy for these channels from both the molecules are shown in Figure 1.
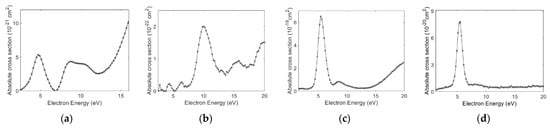
Figure 1.
DEA absolute cross-sections measured for (a) and (b) channels from aniline and (c) and (d) channels from benzylamine as a function of electron energy.
As the figure shows, both the molecules show as the most dominant channel. However, the cross-section for this channel from aniline is two orders of magnitude lower than that for benzylamine. This observation is consistent with the previous measurements of the channel from the saturated and unsaturated amines [7]. Interestingly, the channel also shows a similar trend. The cross-section shows three peaks at 5, 8.7, and 9.9 eV, whereas in the benzylamine, the cross-section peaks at 5.5 and 8.4 eV. The channel peaks at 10 and 5.5 eV, respectively. For benzylamine, the absolute cross-section for also peaks at 5.5 eV, whereas this channel is too weak in aniline. The measured absolute cross-section values for the DEA channels from both the molecules are given in Table 1.

Table 1.
Absolute cross-sections for the dominant channels observed in DEA to aniline and benzylamine. The absolute cross-sections for ammonia and n-propylamine are also given. In all these values, the typical uncertainty is about 15%.
The absolute cross-section values obtained for the benzylamine and aniline follow the trend observed in an earlier work on the saturated and unsaturated amines [7]. We have determined the origin of the hydride ions at various observed peaks by measuring the ion yield curves for the and ions from partially deuterated aniline (C6D5NH2). The results are shown in Figure 2.
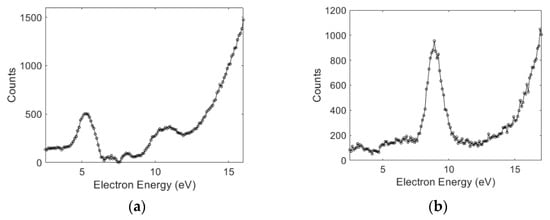
Figure 2.
The ion yield curves as a function of electron energy for (a) and (b) ions from DEA to partially deuterated aniline (C6D5NH2).
As shown in Figure 2a, the 5 and 9.9 eV peaks can be attributed to the site in the molecule. The 8.7 eV peak can be attributed to the phenyl part of the molecule (Figure 2b). This clearly shows the site selectivity observed in the aromatic amine. Interestingly, the presence of the phenyl group does not substantially alter the DEA peak from the site compared to that from ammonia. With the observed analogy with the amine group in the aliphatic compounds, we attribute the 5.5 eV peak in benzylamine to the amine site and that at the 8.4 eV to the phenyl group.
Photodissociation measurements by King et al. [13] on aniline revealed that the second excited singlet electronic state of aniline corresponds to a π → 3s Rydberg electronic transition, calculated to lie at 4.53 eV. They anticipated that the 3s Rydberg electron density in this latter excited state evolves into an σ* character upon the extension of one of the bonds, which shows a conical intersection with the ground state at the stretched N–H bond. It contributes to the direct dissociation of this bond, i.e., the second singlet excited state of aniline is expected to be essentially repulsive and of 1 π σ* character. Hence, this state may act as the parent state for the anion state of aniline, contributing to the channel that peaks at 5 eV. The peak in the hydride ions from benzylamine was at 5.5 eV, which is closer to that obtained in ammonia (5.5 eV) and n-propylamine (5.7 eV).
The first ionization energy of aniline is at 8 eV, and the second is at 9.1 eV. Since the comes at 8.9 eV at the second resonance, we conclude that the excitation of electrons from HOMO may not play any role in this resonance. The HOMO-1 has a contribution largely from the delocalized electrons from the phenyl ring. The 8.9 eV resonance, as observed in aniline and its deuterated form, may arise from the excitation of this HOMO-1 orbital. In an attempt to find the parent state of this resonance, we compared the DEA with the VUV absorption [14] and electron energy loss spectra [15] reported earlier. From these results, we note that the n → 3p Rydberg transition at about 8.5 eV may be the parent state of the DEA resonance at 8.7 eV. Interestingly, DEA to benzene also shows a peak in the channel around 8 eV [16]. Benzylamine also shows a peak at 8.4 eV. We may conclude that the molecule’s phenyl part may play a significant role in this resonance. Benzylamine also shows a very weak shoulder near 10 eV. Based on the comparison with the DEA studies on ammonia and methane, we propose the origin of this shoulder to be from the site, like in ammonia, or from the site of CH2, like in methane.
To probe the effect of the delocalized electrons on the site selectivity of DEA and the underlying dynamics, we performed the angular and kinetic energy distribution measurements of the ions formed by DEA to both the molecules using the VSI technique.
3.2. Angular and Kinetic Energy Distributions
We measured the VSI images of the and ion momentum distribution at various peaks observed in the cross-section measurements of both the molecules. We discuss the details of the kinetic energy and angular distributions obtained from these measurements below.
Ions
The VSIs obtained for the ions near the first peak, i.e., at 4.7 eV in aniline and 5.5 eV in benzylamine, are shown in Figure 3, along with the angular distributions obtained from the images. The VSI obtained for ions from benzylamine shows considerable similarity with that obtained from ammonia [17] and from n-propylamine [6]. However, the image obtained for aniline shows some difference. The corresponding angular distributions are shown in Figure 3c,d. As discussed earlier, the dynamics described for the photodissociation of aniline at 4.53 eV have the origin in the π → 3s Rydberg electronic transition, which further passes on to the σ* orbital. It results in the cleavage of the bond. As the N atom is present next to the phenyl ring, the delocalized π electrons would influence its lone pair of electrons. This, it appears, is not only affecting the cross-section of the hydride ion formation channel through DEA but also the angular distribution.
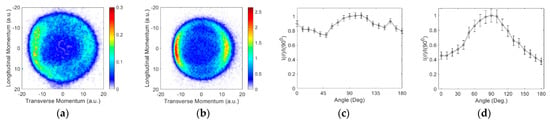
Figure 3.
Momentum images obtained for the ions from (a) aniline at 4.7 eV and (b) benzylamine at 5.5 eV. The incoming electron beam direction is from top to bottom; (c,d) are the respective angular distributions.
The angular distribution obtained for aniline in this channel is considerably flatter, indicating internal dynamics involved in the dissociation process. The dynamics might be in the form of changes in the bond lengths and/or angles to circumvent the barrier present in the direct dissociation path. In comparison, we find that the angular distribution for the ions from benzylamine resembles that for ammonia. The cross-section is also substantially higher than aniline and compares well with n-propylamine [4]. We have also determined the kinetic energy distribution of the ions, as shown in Figure 4.
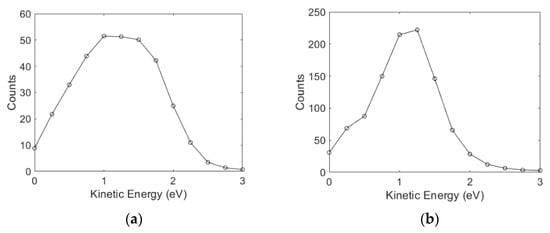
Figure 4.
Kinetic energy distribution of the ions obtained from (a) aniline at 4.7 eV and (b) benzylamine at 5.5 eV electron energy.
The kinetic energy distribution for ions from aniline peaks at 1.25 eV and extends up to 2 eV. The heat of formation of the aniline radical (C6H5NH) can be estimated using its electron affinity (1.61 eV) and the heat to deprotonation reaction of the aniline molecule (1540 kJ/mol) [18]. Based on the heat of formation for H (218 kJ/mol), C6H5NH2 (87 kJ/mol), and C6H5NH (252 kJ/mol) and the electron affinity of H (0.75 eV), we estimate the threshold for the ion formation from the site via two body fragmentation as 3.2 eV. Based on the bond dissociation energy in aniline as 386.2 kJ/mol, the other estimate of the threshold for this channel is 3.25 eV [19]. At the 4.7 eV electron energy, the excess energy in the system would be about 1.5 eV (1.45 eV), out of which the maximum of 1.48 eV (1.43 eV) would appear as the kinetic energy of ion. As most of the excess energy appears as the kinetic energy of the hydride ion, about 0.3 eV energy appears in the internal excitation of the aniline radical. However, the images obtained across the peak do not show a substantial increase in the kinetic energy of the hydride ion, indicating that the excess energy goes as the internal energy of the aniline radical. This is an important feature indicating that there might be a barrier to the direct dissociation channel, and the system undergoes dissociation with a considerable reshaping of the molecule.
For benzylamine, the bond dissociation energy of is 418.3 kJ/mol, which makes the threshold for the ion formation from the site 3.58 eV [20]. Hence, at 5.5 eV electron energy, the fragments would carry a maximum of 1.92 eV energy, and that would translate to the maximum of 1.9 eV as the kinetic energy of the ion. The observed kinetic energy distribution at 5.5 eV peaks around 1.25 eV and extends up to 2 eV. This also clearly indicates the two-body dissociation.
The VSI and the kinetic energy distribution obtained at the second peak in the cross-section for both molecules are shown in Figure 5. The VSIs for both the molecules show a relatively big blob with a considerable intensity throughout. For both the molecules, the kinetic energy distribution obtained is spread over a similar energy range and is flatter for benzylamine than for aniline.
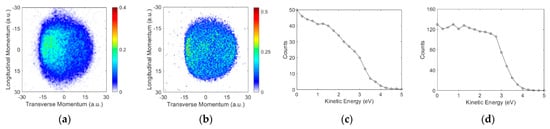
Figure 5.
Momentum images obtained for the ions from (a) aniline at 8.9 eV and (b) benzylamine at 8.4 eV. The incoming electron beam direction is from top to bottom; (c,d) are the respective kinetic energy distributions.
In the absence of the bond dissociation energy for the phenyl part of both compounds, we consider the upper limit for this to be the same as for benzene. The bond dissociation energy in benzene is 474 kJ/mol, making the threshold for this channel 4.2 eV. For benzylamine, the bond dissociation energy of the from the CH2 part is 368 kJ/mol [20]. This makes the threshold for the ion formation from this site 3.06 eV.
Based on these thresholds and the kinetic energy distributions observed in the channel from the site, we conclude that the internal excitation of the molecular neutral fragment is in the range of 2.5 to 4.5 eV, which is sufficient to make this radical break further. The blob observed in the VSI and the overall energetics point to a possible many-body break-up. Moreover, considering the amount of excess energy in the internal excitation, we cannot rule out the ring-breaking dynamics as well. This conclusion is in accordance with the aliphatic compounds where the bond breaks have been associated with the many-body break-up mechanism [21].
Figure 6 shows the VSI obtained for the ion from both molecules. However, although both the molecules show this channel peaking at substantially different electron energies, the momentum images are in the form of a blob. For aniline, the formation is necessarily associated with the ring-breaking many-body dissociation process, whereas for benzylamine, it can arise from the CH2NH2, which is still a many-body break-up mechanism. However, the difference in the resonance energies is indicative of the difference in the origin of this channel. The kinetic energy of this ion in aniline extends up to 0.7 eV peaking at zero. The energy distribution from the benzylamine extends only up to 0.25 eV with a peak at zero. Interestingly, however, the cross-section of this channel in the two molecules differs by more than an order of magnitude.
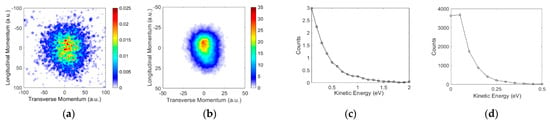
Figure 6.
Momentum images obtained for the ions from (a) aniline at 9.6 eV and (b) benzylamine at 5.5 eV. The incoming electron beam direction is from top to bottom; (c,d) are the respective kinetic energy distributions.
4. Conclusions
To conclude, we have shown that the functional group-dependent site-selective fragmentation observed in aliphatic compounds is also observed in aromatic compounds. The original understanding of the site selectivity due to the excitation of the localized electron cloud from the lone pair of electrons holds for the saturated amine where the dissociation dynamics observed in the ammonia are replicated. However, in the unsaturated amine, the delocalized π electron cloud plays a role in the DEA dynamics. The ion signal from the site peaks around 5 eV. This channel results from the two-body dissociation dynamics. The earlier reasoning for the lower absolute cross-section due to the presence of π electron interaction seems to be reasonable as we have observed an order of magnitude difference in the DEA cross-section between aniline and benzylamine. The ion signal from the site peaks between 8 and 9 eV. Like in aliphatic molecules, in aromatic molecules as well, this channel is associated with the many-body fragmentation dynamics. The channel, which must result from the many-body dissociation, shows different kinetic energy ranges, possibly due to the difference in its origin in the two channels. It would be interesting to compare the dissociation dynamics of these molecules with benzene and other molecules such as pyridine and pyrrol, where the N atom is part of the aromatic ring.
Author Contributions
Conceptualization, V.S.P. and E.K.; data acquisition, V.T., E.K., S.G. and S.D.; analysis, V.T., S.D. and V.S.P.; interpretation, V.S.P.; writing—original draft preparation, V.S.P.; writing—review and editing, V.S.P. and E.K. All authors have read and agreed to the published version of the manuscript.
Funding
Dept. of Atomic Energy, India, under Project Identification No. RTI4002.
Acknowledgments
E.K. acknowledges the Raja Ramanna Fellowship from the Dept. of Atomic Energy, India.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Prabhudesai, V.S.; Kelkar, A.H.; Nandi, D.; Krishnakumar, E. Functional Group Dependent Site Specific Fragmentation of Molecules by Low Energy Electrons. Phys. Rev. Lett. 2005, 95, 143202. [Google Scholar] [CrossRef]
- Ptasińska, S.; Denifl, S.; Grill, V.; Märk, T.D.; Illenberger, E.; Scheier, P. Bond- and Site-Selective Loss of H− from Pyrimidine Bases. Phys. Rev. Lett. 2005, 95, 093201. [Google Scholar] [CrossRef] [PubMed]
- Oster, T.; Kuhn, A.; Illenberger, E. Gas phase negative ion chemistry. Int. J. Mass Spectrom. Ion Processes 1989, 89, 1–72. [Google Scholar] [CrossRef]
- Prabhudesai, V.S.; Nandi, D.; Kelkar, A.H.; Krishnakumar, E. Functional group dependent dissociative electron attachment to simple organic molecules. J. Chem. Phys. 2008, 128, 154309. [Google Scholar] [CrossRef] [PubMed]
- Rescigno, T.N.; Trevisan, C.S.; Orel, A.E.; Slaughter, D.S.; Adaniya, H.; Belkacem, A.; Weyland, M.; Dorn, A.; McCurdy, C.W. Dynamics of dissociative electron attachment to ammonia. Phys. Rev. A 2016, 93, 052704. [Google Scholar] [CrossRef]
- Ram, N.B.; Krishnakumar, E. Dissociative Electron Attachment to Polyatomic Molecules—V: Formic Acid and Propyl Amine. arXiv 2010, arXiv:1007.5169. [Google Scholar]
- Skalicky, T.; Allan, M. The assignment of dissociative electron attachment bands in compounds containing hydroxyl and amino groups. J. Phys. B At. Mol. Opt. Phys. 2004, 37, 4849–4859. [Google Scholar] [CrossRef][Green Version]
- Sanche, L. Nanoscopic aspects of radiobiological damage: Fragmentation induced by secondary low-energy electrons. Mass Spectrom. Rev. 2002, 21, 349–369. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, S.; Illenberger, E. Probing biomolecules: Gas phase experiments and biological relevance. Europhys. News 2002, 33, 207–209. [Google Scholar] [CrossRef][Green Version]
- Gope, K.; Prabhudesai, V.S.; Mason, N.J.; Krishnakumar, E. Dissociation dynamics of transient anion formed via electron attachment to sulfur dioxide. J. Chem. Phys. 2017, 147, 054304. [Google Scholar] [CrossRef]
- Pikhtovnikov, S.V.; Mavrodiev, V.K.; Furley, I.I.; Gataullin, R.R.; Abdrakhmanov, I.B. Resonance electron capture by aniline molecules and its derivatives. High Energy Chem. 2006, 40, 224–229. [Google Scholar] [CrossRef]
- Rawat, P.; Prabhudesai, V.S.; Rahaman, M.A.; Ram, N.B.; Krishnakumar, E. Absolute cross sections for dissociative electron attachment to NH3 and CH4. Int. J. Mass Spectrom. 2008, 277, 96–102. [Google Scholar] [CrossRef]
- King, G.A.; Oliver, T.A.A.; Ashfold, M.N.R. Dynamical insights into 1πσ∗ state mediated photodissociation of aniline. J. Chem. Phys. 2010, 132, 214307. [Google Scholar] [CrossRef] [PubMed]
- Rajasekhar, B.N.; Veeraiah, A.; Sunanda, K.; Jagatap, B.N. Excited states of aniline by photoabsorption spectroscopy in the 30,000–90,000 cm−1 region using synchrotron radiation. J. Chem. Phys. 2013, 139, 064303. [Google Scholar] [CrossRef] [PubMed]
- Ari, T.; Guven, H.; Ecevit, N. Electron energy-loss spectroscopy in monosubstituted benzenes. J. Electron Spectrosc. Relat. Phenom. 1995, 73, 13–23. [Google Scholar] [CrossRef]
- Fenzlaff, H.-P.; Illenberger, E. Low energy electron impact on benzene and the fluorobenzenes: Formation and dissociation of negative ions. Int. J. Mass Spectrom. Ion Processes 1984, 59, 185–202. [Google Scholar] [CrossRef]
- Ram, N.B.; Krishnakumar, E. Dissociative electron attachment resonances in ammonia: A velocity slice imaging based study. J. Chem. Phys. 2012, 136, 164308. [Google Scholar] [CrossRef] [PubMed]
- NIST Chemistry Webbook. Available online: https://webbook.nist.gov/chemistry/ (accessed on 1 September 2022).
- Bordwell, F.G.; Zhang, X.M.; Cheng, J.P. Bond dissociation energies of the nitrogen-hydrogen bonds in anilines and in the corresponding radical anions. Equilibrium acidities of aniline radical cations. J. Org. Chem. 1993, 58, 6410–6416. [Google Scholar] [CrossRef]
- Luo, Y.R. Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Ram, N.B.; Prabhudesai, V.S.; Krishnakumar, E. Velocity imaging of H− from formic acid: Probing functional group dependence in dissociative electron attachment. Eur. Phys. J. D 2020, 74, 49. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).