
13C Tracers for Glucose Degrading Pathway Discrimination in Gluconobacter oxydans 621H

Abstract
:1. Introduction
2. Results
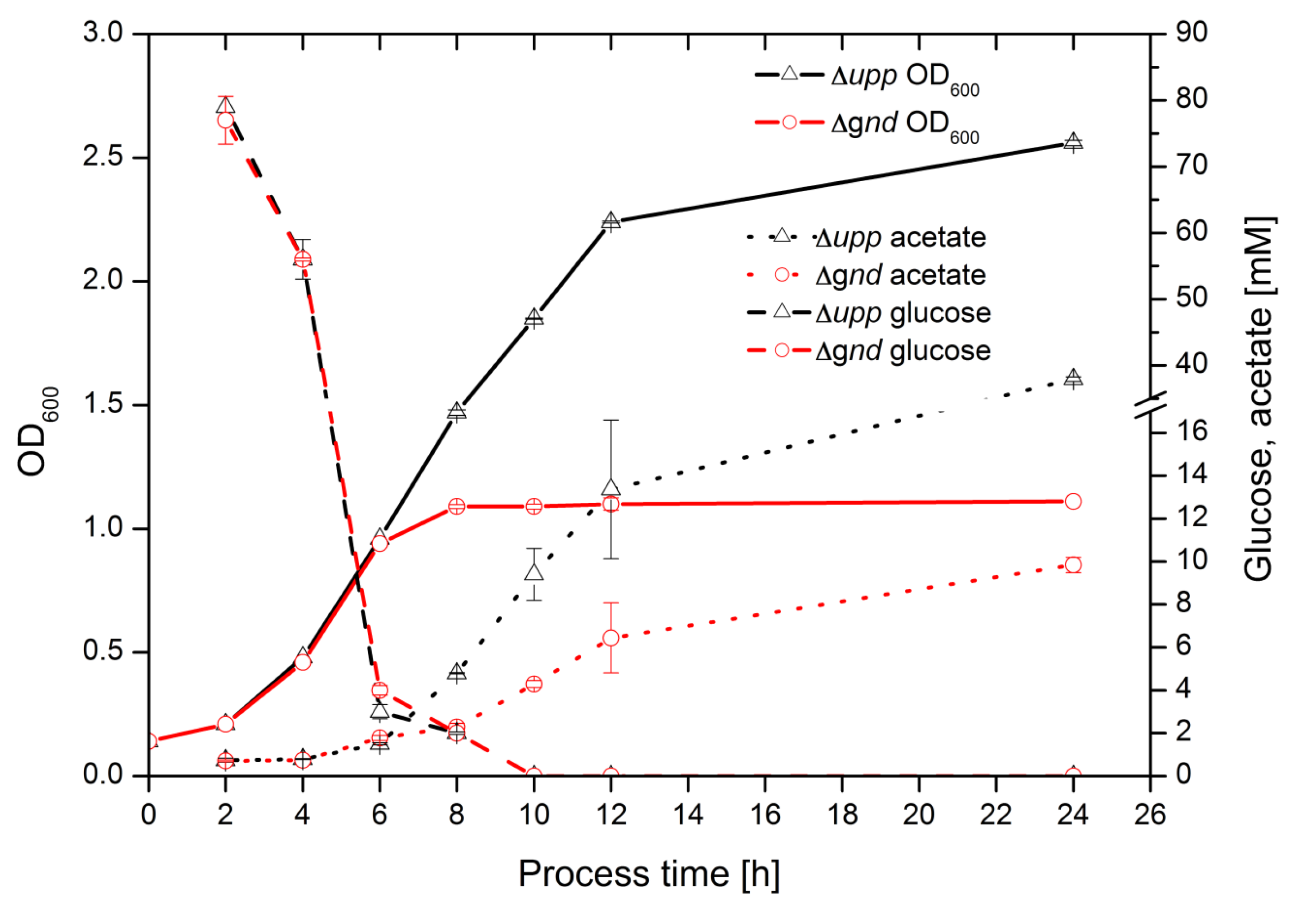
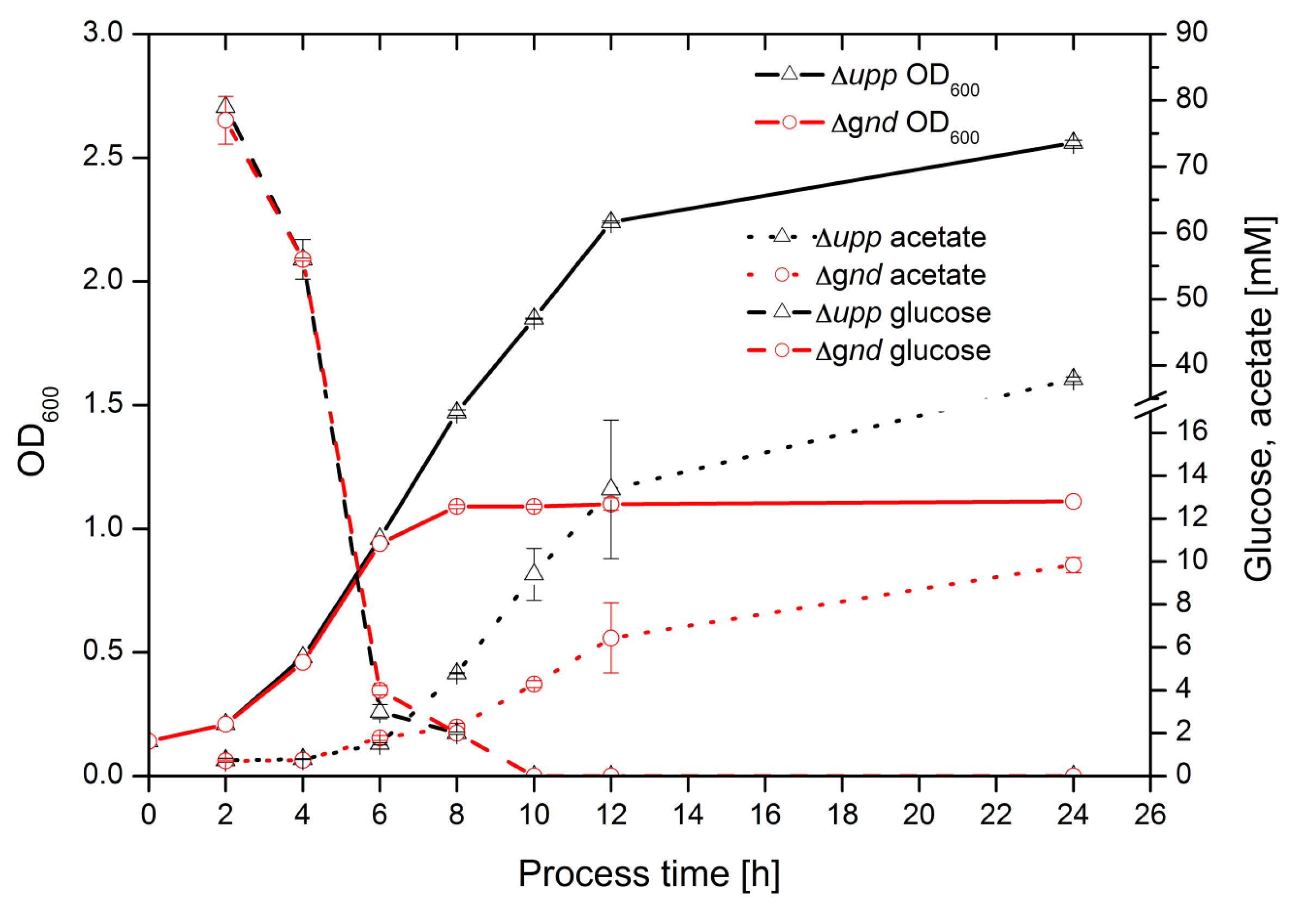
2.1. Cultivation of G. oxydans at a Small Scale
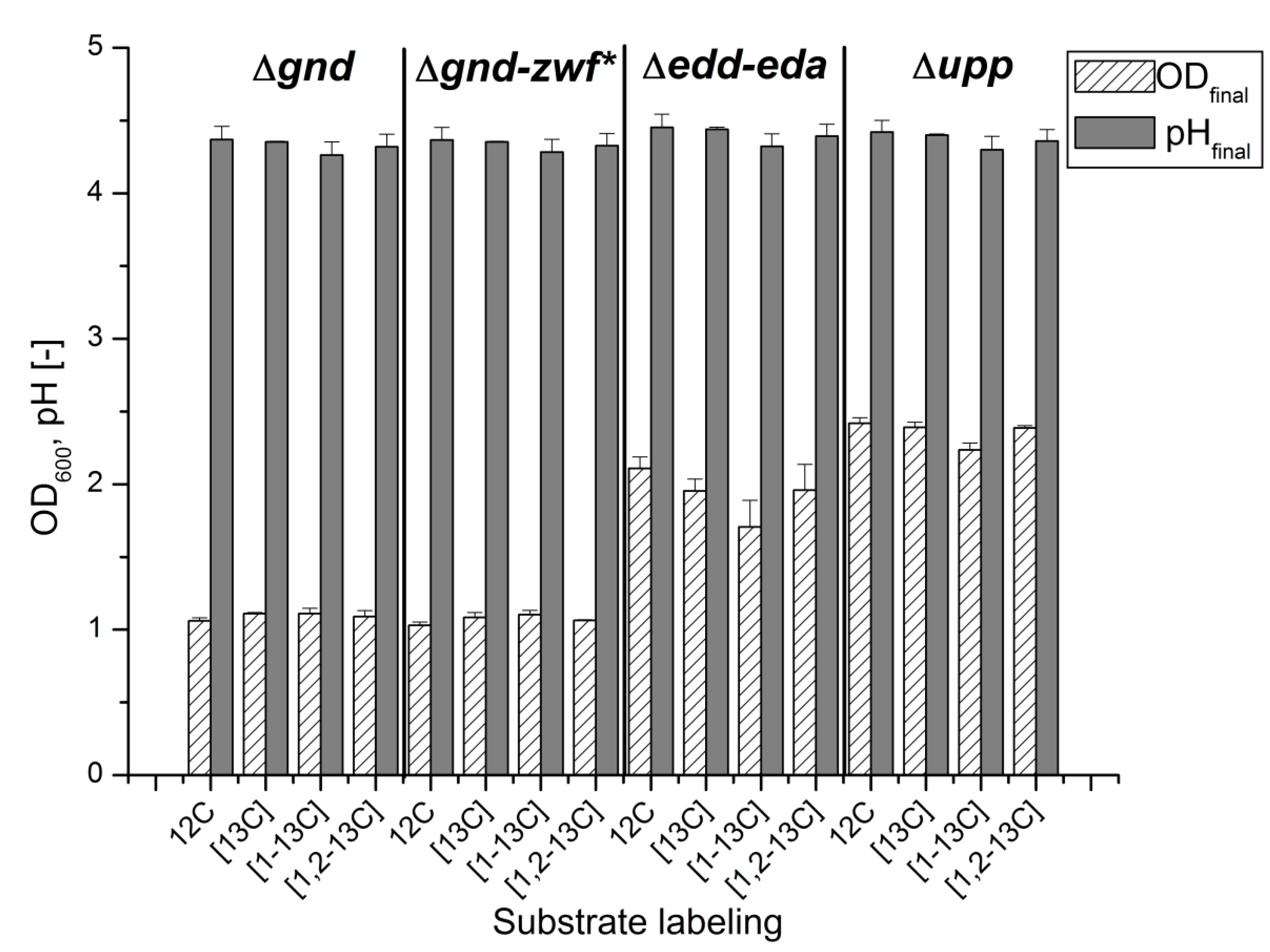
2.2. Transition of 13C Labeling from Glucose to Alanine by G. oxydans Metabolism
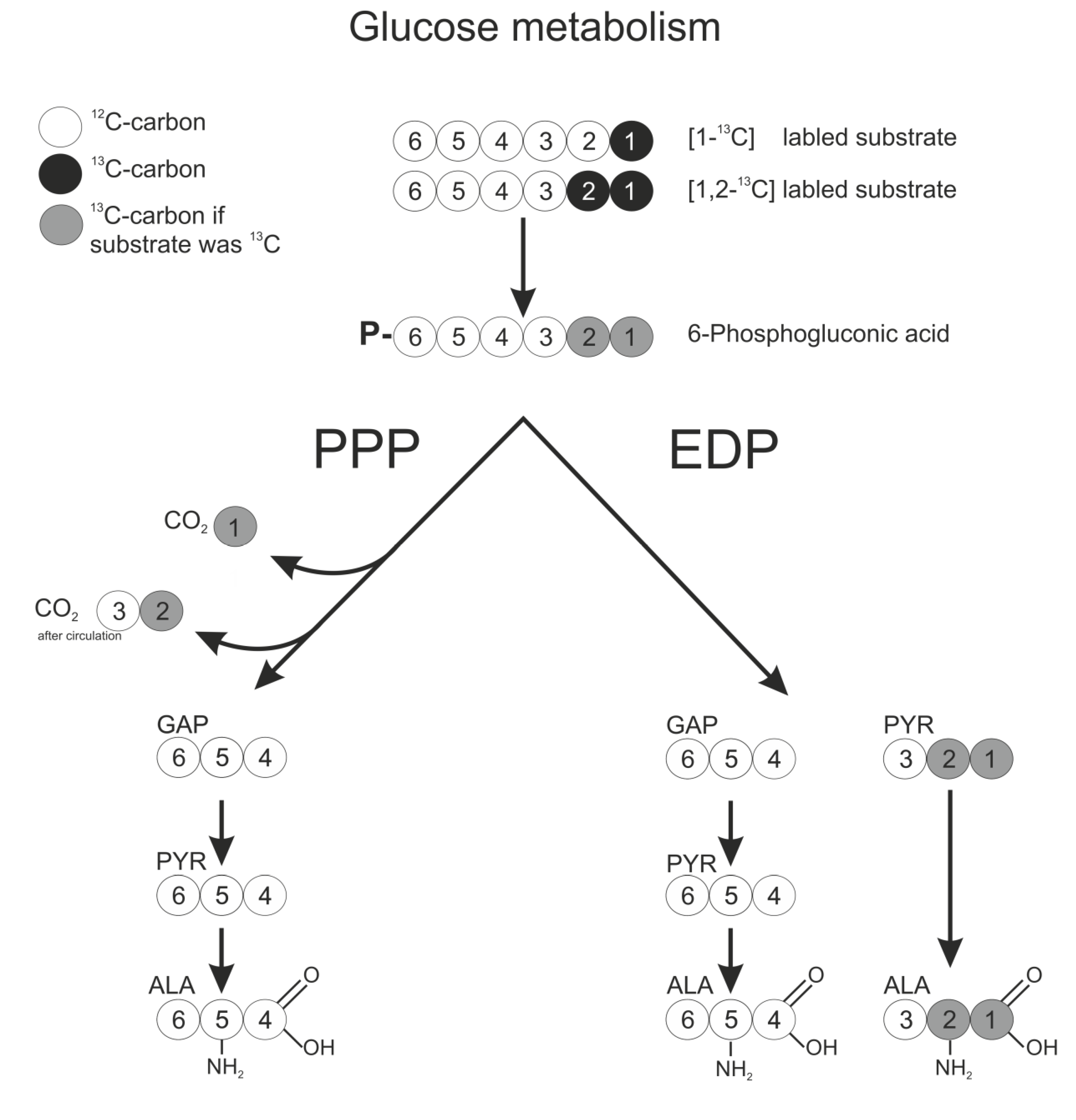
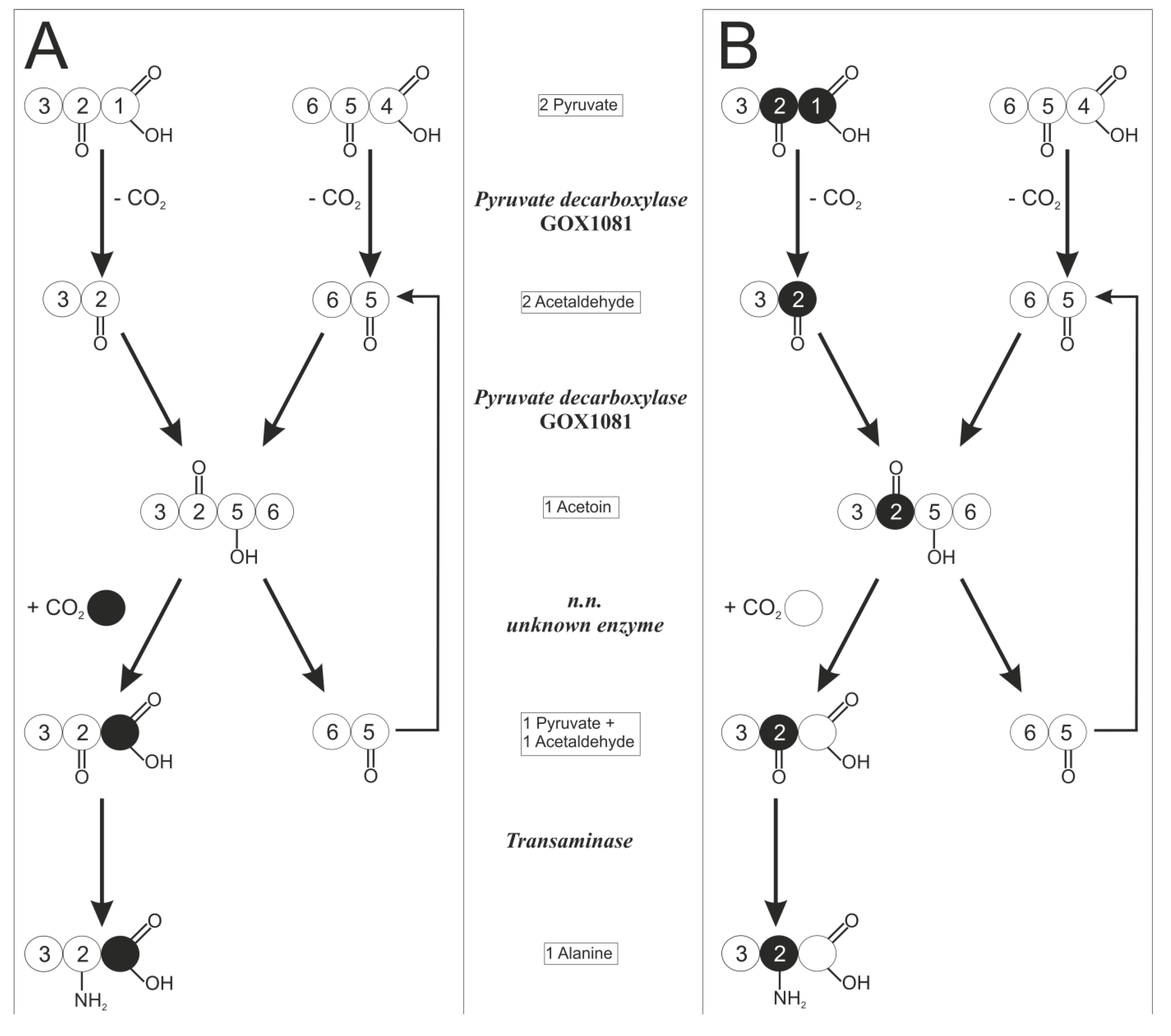
2.3. 13C labeling Experiments for Pathway Discrimination

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mass Spectrometry Signal Resp. Labeling State of Alanine | |||||
|---|---|---|---|---|---|
| Substrate | Strain | m.0 | m.1 | m.2 | m.3 |
| 12C control | Δgnd | 0.96 ± 0.001 | 0.04 ± 0.001 | n.d. | n.d. |
| Δgnd-zwf* | 0.96 ± 0.001 | 0.04 ± 0.001 | n.d. | n.d. | |
| Δedd-eda | 0.96 ± 0.001 | 0.04 ± 0.001 | n.d. | n.d. | |
| Δupp | 0.96 ± 0.001 | 0.04 ± 0.001 | n.d. | n.d. | |
| [13C] control | Δgnd | 0.37 ± 0.014 | 0.02 ± 0.001 | 0.07 ± 0.002 | 0.54 ± 0.015 |
| Δgnd-zwf* | 0.37 ± 0.009 | 0.02 ± 0.001 | 0.08 ± 0.004 | 0.54 ± 0.008 | |
| Δedd-eda | 0.30 ± 0.013 | 0.03 ± 0.002 | 0.08 ± 0.003 | 0.59 ± 0.01 | |
| Δupp | 0.31 ± 0.01 | 0.02 ± 0.008 | 0.08 ± 0.005 | 0.59 ± 0.009 | |
| [1-13C] | Δgnd | 0.51 ± 0.011 | 0.47 ± 0.011 | 0.01 ± 0.000 | n.d. |
| Δgnd-zwf* | 0.50 ± 0.014 | 0.49 ± 0.014 | 0.02 ± 0.000 | n.d. | |
| Δedd-eda | 0.96 ± 0.000 | 0.04 ± 0.000 | n.d. | n.d. | |
| Δupp | 0.76 ± 0.004 | 0.24 ± 0.003 | 0.01 ± 0.000 | n.d. | |
| [1,2-13C] | Δgnd | 0.45 ± 0.016 | 0.09 ± 0.004 | 0.45 ± 0.013 | 0.01 ± 0.000 |
| Δgnd-zwf* | 0.44 ± 0.031 | 0.09 ± 0.006 | 0.46 ± 0.024 | 0.01 ± 0.001 | |
| Δedd-eda | 0.95 ± 0.001 | 0.05 ± 0.001 | n.d. | n.d. | |
| Δupp | 0.64 ± 0.006 | 0.13 ± 0.002 | 0.22 ± 0.004 | 0.01 ± 0.001 | |
| Mass Spectrometry Signal Resp. Labeling State of Alanine | |||||||
|---|---|---|---|---|---|---|---|
| Substrate | Strain | m.0 | m.1.0 | m.1.1 | m.2.1 | m.2.2 | m.3.2 |
| [1-13C] | Δgnd | 0.51 ± 0.011 | 0.45 ± 0.011 | 0.02 ± 0.000 | 0.01 ± 0.000 | n.d. | n.d. |
| Δgnd-zwf* | 0.50 ± 0.014 | 0.47 ± 0.014 | 0.02 ± 0.000 | 0.02 ± 0.000 | n.d. | n.d. | |
| Δedd-eda | 0.96 ± 0.000 | 0.01 ± 0.000 | 0.03 ± 0.000 | n.d. | n.d. | n.d. | |
| Δupp | 0.76 ± 0.004 | 0.21 ± 0.004 | 0.03 ± 0.000 | 0.01 ± 0.000 | n.d. | n.d. | |
| [1,2-13C] | Δgnd | 0.45 ± 0.016 | 0.00 ± 0.002 | 0.08 ± 0.005 | 0.45 ± 0.013 | n.d. | 0.01 ± 0.000 |
| Δgnd-zwf* | 0.44 ± 0.031 | n.d. | 0.08 ± 0.006 | 0.46 ± 0.024 | n.d. | 0.01 ± 0.001 | |
| Δedd-eda | 0.95 ± 0.001 | 0.01 ± 0.000 | 0.04 ± 0.001 | n.d. | n.d. | n.d. | |
| Δupp | 0.64 ± 0.006 | 0.05 ± 0.001 | 0.08 ± 0.001 | 0.21 ± 0.004 | 0.01 ± 0.000 | 0.01 ± 0.001 | |
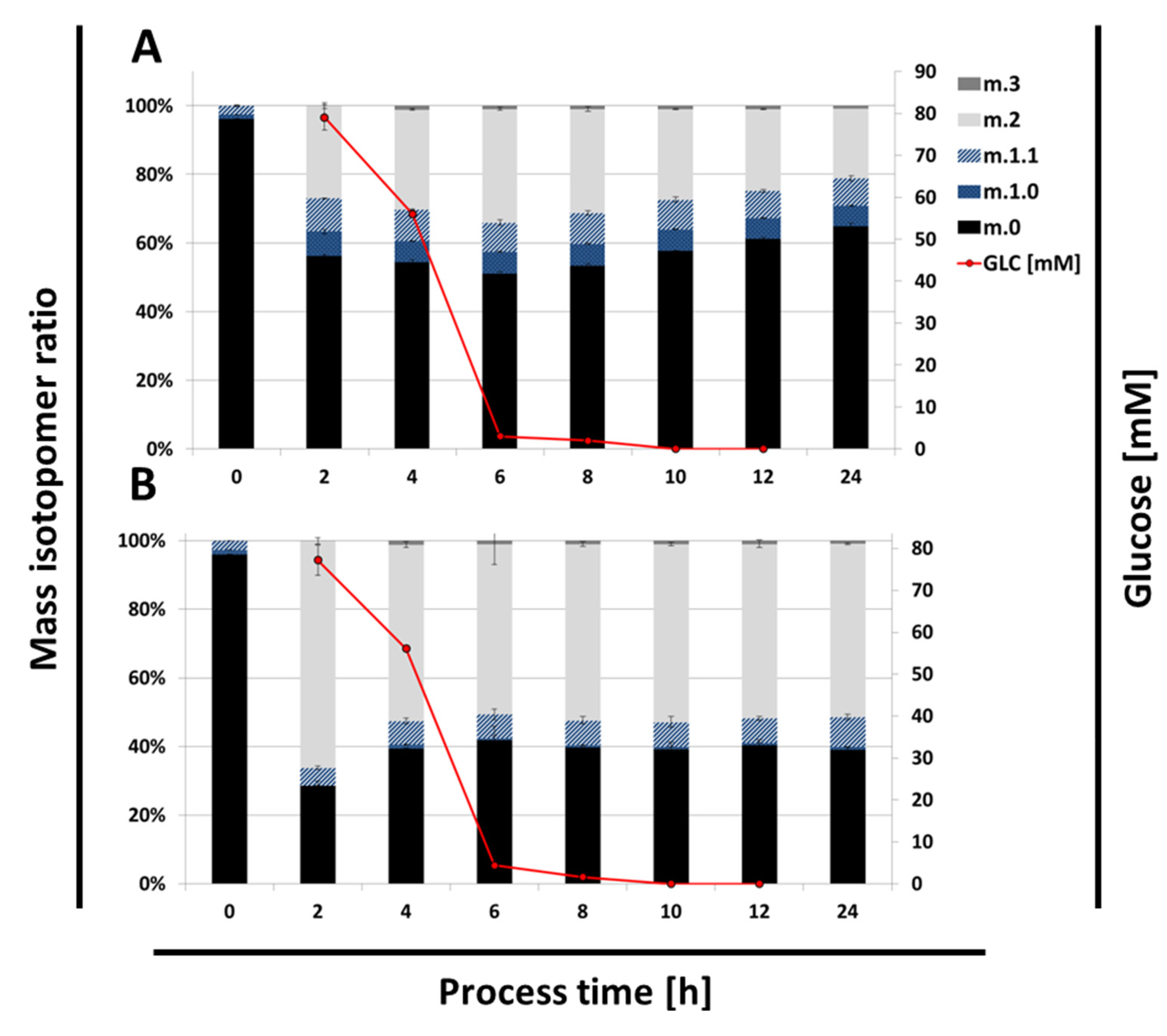
2.4. Time Resolved 13C Labeling Experiments
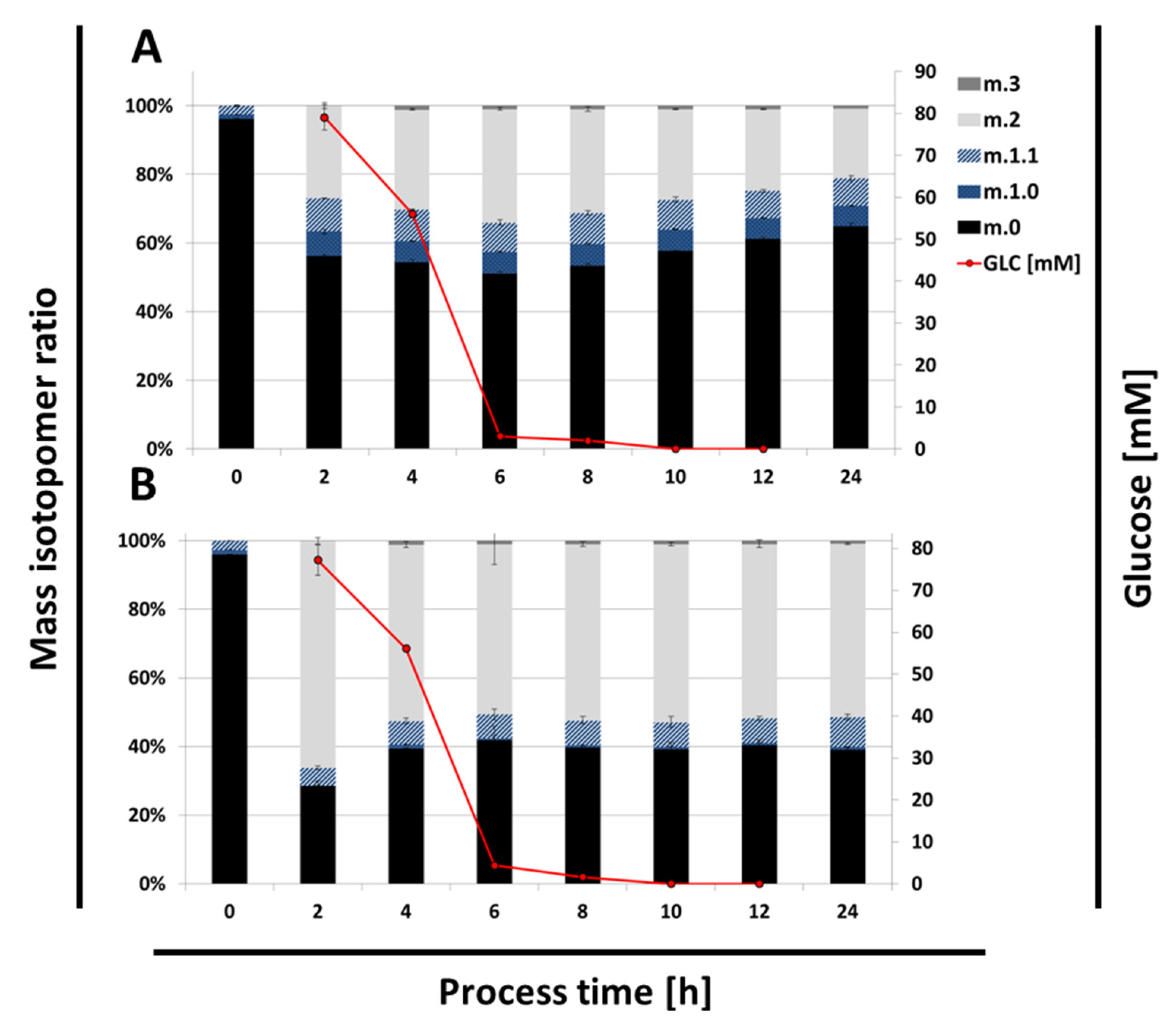
3. Discussion
4. Experimental Section
4.1. Materials
4.2. Bacterial Strains, Media, and Growth Conditions
| Strain (G. oxydans) | Description | Source |
|---|---|---|
| Δupp Δgnd | Δgnd derivative of G. oxydans 621H Δupp | [9] |
| (Deletion of GOX1705) | ||
| Δupp Δgnd-zwf* | Δgnd-zwf* derivative of G. oxydans 621H Δupp | [9] |
| (Deletion of GOX1705) with mutation in zwf (GOX0145) | ||
| Δupp Δedd-eda | Δedd-eda derivative of G. oxydans 621H Δupp | [9] |
| (Deletion of GOX0430 and GOX0431) | ||
| Δupp | Δupp derivative of G. oxydans 621H | [16] |
| (Deletion of GOX0327) |
4.3. Sample Treatment and Analysis
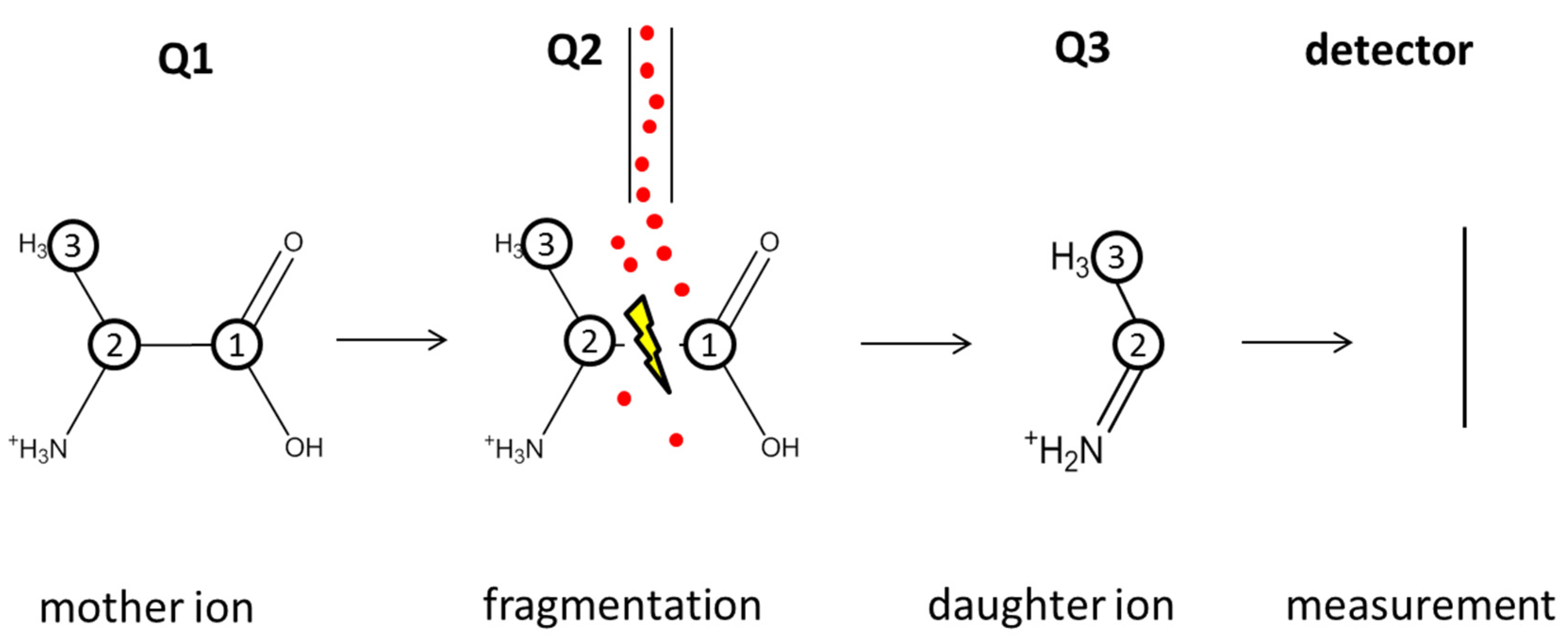
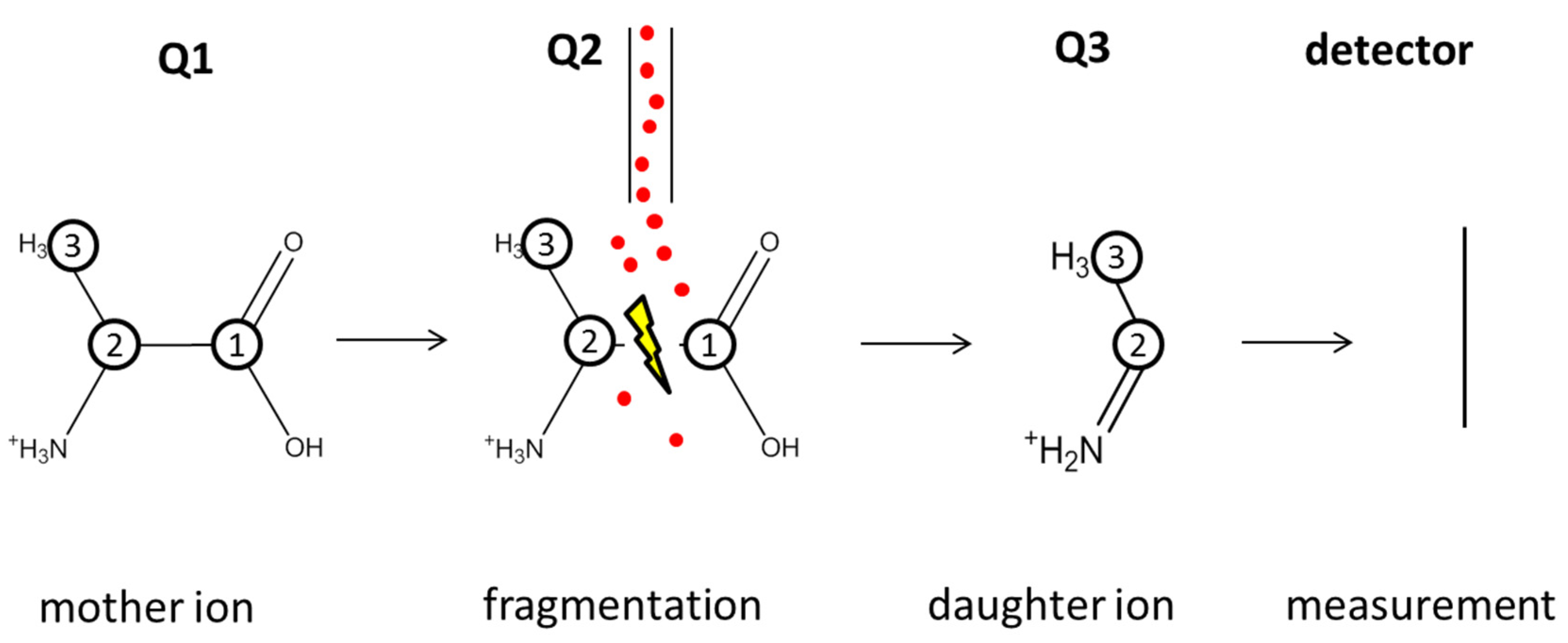
4.4. LC-MS/MS Measurements
4.5. 13C Labeling Nomenclature
4.6. Washout Correction
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gupta, A.; Singh, V.K.; Qazi, G.N.; Kumar, A. Gluconobacter oxydans: Its biotechnological applications. J. Mol. Microbiol. Biotechnol. 2001, 3, 445–456. [Google Scholar] [PubMed]
- Prust, C.; Hoffmeister, M.; Liesegang, H.; Wiezer, A.; Fricke, W.F.; Ehrenreich, A.; Gottschalk, G.; Deppenmeier, U. Complete genome sequence of the acetic acid bacterium Gluconobacter oxydans. Nat. Biotechnol. 2005, 23, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Kersters, K.; Lisdiyanti, P.; Komagata, K.; Swings, J. The family Acetobacteriaceae: The genera Acetobacter, Acidomonas, Asaia, Gluconacetobacter, Gluconobacter, and Kozakia. In The Prokaryotes, 3rd ed.; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer-Verlag GmbH: Heidelberg, Germany, 2006; Volume 5, pp. 163–200. [Google Scholar]
- Raspor, P.; Goranovič, D. Biotechnological applications of acetic acid bacteria. Crit. Rev. Biotechnol. 2008, 28, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.R.; Stokes, J.L. Nutrition of the acetic acid bacteria. J. Bacteriol. 1953, 65, 405–412. [Google Scholar] [PubMed]
- Stokes, J.L.; Larsen, A. Amino acid requirements of Acetobacter suboxydans. J. Bacteriol. 1945, 49, 495–501. [Google Scholar] [PubMed]
- Gossele, F.; Vandenmooter, M.; Verdonck, L.; Swings, J.; Deley, J. The nitrogen requirements of Gluconobacter, Acetobacter and Frateuria. A. Van Leeuw. J. Microb. 1981, 47, 289–296. [Google Scholar] [CrossRef]
- Olijve, W.; Kok, J.J. Analysis of growth of Gluconobacter oxydans in glucose containing media. Arch. Microbiol. 1979, 121, 283–290. [Google Scholar] [CrossRef]
- Richhardt, J.; Bringer, S.; Bott, M. Mutational analysis of the pentose phosphate and entner-doudoroff pathways in Gluconobacter oxydans reveals improved growth of a Δedd Δeda mutant on mannitol. Appl. Environ. Microbiol. 2012, 78, 6975–6986. [Google Scholar] [CrossRef] [PubMed]
- Richhardt, J.; Bringer, S.; Bott, M. Role of the pentose phosphate pathway and the entner-doudoroff pathway in glucose metabolism of Gluconobacter oxydans 621H. Appl. Microbiol. Biotechnol. 2013, 97, 4315–4323. [Google Scholar] [CrossRef] [PubMed]
- Hanke, T.; Nöh, K.; Noack, S.; Polen, T.; Bringer, S.; Sahm, H.; Wiechert, W.; Bott, M. Combined fluxomics and transcriptomics analysis of glucose catabolism via a partially cyclic pentose phosphate pathway in Gluconobacter oxydans 621H. Appl. Environ. Microbiol. 2013, 79, 2336–2348. [Google Scholar] [CrossRef] [PubMed]
- Wiechert, W. Modeling and simulation: Tools for metabolic engineering. J. Biotechnol. 2002, 94, 37–63. [Google Scholar] [CrossRef]
- Christiansen, T.; Christensen, B.; Nielsen, J. Metabolic network analysis of Bacillus clausii on minimal and semirich medium using C-13-Labeled glucose. Metab. Eng. 2002, 4, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Iwatani, S.; van Dien, S.; Shimbo, K.; Kubota, K.; Kageyama, N.; Iwahata, D.; Miyano, H.; Hirayama, K.; Usuda, Y.; Shimizu, K.; et al. Determination of metabolic flux changes during fed-batch cultivation from measurements of intracellular amino acids by LC-MS/MS. J. Biotechnol. 2007, 128, 93–111. [Google Scholar] [CrossRef] [PubMed]
- Thiele, B.; Füllner, K.; Stein, N.; Oldiges, M.; Kuhn, A.J.; Hofmann, D. Analysis of amino acids without derivatization in barley extracts by LC-MS-MS. Anal. Bioanal. Chem. 2008, 391, 2663–2672. [Google Scholar] [CrossRef] [PubMed]
- Klasen, R.; Bringer-Meyer, S.; Sahm, H. Biochemical characterization and sequence analysis of the gluconate:NADP 5-oxidoreductase gene from Gluconobacter oxydans. J. Bacteriol. 1995, 177, 2637–2643. [Google Scholar] [PubMed]
- Droste, P.; Weitzel, M.; Wiechert, W. Visual exploration of isotope labeling networks in 3D. Bioprocess Biosyst. Eng. 2008, 31, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Schneider, B.L.; Reitzer, L. Genetics and regulation of the major enzymes of alanine synthesis in Escherichia coli. J. Bacteriol. 2010, 192, 5304–5311. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.K.; Midgley, M.; Dawes, E.A. The metabolism of 2-oxogluconate by Pseudomonas aeruginosa. J. Gen. Microbiol. 1973, 78, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Whiting, P.H.; Midgley, M.; Dawes, E.A. The role of glucose limitation in the regulation of the transport of glucose, gluconate and 2-oxogluconate, and of glucose metabolism in Pseudomonas aeruginosa. J. Gen. Microbiol. 1976, 92, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Kruger, N.J.; von Schaewen, A. The oxidative pentose phosphate pathway: Structure and organisation. Curr. Opin. Plant Biol. 2003, 6, 236–246. [Google Scholar] [CrossRef]
- Rauch, B.; Pahlke, J.; Schweiger, P.; Deppenmeier, U. Characterization of enzymes involved in the central metabolism of Gluconobacter oxydans. Appl. Microbiol. Biotechnol. 2010, 88, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, A.J. The acetate switch. Microbiol. Mol. Biol. R. 2005, 69, 12–50. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.; Junker, A.; Brauer, K.; Mühlthaler, B.; Kostner, D.; Mientus, M.; Liebl, W.; Ehrenreich, A. Deletion of pyruvate decarboxylase by a new method for efficient markerless gene deletions in Gluconobacter oxydans. Appl. Microbiol. Biotechnol. 2013, 97, 2521–2530. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, H.; Caroline, D.F.; Harding, R.W.; Wagner, R.P. An acetohydroxy acid synthetase from Neurospora crassa. Arch. Biochem. Biophys. 1968, 128, 184–193. [Google Scholar] [CrossRef]
- Siegert, P.; McLeish, M.J.; Baumann, M.; Iding, H.; Kneen, M.M.; Kenyon, G.L.; Pohl, M. Exchanging the substrate specificities of pyruvate decarboxylase from Zymomonas mobilis and benzoylformate decarboxylase from Pseudomonas putida. Protein Eng. Des. Sel. 2005, 18, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Shinagawa, E.; Adachi, O.; Ano, Y.; Yakushi, T.; Matsushita, K. Purification and characterization of membrane-bound 3-dehydroshikimate dehydratase from Gluconobacter oxydans IFO 3244, a new enzyme catalyzing extracellular protocatechuate formation. Biosci. Biotech. Biochem. 2010, 74, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, V.; Simic, P.; Mouncey, N.J.; Bringer, S.; Sahm, H.; Bott, M. Metabolic engineering of Gluconobacter oxydans for improved growth rate and growth yield on glucose by elimination of gluconate formation. Appl. Environ. Microbiol. 2010, 76, 4369–4376. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, P.; Volland, S.; Deppenmeier, U. Overproduction and characterization of two distinct aldehyde-oxidizing enzymes from Gluconobacter oxydans 621H. J. Mol. Microbiol. Biotechnol. 2007, 13, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, P.; Deppenmeier, U. Analysis of aldehyde reductases from Gluconobacter oxydans 621H. Appl. Microbiol. Biotechnol. 2010, 85, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Rohe, P.; Venkanna, D.; Kleine, B.; Freudl, R.; Oldiges, M. An automated workflow for enhancing microbial bioprocess optimization on a novel microbioreactor platform. Microb. Cell Fact. 2012, 11, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Wiechert, W.; Nöh, K. From stationary to instationary metabolic flux analysis. In Advances in Biochemical Engineering/Biotechnology; Springer-Verlag GmbH: Heidelberg, Germany, 2005; Volume 92, pp. 145–172. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ostermann, S.; Richhardt, J.; Bringer, S.; Bott, M.; Wiechert, W.; Oldiges, M. 13C Tracers for Glucose Degrading Pathway Discrimination in Gluconobacter oxydans 621H. Metabolites 2015, 5, 455-474. https://doi.org/10.3390/metabo5030455
Ostermann S, Richhardt J, Bringer S, Bott M, Wiechert W, Oldiges M. 13C Tracers for Glucose Degrading Pathway Discrimination in Gluconobacter oxydans 621H. Metabolites. 2015; 5(3):455-474. https://doi.org/10.3390/metabo5030455
Chicago/Turabian StyleOstermann, Steffen, Janine Richhardt, Stephanie Bringer, Michael Bott, Wolfgang Wiechert, and Marco Oldiges. 2015. "13C Tracers for Glucose Degrading Pathway Discrimination in Gluconobacter oxydans 621H" Metabolites 5, no. 3: 455-474. https://doi.org/10.3390/metabo5030455
APA StyleOstermann, S., Richhardt, J., Bringer, S., Bott, M., Wiechert, W., & Oldiges, M. (2015). 13C Tracers for Glucose Degrading Pathway Discrimination in Gluconobacter oxydans 621H. Metabolites, 5(3), 455-474. https://doi.org/10.3390/metabo5030455