
Identifying the Tautomeric Form of a Deoxyguanosine-Estrogen Quinone Intermediate

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
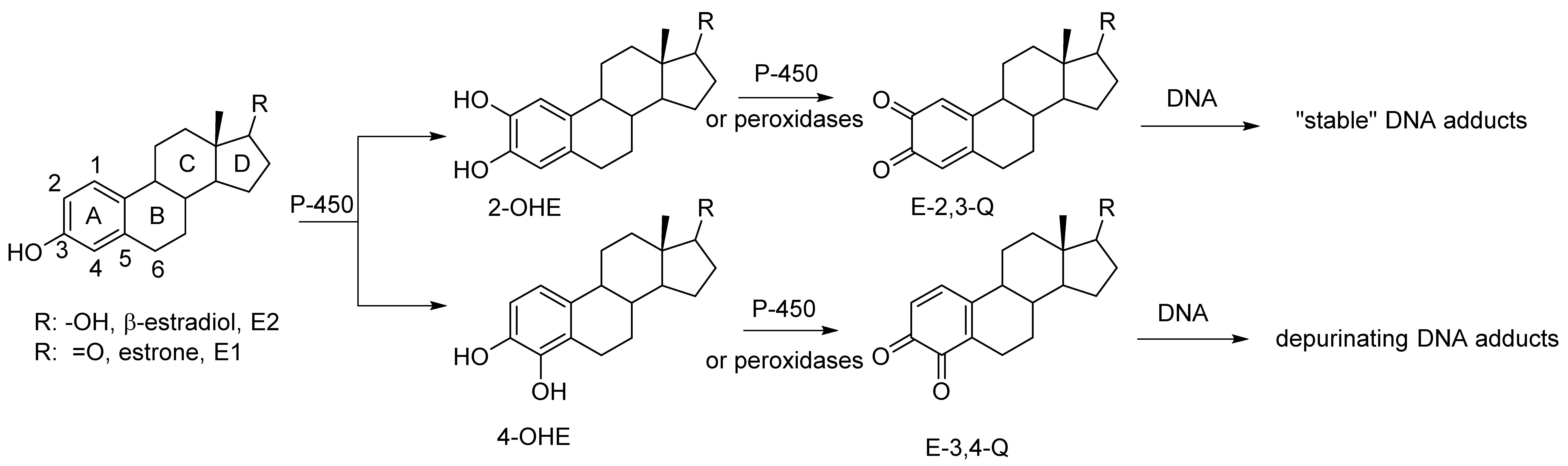
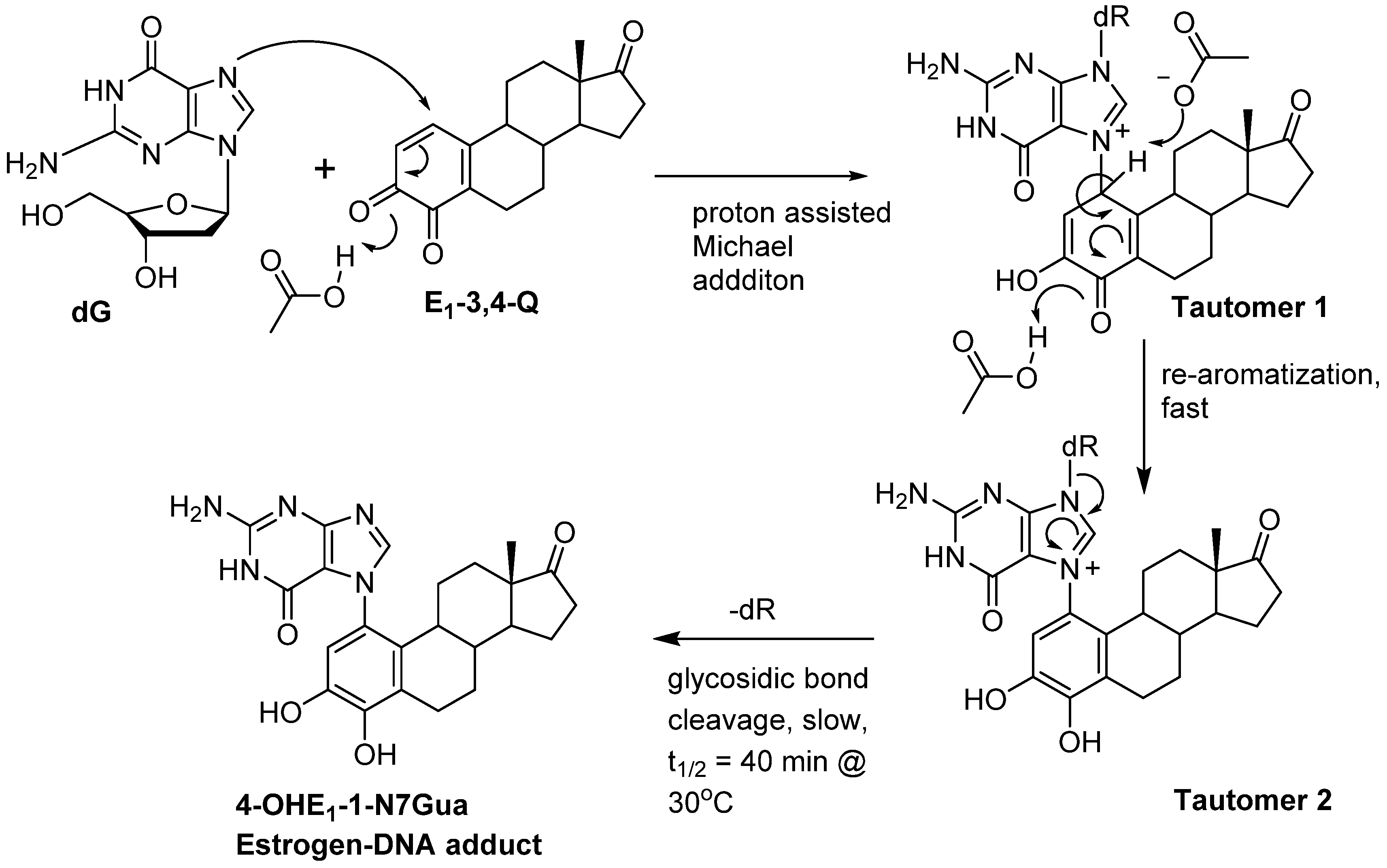
1. Introduction
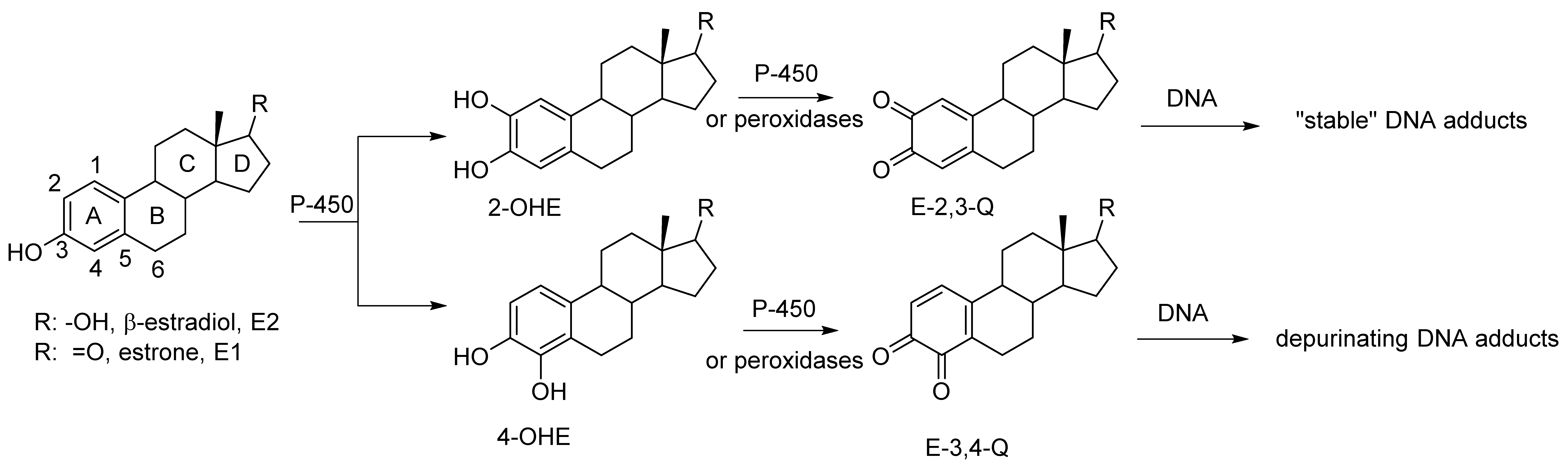
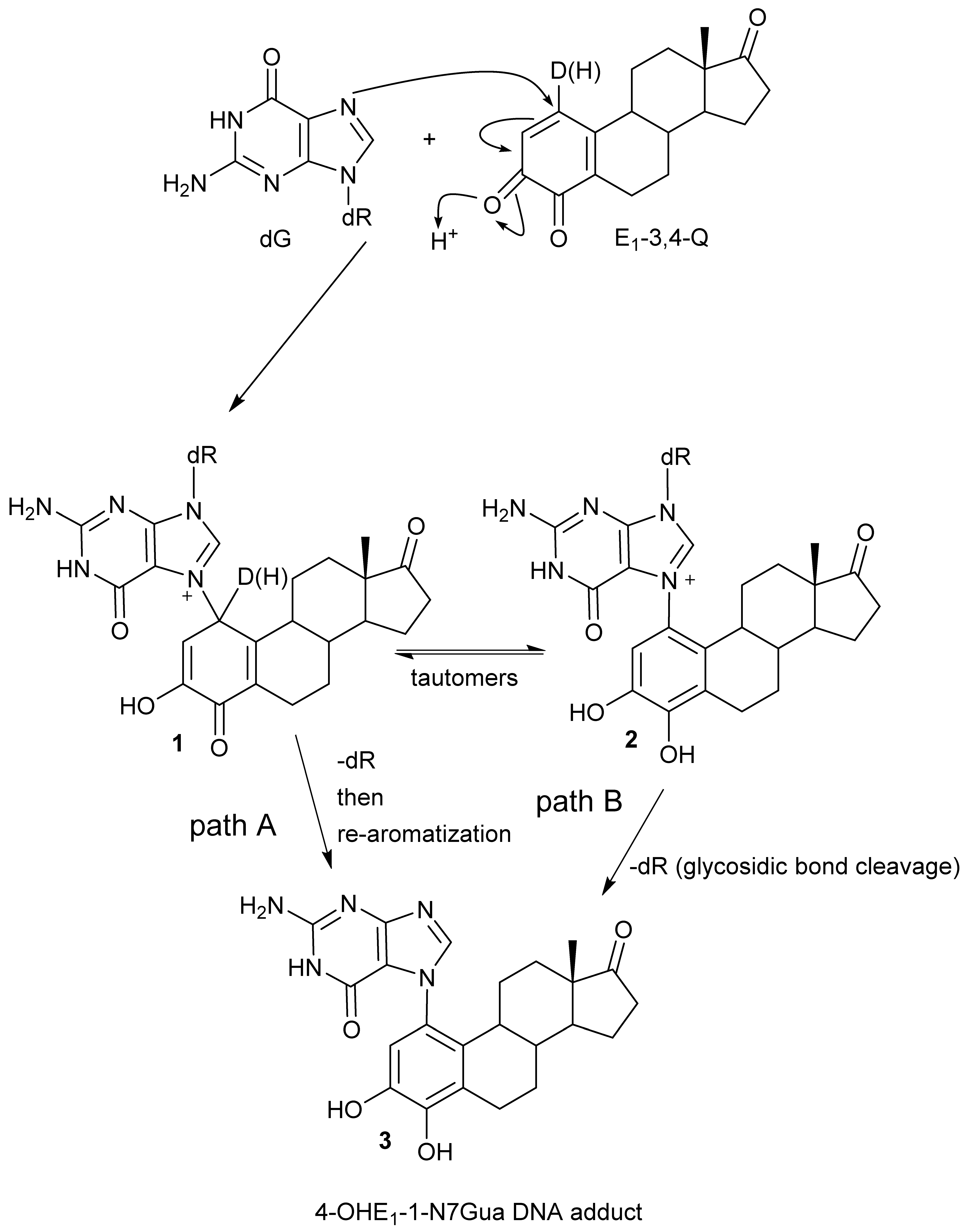
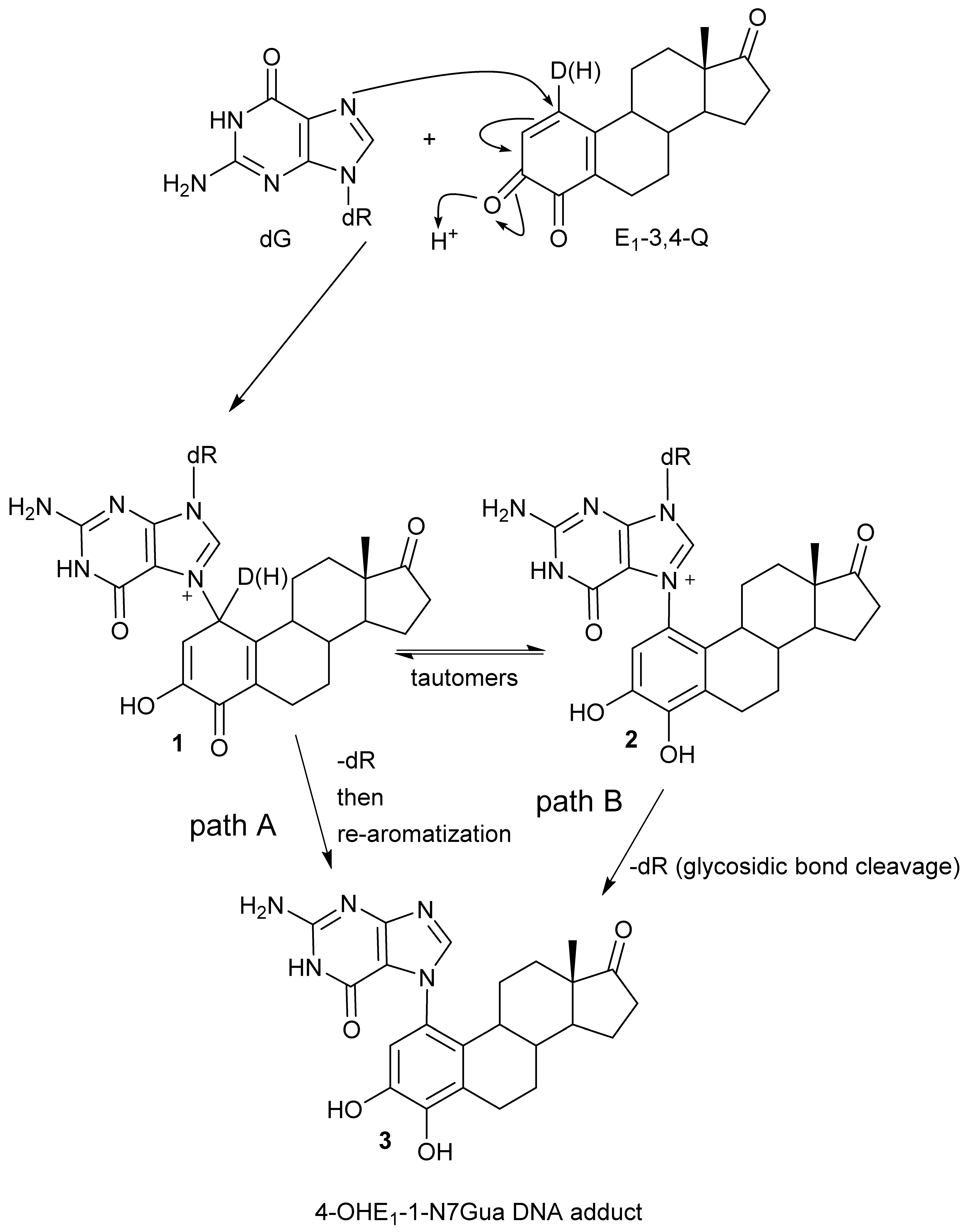
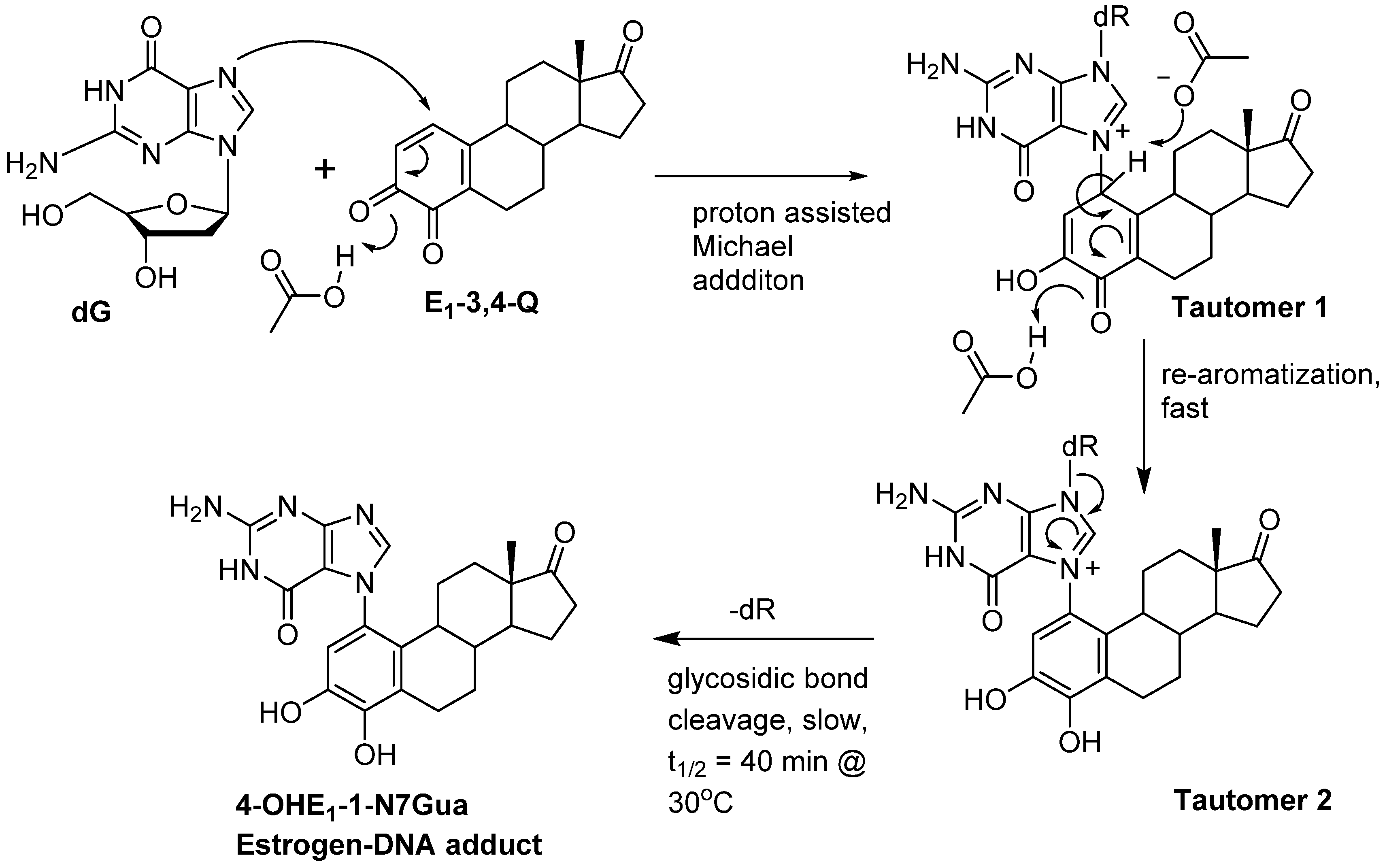
2. Results and Discussion
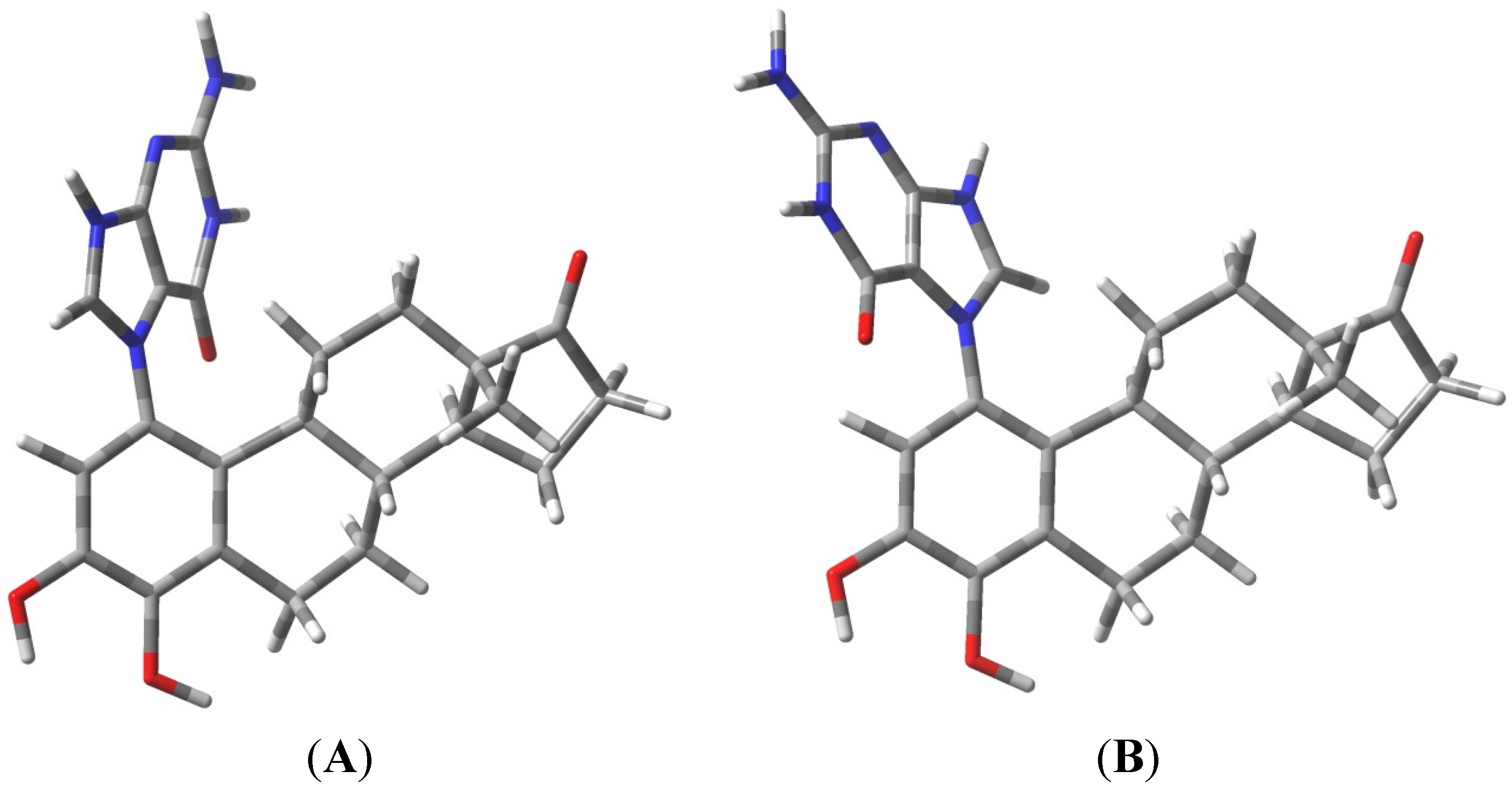
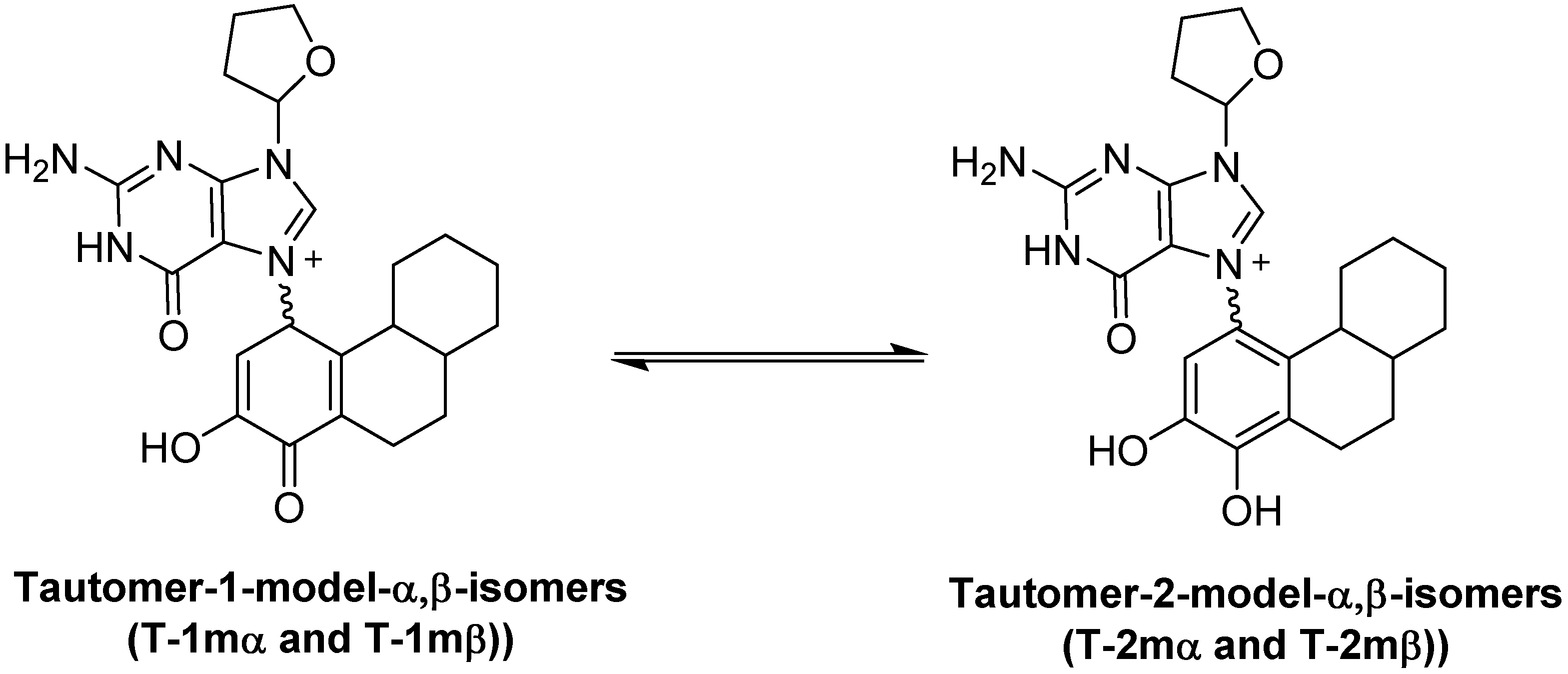
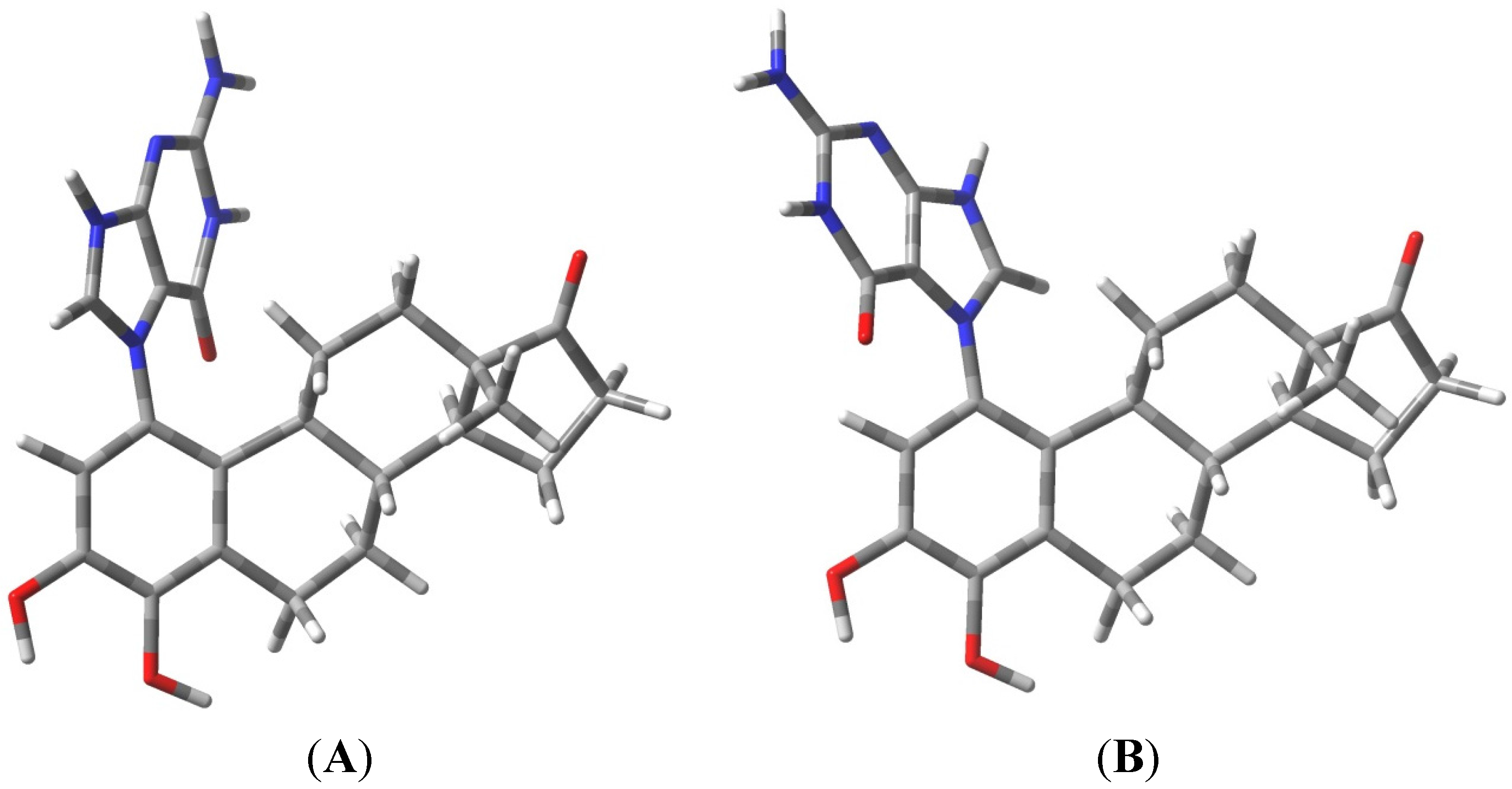
2.1. Thermodynamic Calculations on the Relative Stability of Tautomers 1 and 2
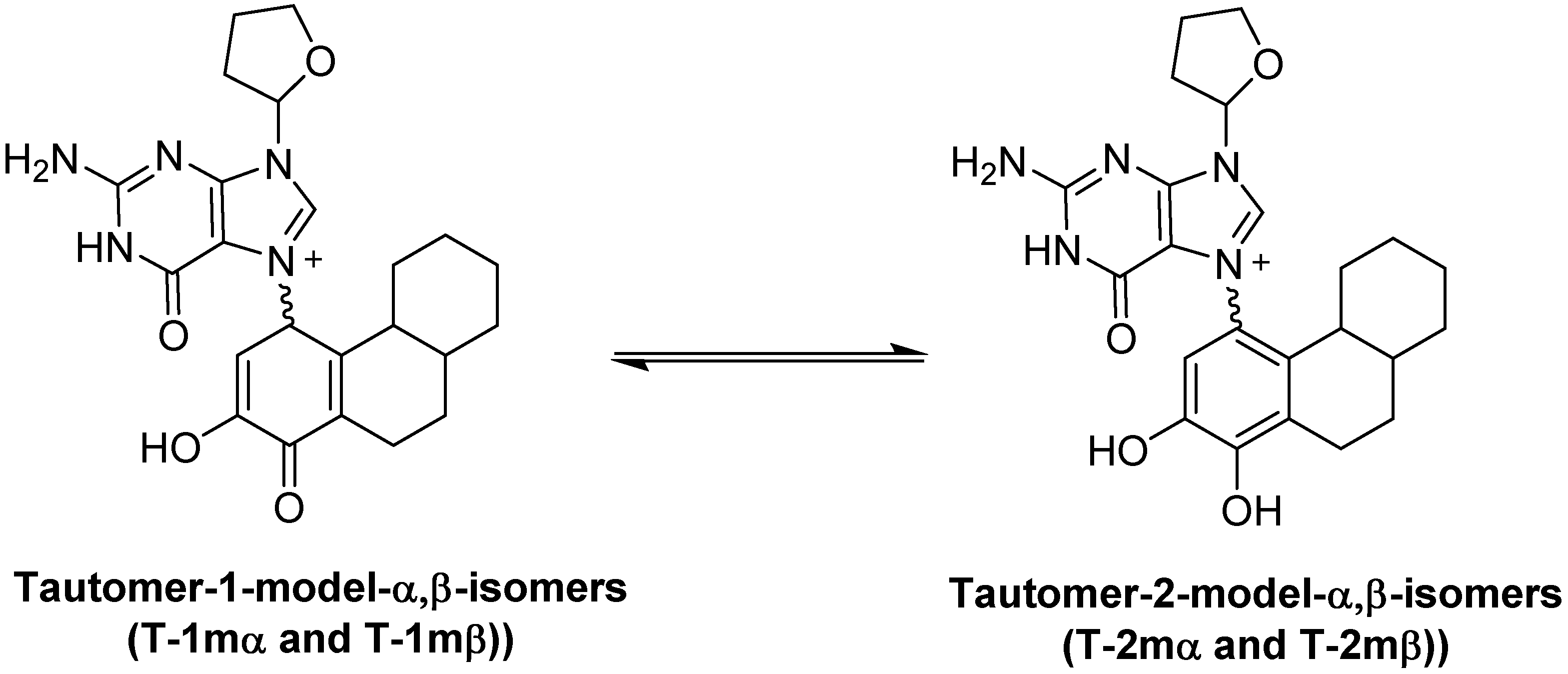
| Compound | Relative Energy (kcal/mol) | N9-C1' Bond Length, (Glycosidic Bond) (Angstroms) | Dipole (Debyes) |
|---|---|---|---|
| T-2mα | 0.00 | 1.4833 | 9.48 |
| T-2mβ | 0.54 | 1.4833 | 10.55 |
| T-1mα | 18.42 a | 1.4909 | 14.12 |
| T-1mβ | 21.34 b | 1.4927 | 13.76 |
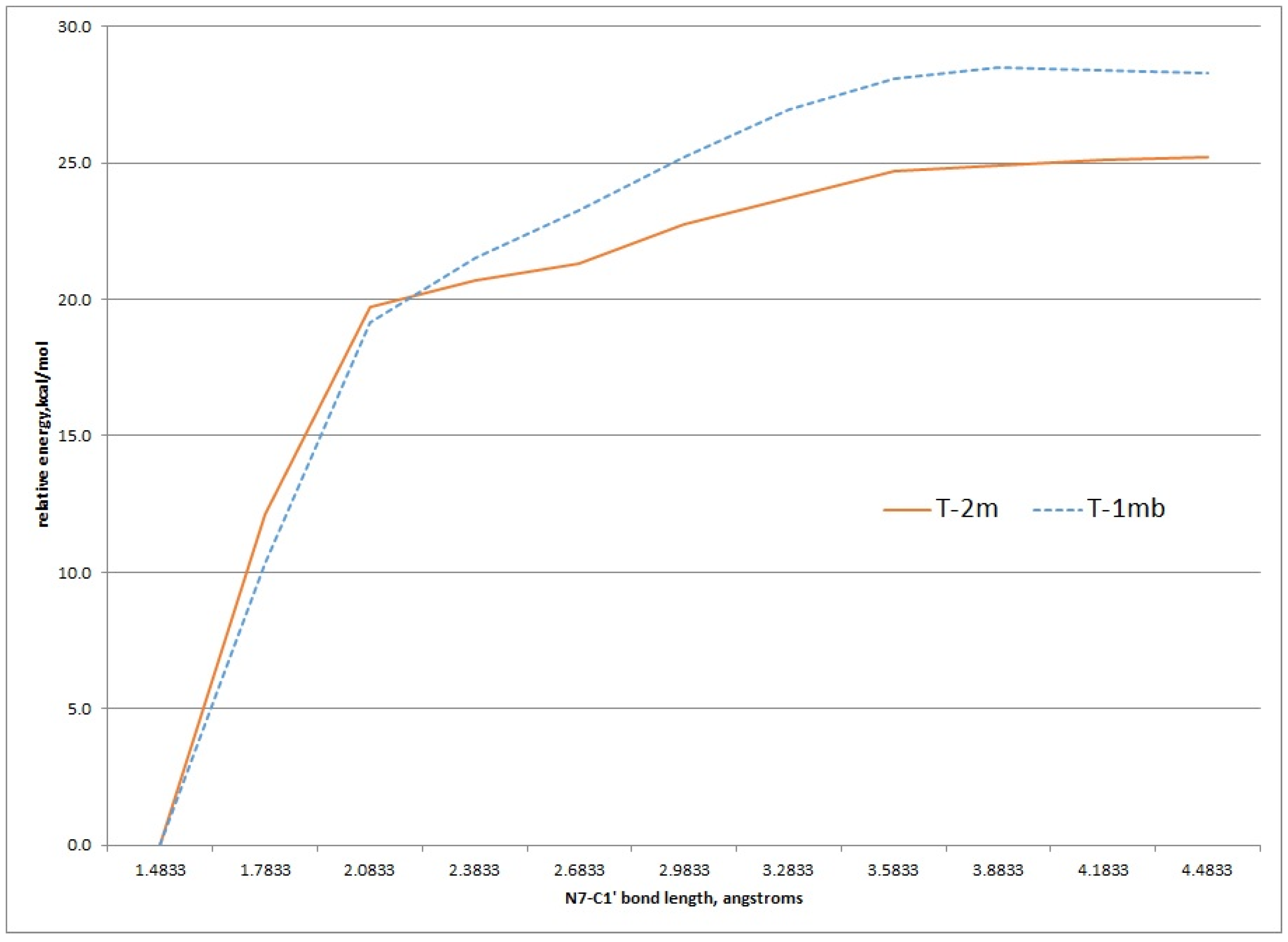
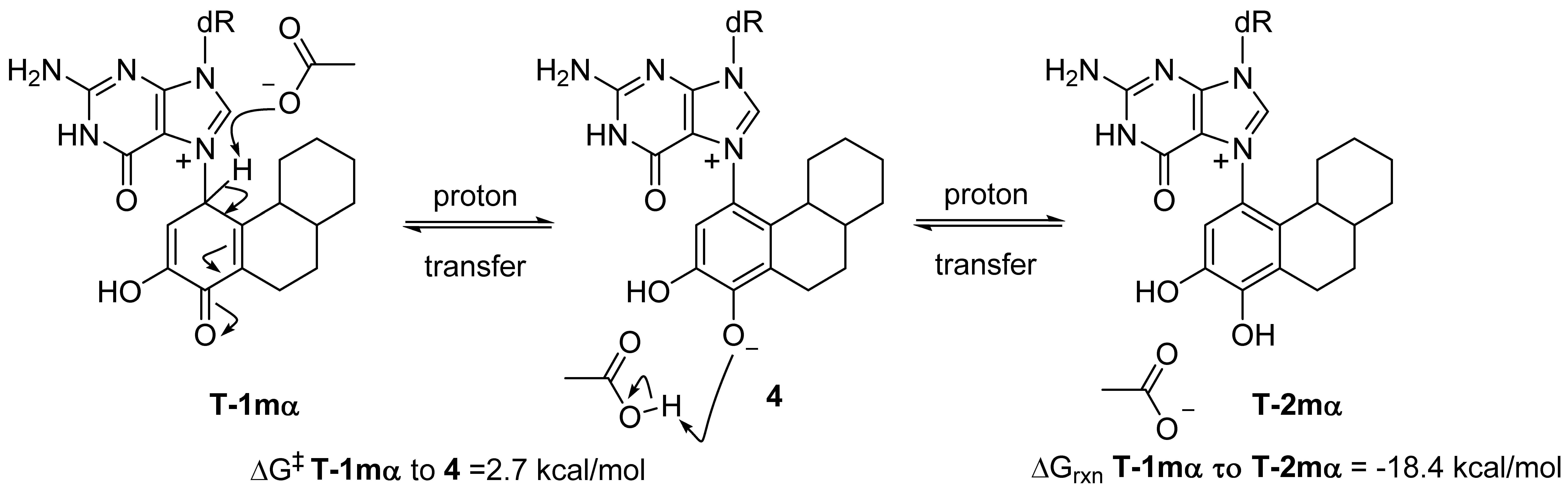
2.2. Kinetic Calculations of Proton Transfer from C1
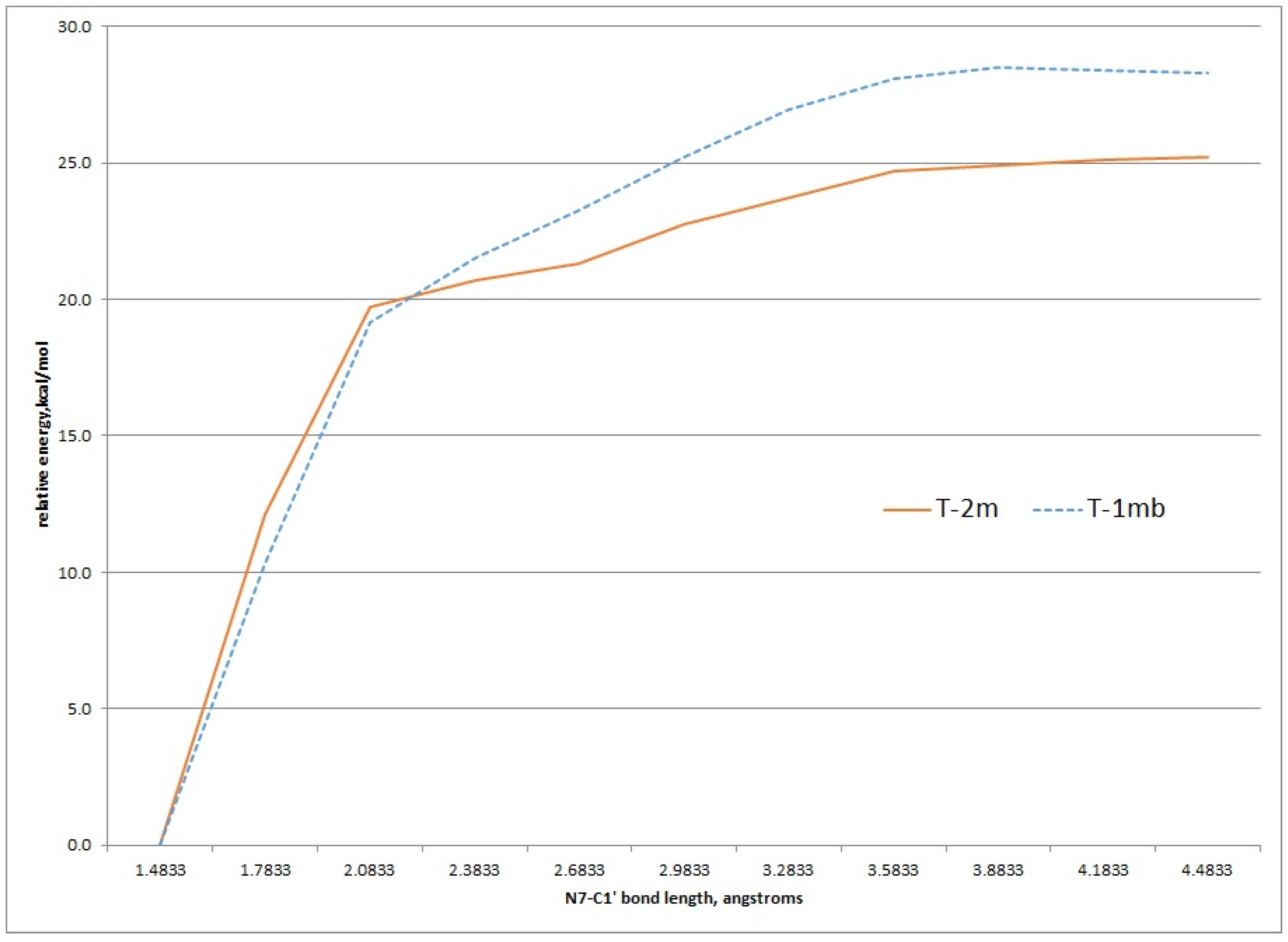
2.3. Kinetic Calculations of Glycosidic Bond Cleavage
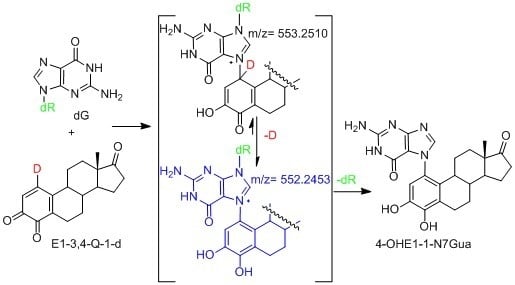
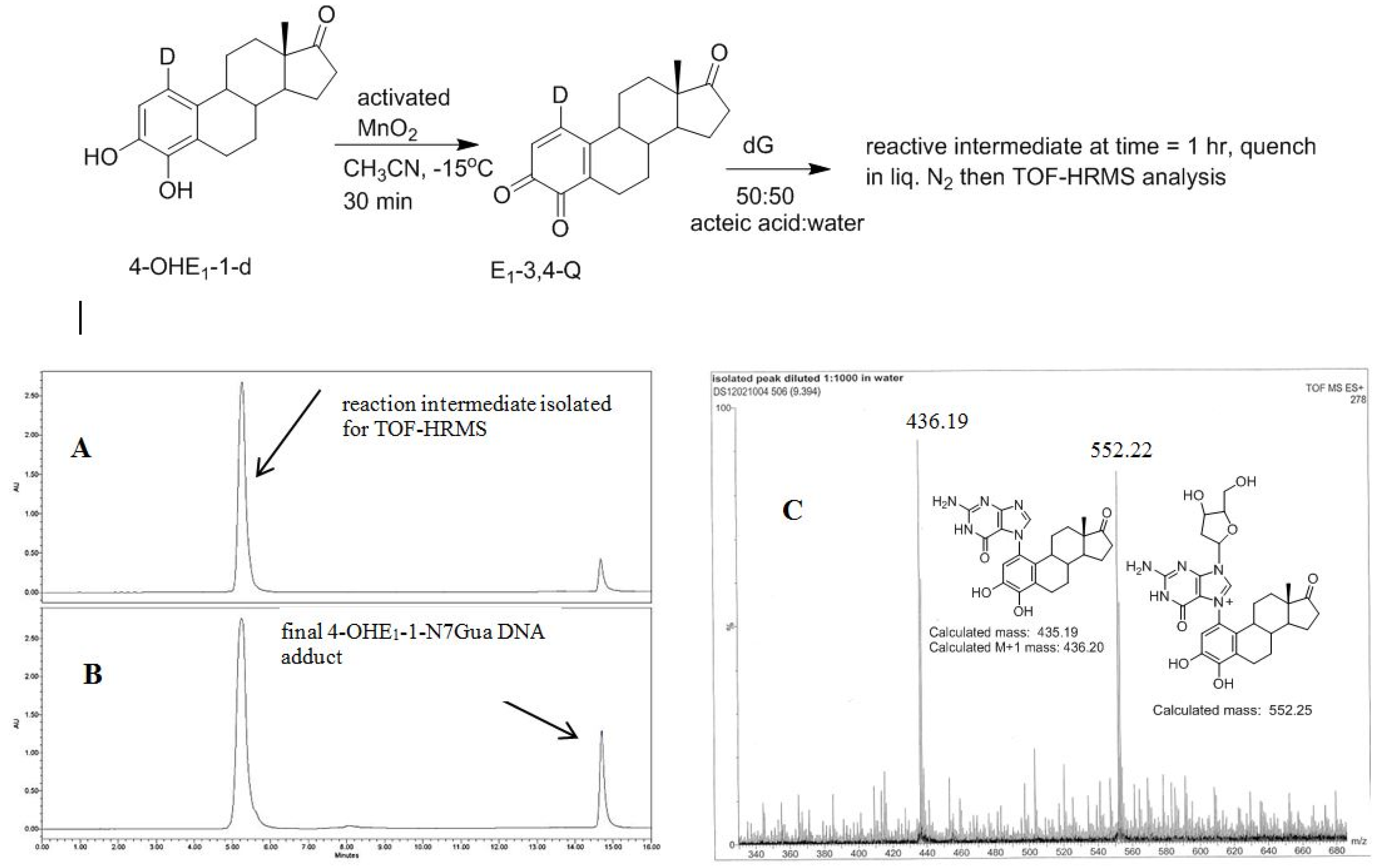
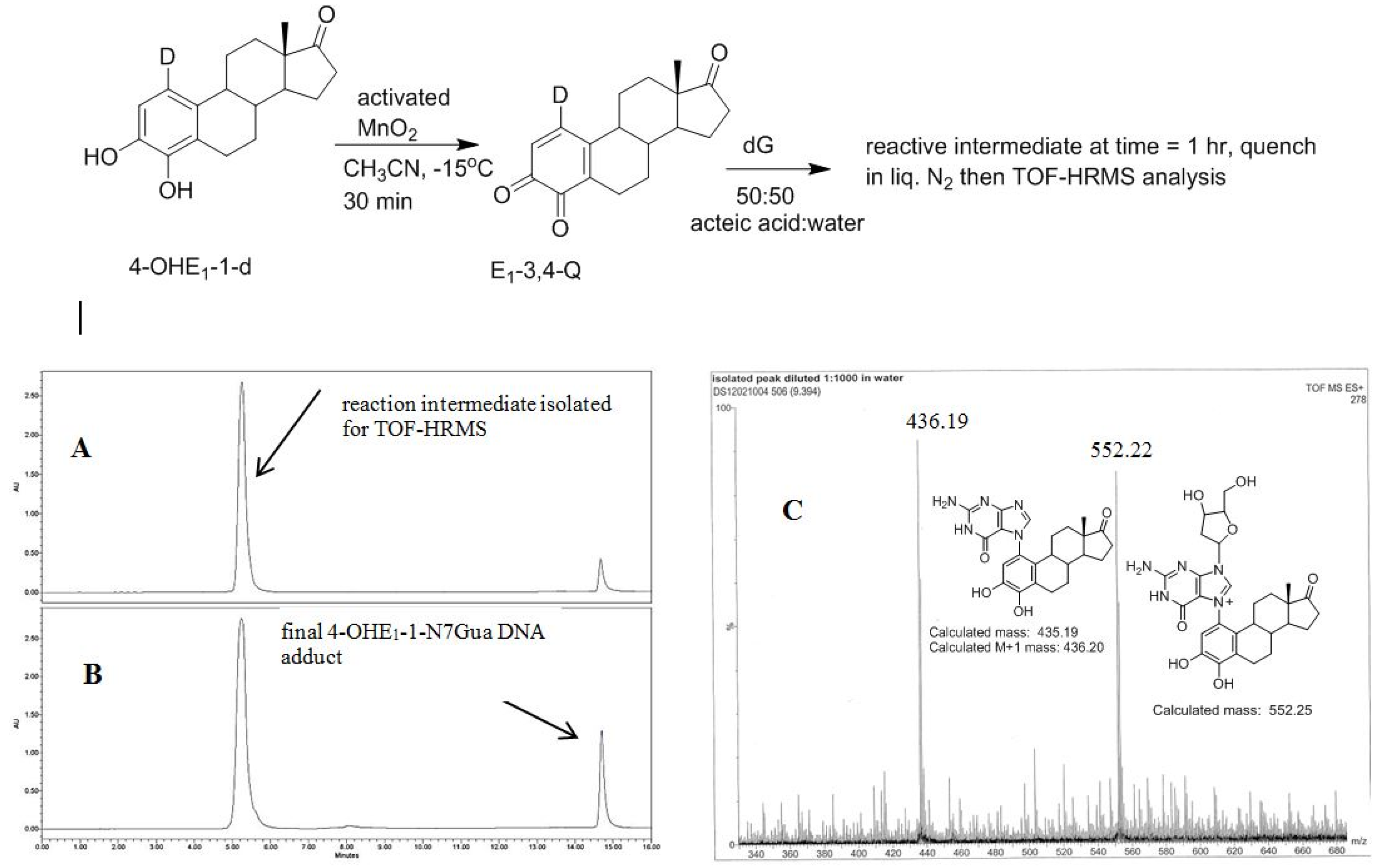
2.4. HPLC-MS Analysis of Deuterium Labeled Reaction Intermediate
3. Experimental Section
3.1. Thermodynamic and Kinetic Calculations
3.2. Chemical and Materials
3.3. Instrumentation
3.4. Procedure for Generating Reactive Intermediate 2 and Determining Deuterium Stability during Estrogen Quinone Formation
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Eliassen, A.H.; Hankinson, S.E. Endogenous hormone levels and risk of breast, endometrial and ovarian cancers: Prospective studies. Adv. Exp. Med. Biol. 2008, 630, 148–165. [Google Scholar] [PubMed]
- Cavalieri, E.; Rogan, E. The molecular etiology and prevention of estrogen-initiated cancers: Ockham’s razor: Pluralitas non est ponenda sine necessitate. Plurality should not be posited without necessity. Mol. Asp. Med. 2014, 36, 1–55. [Google Scholar] [CrossRef] [PubMed]
- Yager, J.D.; Davidson, N.E. Estrogen carcinogenesis in breast cancer. N. Engl. J. Med. 2006, 354, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Stack, D.E.; Byun, J.; Gross, M.L.; Rogan, E.G.; Cavalieri, E.L. Molecular characteristics of catechol estrogen quinones in reactions with deoxyribonucleosides. Chem. Res. Toxicol. 1996, 9, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Characterization of human microsomal cytochrome p-450 enzymes. Annu. Rev. Pharmacol. Toxicol. 1989, 29, 241–264. [Google Scholar] [CrossRef] [PubMed]
- Badawi, A.F.; Cavalieri, E.L.; Rogan, E.G. Role of human cytochrome p450 1a1, 1a2, 1b1, and 3a4 in the 2-, 4-, and 16a-hydroxylation of 17b-estradiol. Metab. Clin. Exp. 2001, 50, 1001–1003. [Google Scholar] [CrossRef] [PubMed]
- Stack, D.E.; Cavalieri, E.L.; Rogan, E.G. Catecholestrogens as procarcinogens: Depurinating adducts and tumor initiation. Adv. Pharmacol. 1998, 42, 833–836. [Google Scholar] [PubMed]
- Cavalieri, E.L.; Stack, D.E.; Devanesan, P.D.; Todorovic, R.; Dwivedy, I.; Higginbotham, S.; Johansson, S.L.; Patil, K.D.; Gross, M.L.; Gooden, J.K.; et al. Molecular origin of cancer: Catechol estrogen-3,4-quinones as endogenous tumor initiators. Proc. Natl. Acad. Sci. USA 1997, 94, 10937–10942. [Google Scholar] [CrossRef] [PubMed]
- Zahid, M.; Kohli, E.; Saeed, M.; Rogan, E.; Cavalieri, E. The greater reactivity of estradiol-3,4-quinone vs. Estradiol-2,3-quinone with DNA in the formation of depurinating adducts: Implications for tumor-initiating activity. Chem. Res. Toxicol. 2006, 19, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Li, S.A. Estrogen carcinogenesis in syrian hamster tissues: Role of metabolism. Fed. Proc. 1987, 46, 1858–1863. [Google Scholar] [PubMed]
- Liehr, J.G.; Fang, W.F.; Sirbasku, D.A.; Ari-Ulubelen, A. Carcinogenicity of catechol estrogens in syrian hamsters. J. Steroid Biochem. 1986, 24, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Newbold, R.R.; Liehr, J.G. Induction of uterine adenocarcinoma in cd-1 mice by catechol estrogens. Cancer Res. 2000, 60, 235–237. [Google Scholar] [PubMed]
- Liehr, J.G.; Ricci, M.J. 4-hydroxylation of estrogens as marker of human mammary tumors. Proc. Natl. Acad. Sci. USA 1996, 93, 3294–3296. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Chakravarti, D.; Edney, J.A.; Hollins, R.R.; Johnson, P.J.; West, W.W.; Higginbotham, S.M.; Cavalieri, E.L.; Rogan, E.G. Relative imbalances in the expression of estrogen-metabolizing enzymes in the breast tissue of women with breast carcinoma. Oncol. Rep. 2005, 14, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, N.W.; Yang, L.; Pruthi, S.; Ingle, J.N.; Sandhu, N.; Rogan, E.G.; Cavalieri, E.L. Urine biomarkers of risk in the molecular etiology of breast cancer. Breast Cancer Basic Clin. Res. 2009, 3, 1–8. [Google Scholar]
- Stack, D.E.; Li, G.; Hill, A.; Hoffman, N. Mechanistic insights into the michael addition of deoxyguanosine to catechol estrogen-3,4-quinones. Chem. Res. Toxicol. 2008, 21, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Stack, D.E.; Ritonya, J.; Jakopovic, S.; Maloley-Lewis, B. Regioselective deuterium labeling of estrone and catechol estrogen metabolites. Steroids 2014, 92, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Peverati, R.; Truhlar, D.G. Quest for a universal density functional: The accuracy of density functionals across a broad spectrum of databases in chemistry and physics. Philos. Trans. A Math. Phys. Eng. Sci. 2014, 372, 20120476. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The m06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four m06-class functionals and 12 other functionals. Theor. Chem. Acc. 2007, 120, 215–241. [Google Scholar]
- Wirz, J. Kinetics of proton transfer reactions involving carbon. Pure Appl. Chem. 1998, 70, 2221–2232. [Google Scholar] [CrossRef]
- Easton, R.E.; Giesen, D.; Welch, A.; Cramer, C.; Truhlar, D. The midi! Basis set for quantum mechanical calculations of molecular geometries and partial charges. Theor. Chim. Acta 1996, 93, 281–301. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cancès, E. The ief version of the pcm solvation method: An overview of a new method addressed to study molecular solutes at the qm ab initio level. J. Mol. Struct.THEOCHEM 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Weigend, F. Accurate coulomb-fitting basis sets for h to rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Pezzella, A.; Lista, L.; Napolitano, A.; D’Ischia, M. An expedient one-pot entry to catecholestrogens and other catechol compounds via ibx-mediated phenolic oxygenation. Tetrahedron Lett. 2005, 46, 3541–3544. [Google Scholar] [CrossRef]
- Gates, K.S.; Nooner, T.; Dutta, S. Biologically relevant chemical reactions of n7-alkylguanine residues in DNA. Chem. Res. Toxicol. 2004, 17, 839–856. [Google Scholar] [CrossRef] [PubMed]
- Drohat, A.C.; Maiti, A. Mechanisms for enzymatic cleavage of the n-glycosidic bond in DNA. Org. Biomol. Chem 2014, 12, 8367–8378. [Google Scholar] [CrossRef] [PubMed]
- Li, K.-M.; Todorovic, R.; Devanesan, P.; Higginbotham, S.; Koefeler, H.; Ramanathan, R.; Gross, M.L.; Rogan, E.G.; Cavalieri, E.L. Metabolism and DNA binding studies of 4-hydroxyestradiol and estradiol-3,4-quinone in vitro and in female aci rat mammary gland in vivo. Carcinogenesis 2004, 25, 289–297. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stack, D.E. Identifying the Tautomeric Form of a Deoxyguanosine-Estrogen Quinone Intermediate. Metabolites 2015, 5, 475-488. https://doi.org/10.3390/metabo5030475
Stack DE. Identifying the Tautomeric Form of a Deoxyguanosine-Estrogen Quinone Intermediate. Metabolites. 2015; 5(3):475-488. https://doi.org/10.3390/metabo5030475
Chicago/Turabian StyleStack, Douglas E. 2015. "Identifying the Tautomeric Form of a Deoxyguanosine-Estrogen Quinone Intermediate" Metabolites 5, no. 3: 475-488. https://doi.org/10.3390/metabo5030475
APA StyleStack, D. E. (2015). Identifying the Tautomeric Form of a Deoxyguanosine-Estrogen Quinone Intermediate. Metabolites, 5(3), 475-488. https://doi.org/10.3390/metabo5030475