
Early Diagnosis of Bloodstream Infections Using Serum Metabolomic Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Blood Sample Collection
2.2. Bacterial Identification and Antibiotic Susceptibility Testing (AST)
2.3. Sample Preparation for Metabolomic Analysis
2.4. Metabolomic Analysis
2.5. Metabolomic Data Processing
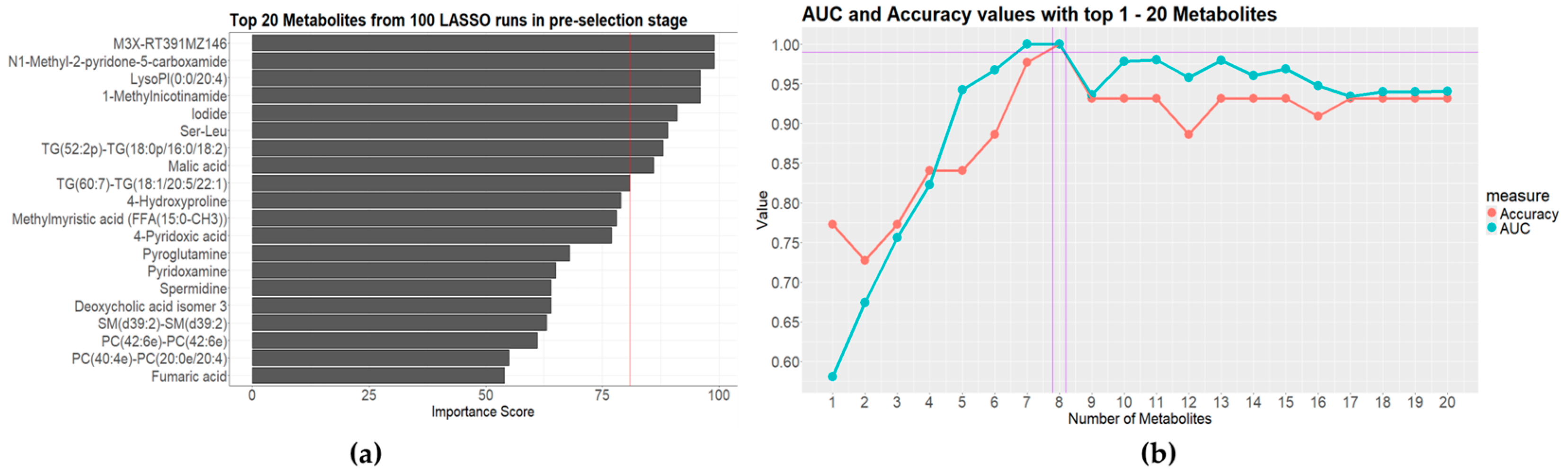
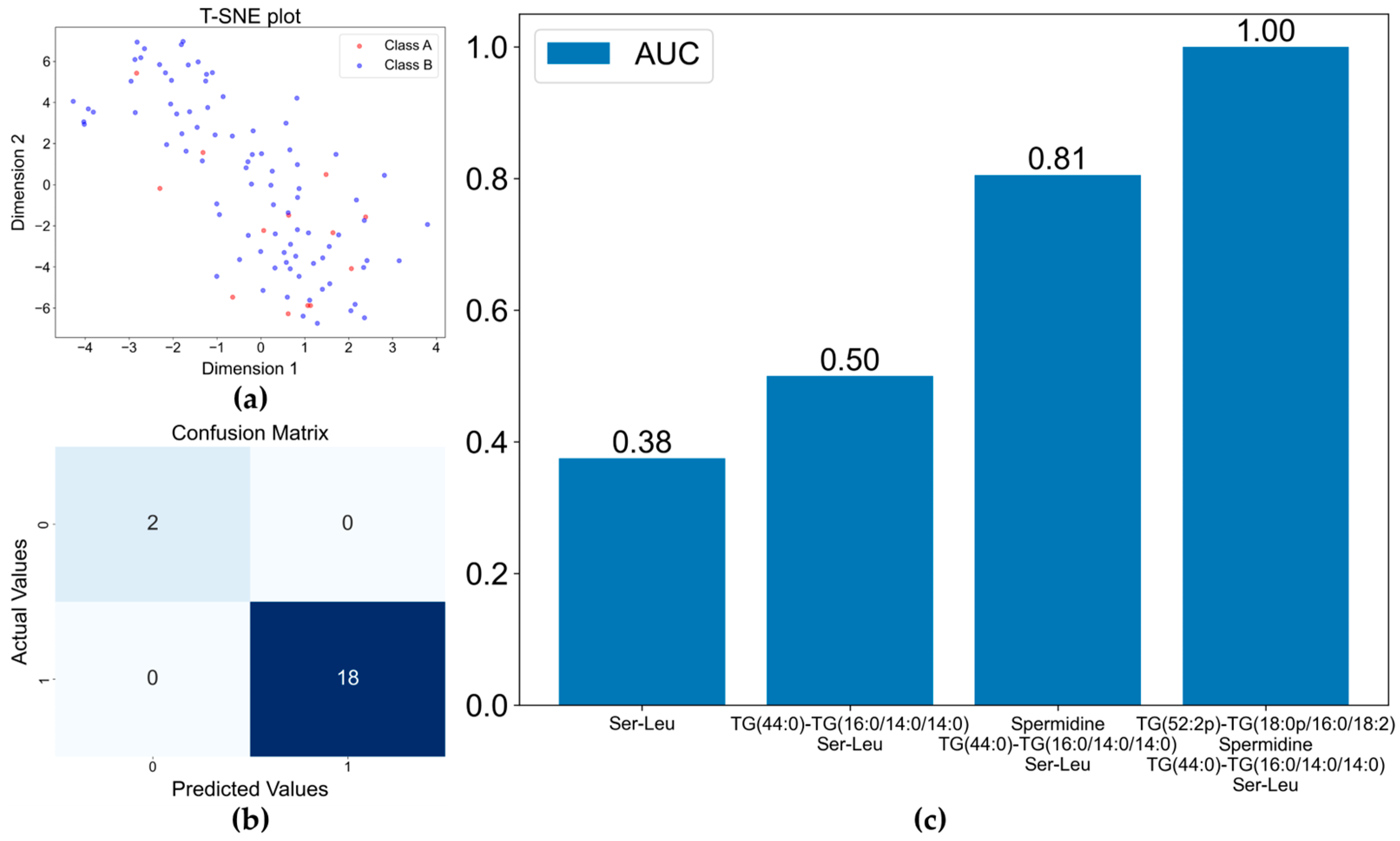
2.6. Statistical Analysis
3. Results
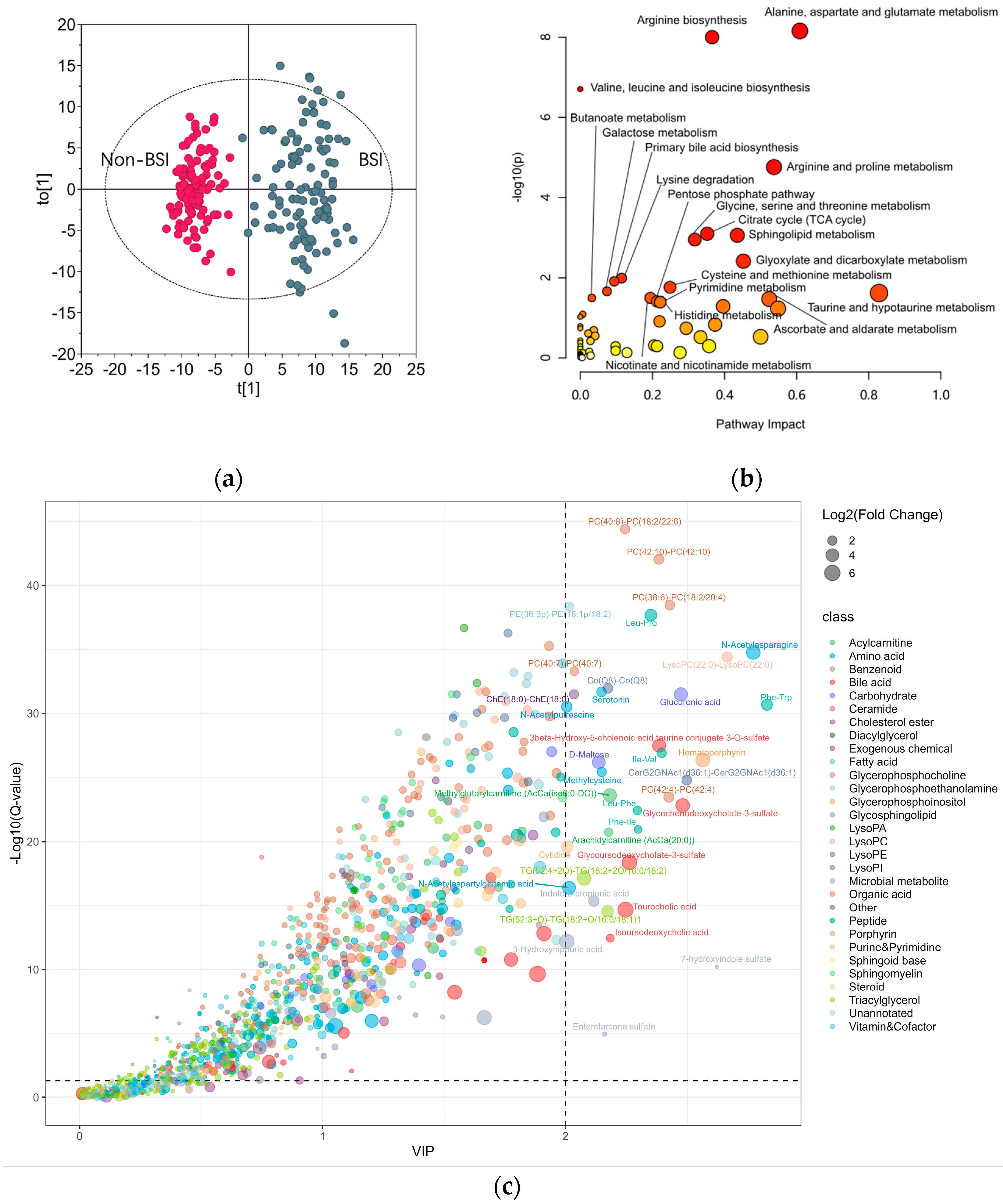
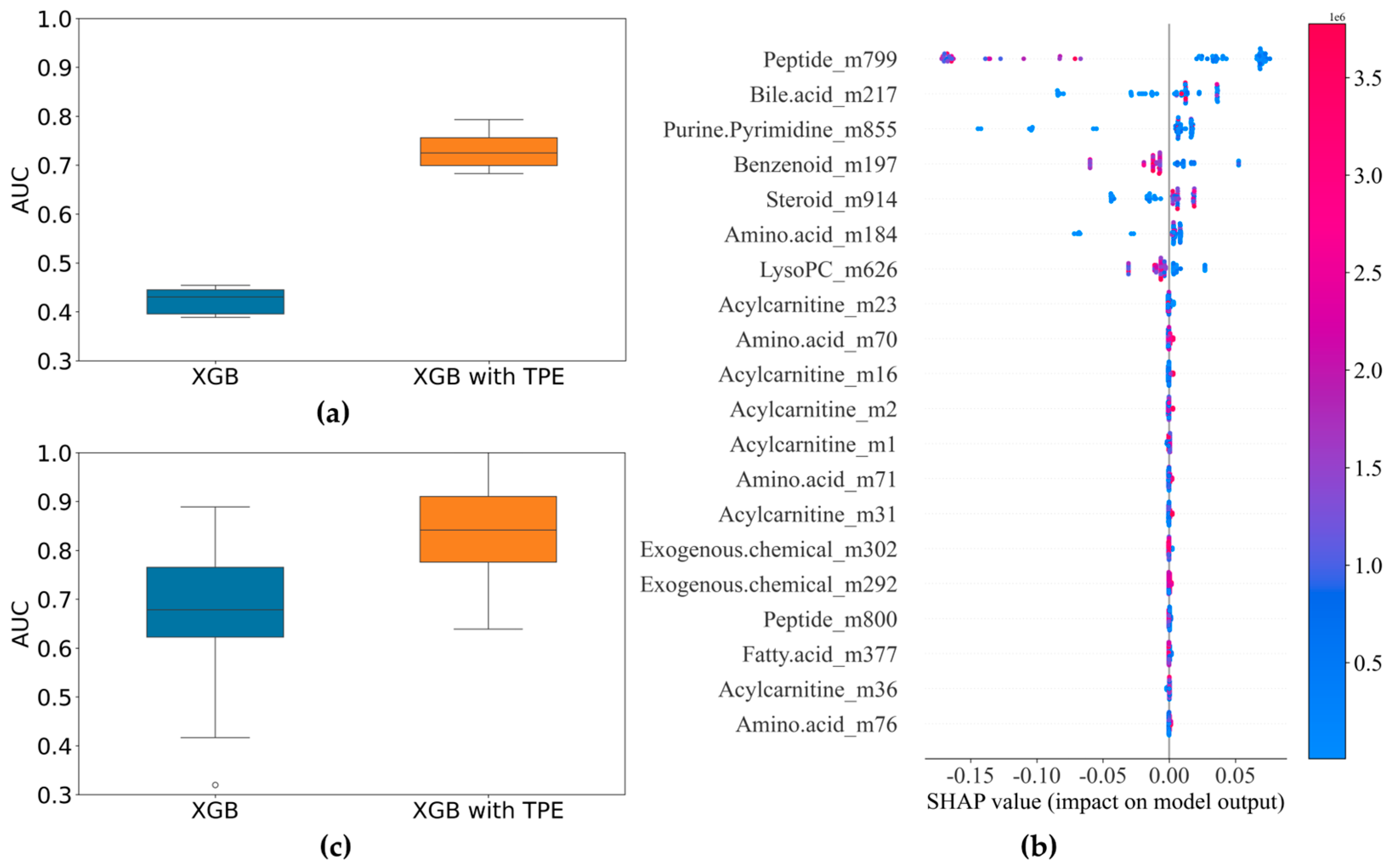
3.1. Differentation Between BSI and Non-BSI Cases
3.2. Subgroup Differentiation of BSI Cases
3.2.1. Classification of the Pathogens into Four Groups
3.2.2. Differentiation Between Gram-Positive and Gram-Negative Bacteria
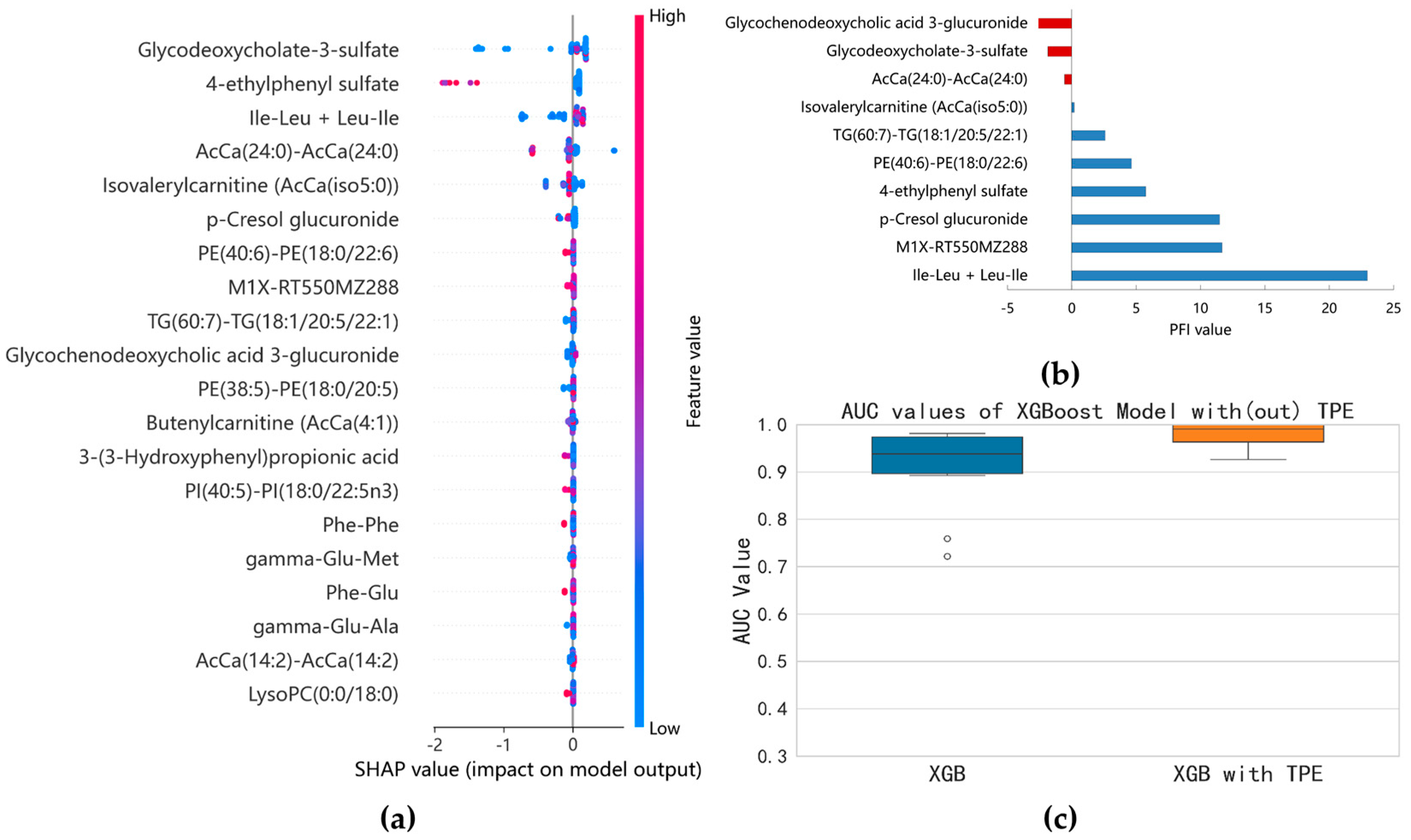
3.2.3. Identification of E. coli and K. pneumoniae BSIs
3.2.4. Identification of ESBLs(−) and ESBLs(+) Cases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Q.; Liu, X.; Hu, B.; Xu, H.; Sun, R.; Li, P.; Zhang, Y.; Yang, H.; Ma, N.; Sun, X. Diagnostic performance and clinical impact of blood metagenomic next-generation sequencing in ICU patients suspected monomicrobial and polymicrobial bloodstream infections. Front. Cell. Infect. Microbiol. 2023, 13, 1192931. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Shi, W.; Wen, Y.; Mao, E.; Ni, T. Comparison of pathogen detection consistency between metagenomic next-generation sequencing and blood culture in patients with suspected bloodstream infection. Sci. Rep. 2023, 13, 9460. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, O.; De Geer, L.; Chew, M.S. Nonadherence to antibiotic guidelines in patients admitted to ICU with sepsis is associated with increased mortality: A registry-based, retrospective cohort study. Eur. J. Anaesthesiol. 2020, 37, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Al-Hasan, M.N. Overall burden of bloodstream infection and nosocomial bloodstream infection in North America and Europe. Clin. Microbiol. Infect. 2013, 19, 501–509. [Google Scholar] [CrossRef]
- Kern, W.V.; Rieg, S. Burden of bacterial bloodstream infection-a brief update on epidemiology and significance of multidrug-resistant pathogens. Clin. Microbiol. Infect. 2020, 26, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Opota, O.; Croxatto, A.; Prod’hom, G.; Greub, G. Blood culture-based diagnosis of bacteraemia: State of the art. Clin. Microbiol. Infect. 2015, 21, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Rutanga, J.P.; Nyirahabimana, T. Clinical Significance of Molecular Diagnostic Tools for Bacterial Bloodstream Infections: A Systematic Review. Interdiscip. Perspect. Infect. Dis. 2016, 2016, 6412085. [Google Scholar] [CrossRef]
- Weinstein, M.P.; Towns, M.L.; Quartey, S.M.; Mirrett, S.; Reimer, L.G.; Parmigiani, G.; Reller, L.B. The clinical significance of positive blood cultures in the 1990s: A prospective comprehensive evaluation of the microbiology, epidemiology, and outcome of bacteremia and fungemia in adults. Clin. Infect.Dis. Off. Publ. Infect. Dis. Soc. Am. 1997, 24, 584–602. [Google Scholar] [CrossRef]
- Hou, T.Y.; Chiang-Ni, C.; Teng, S.H. Current status of MALDI-TOF mass spectrometry in clinical microbiology. J. Food Drug Anal. 2019, 27, 404–414. [Google Scholar] [CrossRef]
- Peri, A.M.; Harris, P.N.A.; Paterson, D.L. Culture-independent detection systems for bloodstream infection. Clin. Microbiol. Infect. 2022, 28, 195–201. [Google Scholar] [CrossRef]
- Campion, M.; Scully, G. Antibiotic Use in the Intensive Care Unit: Optimization and De-Escalation. J. Intensive Care Med. 2018, 33, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.; Shani, V.; Muchtar, E.; Kariv, G.; Robenshtok, E.; Leibovici, L. Systematic review and meta-analysis of the efficacy of appropriate empiric antibiotic therapy for sepsis. Antimicrob. Agents Chemother. 2010, 54, 4851–4863. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, R.P.; Levy, M.M.; Rhodes, A.; Annane, D.; Gerlach, H.; Opal, S.M.; Sevransky, J.E.; Sprung, C.L.; Douglas, I.S.; Jaeschke, R.; et al. Surviving Sepsis Campaign: International guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med. 2013, 39, 165–228. [Google Scholar] [CrossRef] [PubMed]
- Opota, O.; Jaton, K.; Greub, G. Microbial diagnosis of bloodstream infection: Towards molecular diagnosis directly from blood. Clin. Microbiol. Infect. 2015, 21, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Samuel, L. Direct-from-Blood Detection of Pathogens: A Review of Technology and Challenges. J. Clin. Microbiol. 2023, 61, e0023121. [Google Scholar] [CrossRef] [PubMed]
- Dubourg, G.; Raoult, D. Emerging methodologies for pathogen identification in positive blood culture testing. Expert Rev. Mol. Diagn. 2016, 16, 97–111. [Google Scholar] [CrossRef]
- Samuel, L. Direct Detection of Pathogens in Bloodstream During Sepsis: Are We There Yet? J. Appl. Lab. Med. 2019, 3, 631–642. [Google Scholar] [CrossRef]
- Peng, J.M.; Du, B.; Qin, H.Y.; Wang, Q.; Shi, Y. Metagenomic next-generation sequencing for the diagnosis of suspected pneumonia in immunocompromised patients. J. Infect. 2021, 82, 22–27. [Google Scholar] [CrossRef]
- Su, L.D.; Chiu, C.Y.; Gaston, D.; Hogan, C.A.; Miller, S.; Simon, D.W.; Thakur, K.T.; Yang, S.; Piantadosi, A. Clinical Metagenomic Next-Generation Sequencing for Diagnosis of Central Nervous System Infections: Advances and Challenges. Mol. Diagn. Ther. 2024, 28, 513–523. [Google Scholar] [CrossRef]
- Kontula, K.S.K.; Skogberg, K.; Ollgren, J.; Järvinen, A.; Lyytikäinen, O. Population-Based Study of Bloodstream Infection Incidence and Mortality Rates, Finland, 2004–2018. Emerg. Infect. Dis. 2021, 27, 2560–2569. [Google Scholar] [CrossRef]
- Peker, N.; Couto, N.; Sinha, B.; Rossen, J.W. Diagnosis of bloodstream infections from positive blood cultures and directly from blood samples: Recent developments in molecular approaches. Clin. Microbiol. Infect. 2018, 24, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Heron, M. Deaths: Leading Causes for 2014. Natl. Vital Stat. Rep. Cent. Dis. Control Prev. Natl. Cent. Health Stat. Natl. Vital Stat. Syst. 2016, 65, 1–96. [Google Scholar]
- Mickiewicz, B.; Thompson, G.C.; Blackwood, J.; Jenne, C.N.; Winston, B.W.; Vogel, H.J.; Joffe, A.R. Development of metabolic and inflammatory mediator biomarker phenotyping for early diagnosis and triage of pediatric sepsis. Crit. Care 2015, 19, 320. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Li, W.; Sun, L.; Lin, Z.; Jiang, Y.; Ling, Y.; Lin, X. Comparative metabolomics shows the metabolic profiles fluctuate in multi-drug resistant Escherichia coli strains. J. Proteom. 2019, 207, 103468. [Google Scholar] [CrossRef]
- Jaurila, H.; Koivukangas, V.; Koskela, M.; Gäddnäs, F.; Myllymaa, S.; Kullaa, A.; Salo, T.; Ala-Kokko, T.I. 1H NMR Based Metabolomics in Human Sepsis and Healthy Serum. Metabolites 2020, 10, 70. [Google Scholar] [CrossRef]
- Mayers, J.R.; Varon, J.; Zhou, R.R.; Daniel-Ivad, M.; Beaulieu, C.; Bhosle, A.; Glasser, N.R.; Lichtenauer, F.M.; Ng, J.; Vera, M.P.; et al. A metabolomics pipeline highlights microbial metabolism in bloodstream infections. Cell 2024, 187, 4095–4112.e4021. [Google Scholar] [CrossRef]
- Du, B.; Ding, D.; Ma, C.; Guo, W.; Kang, L. Locust density shapes energy metabolism and oxidative stress resulting in divergence of flight traits. Proc. Natl. Acad. Sci. USA 2022, 119, e2115753118. [Google Scholar] [CrossRef]
- Jia, L.; Yang, J.; Zhu, M.; Pang, Y.; Wang, Q.; Wei, Q.; Li, Y.; Li, T.; Li, F.; Wang, Q.; et al. A metabolite panel that differentiates Alzheimer’s disease from other dementia types. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2022, 18, 1345–1356. [Google Scholar] [CrossRef]
- Burnham, C.D.; Yarbrough, M.L. Best Practices for Detection of Bloodstream Infection. J. Appl. Lab. Med. 2019, 3, 740–742. [Google Scholar] [CrossRef]
- Saifi, S.; Ashraf, A.; Hasan, G.M.; Shamsi, A.; Hassan, M.I. Insights into the preventive actions of natural compounds against Klebsiella pneumoniae infections and drug resistance. Fitoterapia 2024, 173, 105811. [Google Scholar] [CrossRef]
- Perez-Nadales, E.; Fernandez-Ruiz, M.; Gutierrez-Gutierrez, B.; Pascual, A.; Rodriguez-Bano, J.; Martinez-Martinez, L.; Aguado, J.M.; Torre-Cisneros, J. Extended-spectrum beta-lactamase-producing and carbapenem-resistant Enterobacterales bloodstream infection after solid organ transplantation: Recent trends in epidemiology and therapeutic approaches. Transpl. Infect. Dis. 2022, 24, e13881. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xiao, C.; Guan, P.; Chen, Q.; You, L.; Kong, H.; Qin, W.; Dou, P.; Li, Q.; Li, Y.; et al. Metabolomics acts as a powerful tool for comprehensively evaluating vaccines approved under emergency: A CoronaVac retrospective study. Front. Immunol. 2023, 14, 1168308. [Google Scholar] [CrossRef] [PubMed]
- Pudjihartono, N.; Fadason, T.; Kempa-Liehr, A.W.; O’Sullivan, J.M. A Review of Feature Selection Methods for Machine Learning-Based Disease Risk Prediction. Front. Bioinform. 2022, 2, 927312. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Mansmann, U.; Du, S.; Hornung, R. Benchmark study of feature selection strategies for multi-omics data. BMC Bioinform. 2022, 23, 412. [Google Scholar] [CrossRef]
- Huang, D.; Li, R.; Wang, H. Feature Screening for Ultrahigh Dimensional Categorical Data with Applications. J. Bus. Econ. Stat. 2014, 32, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Samworth, R.; Wu, Y. Ultrahigh dimensional feature selection: Beyond the linear model. J. Mach. Learn. Res. 2009, 10, 2013–2038. [Google Scholar]
- Gatti, M.; Bonazzetti, C.; Tazza, B.; Pascale, R.; Miani, B.; Malosso, M.; Beci, G.; Marzolla, D.; Rinaldi, M.; Viale, P.; et al. Impact on clinical outcome of follow-up blood cultures and risk factors for persistent bacteraemia in patients with gram-negative bloodstream infections: A systematic review with meta-analysis. Clin. Microbiol. Infect. 2023, 29, 1150–1158. [Google Scholar] [CrossRef]
- Nishio, A.; Rehermann, B. Virus-Induced Interferon Regulates the Urea Cycle. Immunity 2019, 51, 975–977. [Google Scholar] [CrossRef]
- Sanchez-Garcia, F.J.; Perez-Hernandez, C.A.; Rodriguez-Murillo, M.; Moreno-Altamirano, M.M.B. The Role of Tricarboxylic Acid Cycle Metabolites in Viral Infections. Front. Cell. Infect. Microbiol. 2021, 11, 725043. [Google Scholar] [CrossRef]
- Dawrs, S.N.; Virdi, R.; Islam, M.N.; Hasan, N.A.; Norton, G.J.; Crooks, J.L.; Parr, J.; Heinz, D.; Cool, C.D.; Belisle, J.T.; et al. Immunological and metabolic characterization of environmental Mycobacterium chimaera infection in a murine model. Microbes Infect. 2023, 25, 105184. [Google Scholar] [CrossRef]
- Mohammed, S.; Bindu, A.; Viswanathan, A.; Harikumar, K.B. Sphingosine 1-phosphate signaling during infection and immunity. Prog. Lipid Res. 2023, 92, 101251. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xie, Y.; Gao, P.; Zhang, S.; Tan, H.; Yang, F.; Lian, R.; Tian, J.; Xu, G. A metabolomics-based method for studying the effect of yfcC gene in Escherichia coli on metabolism. Anal. Biochem. 2014, 451, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, S.A.; Gardner, C.; Das, S. Diagnosis and Management of Bloodstream Infections With Rapid, Multiplexed Molecular Assays. Front. Cell. Infect. Microbiol. 2022, 12, 859935. [Google Scholar] [CrossRef] [PubMed]
- Walczak-Skierska, J.; Monedeiro, F.; Maslak, E.; Zloch, M. Lipidomics Characterization of the Microbiome in People with Diabetic Foot Infection Using MALDI-TOF MS. Anal. Chem. 2023, 95, 16251–16262. [Google Scholar] [CrossRef]
- Salawudeen, A.; Raji, Y.E.; Jibo, G.G.; Desa, M.N.M.; Neoh, H.M.; Masri, S.N.; Di Gregorio, S.; Jamaluddin, T. Epidemiology of multidrug-resistant Klebsiella pneumoniae infection in clinical setting in South-Eastern Asia: A systematic review and meta-analysis. Antimicrob. Resist. Infect. Control 2023, 12, 142. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, S.; Li, R.; Wang, H.; Wang, L.; Gao, Y.; Wen, Y.; Gong, T.; Ruan, S.; Li, H.; Gao, P. Early Diagnosis of Bloodstream Infections Using Serum Metabolomic Analysis. Metabolites 2024, 14, 685. https://doi.org/10.3390/metabo14120685
Han S, Li R, Wang H, Wang L, Gao Y, Wen Y, Gong T, Ruan S, Li H, Gao P. Early Diagnosis of Bloodstream Infections Using Serum Metabolomic Analysis. Metabolites. 2024; 14(12):685. https://doi.org/10.3390/metabo14120685
Chicago/Turabian StyleHan, Shuang, Ruihua Li, Hao Wang, Lin Wang, Yiming Gao, Yaolin Wen, Tianyang Gong, Shiyu Ruan, Hui Li, and Peng Gao. 2024. "Early Diagnosis of Bloodstream Infections Using Serum Metabolomic Analysis" Metabolites 14, no. 12: 685. https://doi.org/10.3390/metabo14120685
APA StyleHan, S., Li, R., Wang, H., Wang, L., Gao, Y., Wen, Y., Gong, T., Ruan, S., Li, H., & Gao, P. (2024). Early Diagnosis of Bloodstream Infections Using Serum Metabolomic Analysis. Metabolites, 14(12), 685. https://doi.org/10.3390/metabo14120685