Abstract
Hypertrophic (HCM) and dilated (DCM) cardiomyopathies are among the leading causes of sudden cardiac death. We identified 38 pathogenic or likely pathogenic variant carriers for HCM in three sarcomere genes (MYH7, MYBPC3, TPMI) among 9.928 participants of the METSIM Study having whole exome sequencing data available. Eight of them had a clinical diagnosis of HCM. We also identified 20 pathogenic or likely pathogenic variant carriers for DCM in the TTN gene, and six of them had a clinical diagnosis of DCM. The aim of our study was to investigate the metabolite signature in the carriers of the pathogenic or likely pathogenic genetic variants for HCM and DCM, compared to age- and body-mass-index-matched controls. Our novel findings were that the carriers of pathogenic or likely pathogenic variants for HCM had significantly increased concentrations of bradykinin (des-arg 9), vanillactate, and dimethylglycine and decreased concentrations of polysaturated fatty acids (PUFAs) and lysophosphatidylcholines compared with the controls without HCM. Additionally, our novel findings were that the carriers of pathogenic or likely pathogenic variants for DCM had significantly decreased concentrations of 1,5-anhydrogluticol, histidine betaine, N-acetyltryptophan, and methylsuccinate and increased concentrations of trans-4-hydroxyproline compared to the controls without DCM. Our population-based study shows that the metabolite signature of the genetic variants for HCM and DCM includes several novel metabolic pathways not previously described.
1. Introduction
Cardiomyopathies are among the leading causes of sudden cardiac death. The prevalence of hypertrophic cardiomyopathy (HCM) has been estimated to be 1 in 500 and dilated cardiomyopathy (DCM) 1 in 2500 individuals worldwide [1,2,3]. The definition of HCM is based on an increase in the left ventricular (LV) wall thickness ≥15 mm in the absence of other causes of LV hypertrophy such as hypertension, valvular diseases, or coronary artery disease [3]. DCM is defined as a left ventricular dilatation that leads to systolic dysfunction in the absence of any other abnormal loading conditions or coronary artery disease [3].
HCM and DCM have autosomal dominant inheritance with strong phenotypic heterogeneity and incomplete penetrance [4,5]. Mutations in the genes encoding sarcomeric proteins (ACTC1, MYBPC3, MYH7, MYL2, MYL3, TNNI3, TNNT2, TPM1) are the most important genes for HCM. Very rarely (<2%) mutations in non-sarcomeric genes cause HCM [6]. We previously found pathogenic or likely pathogenic mutations in 38% of 382 clinically diagnosed patients with HCM [7]. Other studies have found causal variants in 30–60% of the patients with HCM [8]. Finnish three founder mutations, Gln1061Ter of MYBPC3, Arg1053Gln of MYH7 and Asp175Asn of TPM1, and a prevalent mutation Val606Met of MYH7 account for 28% of HCM cases in Finns [7]. Genes that are causing DCM encode components of sarcomere (TTN, MYH7, TNNT2, TNNI3, TPM1, ACTC1), sarcomere associated proteins (PLN, BAG3), nuclear membrane (LMNA, EMD), cytoskeleton (DES), outer cellular membrane, extracellular matrix (DMD), ion channels (SCN5A), mitochondrial proteins (TAZ, DNAJC19), and splice-regulating proteins (RBM20) [8]. Pathogenic or likely pathogenic variants explain about 30% of the cases of DCM. Importantly, HCM and DCM are not defined by specific genetic mutations, but by specific morphological and functional alterations in the heart [9,10].
Myocardial energy metabolism is changed in cardiomyopathies. HCM is caused by altered biophysical properties of cardiomyocytes, disturbed calcium handling and abnormal cellular metabolism [11,12]. The primary defect is a sarcomere mutation, but clinical phenotypes are determined by genetic, epigenetic, and environmental factors [9]. A hallmark of pathological cardiac hypertrophy in patients with HCM is the reversion to fetal gene expression associated with reductions in fatty acid oxidation and an increase in glucose utilization by an enhancement of glucose uptake and glycolysis [12].
Metabolomics allows a detailed characterization of metabolic phenotypes and enables precision medicine approach including the characterization of metabolic derangements that underlie disease, discovery of new therapeutic targets and biomarkers that may be used to diagnose a disease or monitor the course of therapy [9,13,14]. Application of metabolomics in studies of cardiomyopathies is still limited although the current methods based on mass spectrometry allow the screening over 1000 plasma metabolites. An advantage of studying plasma metabolites as biomarkers for HCM and DCM is that cardiomyopathies have a well-established genetic basis, and that cardiovascular risk factors do not play a major role in the risk of these diseases. Our large Finnish population-based cohort the Metabolic Syndrome In Men (METSIM) Study [15] including 10,197 men is ideal to investigate metabolite signature in carriers of pathogenic variants for cardiomyopathies.
2. Results
2.1. Identification of Genetic Variants for HCM and DCM
We identified the pathogenic or likely pathogenic variants for HCM in our exome sequencing data in the following eight genes associated with HCM based on previous studies: ACTC1, MYBPC3, MYH7, MYL2, MYL3, TNNI3, TNNT2, and TPM1 [16]. We found 38 pathogenic or likely pathogenic genetic variants for HCM in three sarcomere genes. For each carrier of pathogenic or likely pathogenic genetic variants for HCM, we selected five age- and BMI-matched controls.
There were no statistically significant differences in clinical and laboratory measurements between cases and controls (Table 1). The prevalence of the pathogenic variants for HCM was 0.37% of the participants, suggesting that 1 of 270 participants was a carrier of a pathogenic variant for HCM in our population (Table 2). The most frequent pathogenic genetic variant was Arg1053Gln of MYH7, found in 24 participants. The frequencies of other pathogenic variant carriers were low: Arg941His of MYH7 (n = 4), Gln1061Ter of MYBPC3 (n = 3), c.655-2A > C of MYBPC3 (n = 4), Gly853fs of MYBPC3 (n = 1), and Asp175Asn of TPM1 (n = 2).

Table 1.
Clinical and laboratory measurements in the participants with HCM and DCM and corresponding controls.
Table 2.
Pathogenic or likely pathogenic genetic variants for HCM and DCM in the METSIM study.
Eight of thirty-six participants had a clinical diagnosis of HCM based on Kuopio University Hospital medical records. We compared the metabolite signature between participants having a clinical diagnosis of HCM (n = 8) with participants who did not have a clinical diagnosis of HCM (n = 28). We did not find statistically significant differences in metabolite concentrations between these groups.
We screened the pathogenic or likely pathogenic variants for DCM in our exome sequencing data in the following sixteen genes associated with DCM in previous studies: ACTC1, BAG3, DES, DMD, DNAJC19, EMD, LMNA, MYH7, PLN, RBM20, SCN5A, TAZ, TNNI3, TNNT2, TPM1, and TTN [8,17]. We included in our analyses only the variants located in the protein coding regions or in canonical splice sites. We found 20 pathogenic or likely pathogenic genetic variants only in the TTN gene (Table 2). These variants caused terminal codon in fifteen cases; one was a splice variant, and four were pathogenic duplications. For each carrier of the pathogenic or likely pathogenic genetic variants for DCM, we selected five age- and BMI-matched controls. There were no statistically significant differences in clinical and laboratory measurements between the cases and controls (Table 1). The prevalence of pathogenic variants for DCM was 0.20%, suggesting that 1 of 500 participants was a carrier of a pathogenic variant.
2.2. Identification of Metabolites Associated with HCM Pathogenic Variants
We found 23 novel metabolites associated with the HCM pathogenic variants. Compared to the age- and BMI-matched controls, the carriers of these variants had increased concentrations of metabolites in the amino acid pathway, including dimethylglycine, vanillactate, and des-Arg 9-bradykinin. We also found that the carriers of the HCM variants had decreased concentrations of 17 lipids, especially fatty acids and lysophosphatidylcholines, compared to controls (Table 3).
Table 3.
Statistically significant differences in metabolite concentrations between the carriers (Cases) and non-carriers (Controls) of the pathogenic or likely pathogenic HCM variants.
2.3. Identification of Metabolites Associated with DCM Pathogenic Variants
We found that the carriers of the DCM pathogenic variants had six novel associations with metabolites belonging to the carbohydrate, lipid, and amino acid pathways. We found decreased concentrations of 1,5-anhydrogluticol (1,5-AG, glycolysis pathway), bilirubin (lipid pathway), histidine betaine, N-acetyltryptophan, and methylsuccinate (amino acid pathway) in the carriers of the DCM pathogenic variants compared to the correspondent controls. We also found increased concentrations of trans-4-hydroxyproline and confirmed a previously reported association with homoarginine in the carriers of DCM pathogenic variants compared to the controls (Table 4).
Table 4.
Statistically significant differences in metabolite concentrations between the carriers (Cases) and non-carriers (Controls) of the pathogenic or likely pathogenic DCM variants.
Six of twenty participants had a clinical diagnosis of DCM based on Kuopio University Hospital medical records. We compared the metabolite signature between participants with a clinical diagnosis of DCM (n = 8) with participants without a clinical diagnosis of DCM (n = 28) but did not find statistically significant differences in metabolite concentrations between these groups.
3. Discussion
We identified 38 pathogenic or likely pathogenic genetic variants for HCM in three sarcomere genes (MYH7 MYBPC3, TPM1) among 9928 participants in a population-based METSIM study. Only eight of them had clinically diagnosed HCM based on Kuopio University Hospital medical records. We found 20 pathogenic or likely pathogenic genetic variants in the TTN gene for DCM, and almost all of them were stop codon or frameshift variants, in agreement with previously published studies [18,19]. Six of twenty participants had a clinical diagnosis of DCM.
Our study was based only on the carriers of pathogenic or likely pathogenic genetic variants for HCM and DCM, and therefore, it is likely that these individuals had clinically mild cases of cardiomyopathies. This makes our study population more homogenous than studies based on the clinical diagnosis of cardiomyopathies and gives an excellent opportunity to investigate the effects of HCM and DCM variants on the metabolic signature of these diseases.
Previous metabolomics studies in HCM have been heterogenous, comparing the metabolite signature between obstructive and nonobstructive HCM [20,21], or focusing on myocardial biopsies [12] or the metabolomics profile of the carriers of a single mutation [22]. Our study focuses on the carriers of pathogenic variants causing HCM who were carefully matched for the controls with respect to age and BMI. Our control groups were five times larger than cardiomyopathy groups, increasing the power of the statistical analyses.
We found statistically significant differences between the HCM variant carriers and controls in several metabolites belonging to the amino acid, lipid, and carbohydrate classes (Figure 1). Our novel findings were that we found increased concentrations of des-arg(9)-bradykinin, vanillactate, and dimethylglycine in participants with the pathogenic HCM variants compared to the controls. Des-arg(9)-bradykinin is an active metabolite of bradykinin and stimulates β1 receptors [23]. The β-adrenergic signaling pathway is one of the pathways that mediates cardiac hypertrophy and plays a central role in the pathophysiology of heart failure [24,25]. Chronic stimulation of β1 receptors induces cardiac hypertrophy in in vitro experiments and in animal studies by activating adenylyl cyclase via Gs proteins [26]. Consequently, the cellular concentration of cyclic AMP (cAMP) increases and stimulates protein kinase A, which modulates the calcium channels [27]. Human studies have shown that chronically elevated adrenergic signaling leads to altered calcium homeostasis in the failing hypertrophied cardiac myocyte and results in an increase in intracellular calcium concentrations [28,29]. Studies based on human heart biopsies have shown that calcium-activated gene expression leads to an induction of a ‘fetal’ gene expression profile for the contractile proteins and directly contributes to contractile dysfunction and pathologic hypertrophy in the heart [28,29].
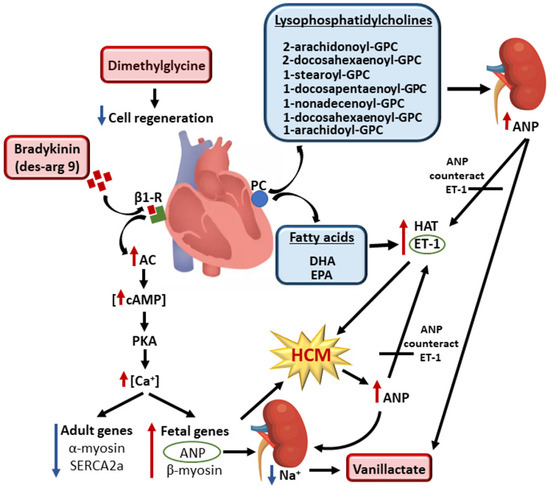
Figure 1.
Metabolites involved in the cell signaling that regulates hypertrophy of cardiomyocytes in the carriers of pathogenic or likely pathogenic genetic variants for HCM. Red boxes indicate increased concentrations and blue boxes decreased concentrations of the metabolites in the carriers of genetic variants compared to the controls. Bradykinin activates β1-receptors, resulting in high intracellular concentrations of calcium and suppression of adult and activation of fetal gene expression. This leads to enhanced cardiomyocyte contractility, impaired relaxation, induced hypertrophic remodeling, and a release of ANP as a response to hemodynamic loading. Increased ANP concentrations lead to an increase in sodium excretion, which increases vanillactate concentrations. Hydrolysis of PC generates lysophosphatidylcholines (LPC) and fatty acids. A decrease in lysophosphatidylcholine concentration increases ANP concentrations. Low concentrations of EPA and DHA activate HAT and ET-1, triggering hypertrophy of cardiomyocytes. Released ANP antagonizes ET-1 and restores hemodynamic stability. Dimethylglycine impairs cell regeneration and contributes to the development of HCM. Abbreviations: AC, adenyl cyclase; ANP, atrial natriuretic peptide; β1-R, β1 receptor; Ca, calcium; cAMP, cyclic adenosine monophosphate; DHA, docosahexaenoate; EPA, eicosapentaenoate; ET-1, endothelin 1; GPC, glycerophospatidylcholine; HAT, histone acyltransferase; Na, sodium; LPC, lysophosphatidylcholine; PC, phosphatidylcholine; PKA, protein kinase A; SERCA2a, Ca2+-ATPase 2a.
The fetal pattern of gene expression includes an upregulation of the fetal genes, β-myosin heavy chain and atrial natriuretic peptide (ANP), and a downregulation of the adult genes, α-myosin heavy chain and sarcoendoplasmic reticulum Ca2+-ATPase 2a (SERCA2a) [29,30]. ANP is secreted by the heart in response to volume expansion and leads to an increase in sodium excretion [31]. In agreement with this notion, we found increased concentrations of vanillactate, a metabolite that has previously been shown to be upregulated by sodium reduction in hypertensive subjects [32]. We also found elevated concentrations of dimethylglycine (DMG) in participants carrying pathogenic variants for HCM (Figure 1). Apoptosis of cardiomyocytes seems partly to depend on mitochondrial processes [33], and DMG metabolism inside the mitochondria may influence the production of nucleotides [34], potentially affecting the cell’s regenerative abilities.
We found that participants carrying the pathogenic HCM genetic variants showed predominantly decreased concentrations of long-chain polyunsaturated fatty acids (PUFAs) and lysophosphatidylcholines (LPC) containing PUFAs in their acyl chain (Figure 1). We also found decreased concentrations of the PUFAs eicosapentaenoate (EPA), arachidonate, and docosahexaenoate (DHA) in participants carrying pathogenic HCM genetic variants. In vitro studies have shown that EPA and DHA repress hypertrophic responses in cardiomyocytes by the direct inhibition of histone acetyltransferase activity, and that EPA attenuates endothelin 1 (ET-1)-induced cardiomyocyte hypertrophy [35,36].
We also found decreased concentrations of LPCs in the participants carrying pathogenic genetic variants for HCM (Figure 1). Hydrolysis of phosphatidylcholine by phospholipase A2 generates LPC and a fatty acid [37]. LPCs have potent cardiac effects, including inhibition of the release of atrial natriuretic peptide (ANP) [38]. Cardiac hypertrophy is a fundamental process of adaptation to an increased workload due to hemodynamic overload and is known to activate the cardiac ANP system, with a subsequent high plasma concentration as one of the cardiac compensatory mechanisms [31]. Ca2+, together with calcium-calmodulin kinase II, may be one of the most important factors affecting ANP secretion [39]. An increase in intracellular calcium, combined with an increased generation of reactive oxygen species (ROS), leads to the activation of Ca2+/calmodulin kinase II in the hypertrophied heart, triggering the structural deterioration of the myocardium and resulting in contractile dysfunction and arrhythmia in the failing heart [40].
We found that the participants carrying pathogenic genetic variants for DCM had decreased concentrations of 1,5-anhydrogluticol (1,5-AG), a metabolite belonging to the carbohydrate pathway (Figure 2). Previous studies have shown that 1,5-AG concentrations are associated with vascular endothelial dysfunction [41] and that endothelial dysfunction is associated with the development of DCM [42]. We found that bilirubin concentration was decreased in carriers of the pathogenic DCM variants [43]. In agreement with our results, previous studies have shown that bilirubin concentrations are significantly decreased in patients with heart failure [44,45].

Figure 2.
Metabolites involved in the generation of reactive oxygen species, collagen degradation, and cardiac remodeling in the carriers of pathogenic DCM variants. Red boxes indicate increased concentrations and blue boxes decreased concentrations of the metabolites in the carriers of the pathogenic genetic variants compared to the controls. Collagen degradation increases 4-trans-hydroxyproline concentrations and increases the rate of cardiac remodeling, leading to DCM. Decreased levels of histidine betaine lead to low concentrations of antioxidant ergothioneine, which increases the generation of ROS. ROS, in turn, increases collagen degradation and plays a role in endothelial dysfunction, which is also aggravated by decreased concentrations of 1,5 AG. Low concentrations of homoarginine decreases cardiomyocytes function and increases the rate of cardiac remodeling, triggering the development of DCM. Abbreviations: 1,5 AG, 1,5-anhydroglucitol; DCM, dilated cardiomyopathy; ROS, reactive oxygen species.
In our study, the carriers of a pathogenic DCM variant had decreased concentrations of metabolites belonging to the amino acid pathway, namely, homoarginine, histidine betaine, N-acetyltryptophan, and methylsuccinate (Figure 2). Decreased homoarginine concentration has been associated with dilatation and decreased function of the left ventricle in the general population [46]. Additionally, a previous study in mice has shown that decreased homoarginine concentrations may impair cardiomyocyte function [47]. Histidine betaine is a downstream metabolite of histidine generated by gut bacteria and a precursor of an antioxidant ergothioneine [48,49]. A previous study reported that plasma concentrations of histidine were reduced in patients with primary DCM, as compared to corresponding controls [43].
We found increased concentrations of trans-4-hydroxyproline in the carriers of pathogenic DCM variants. Trans-4-hydroxyproline is a major component of collagen [37], and trans-4-hydroxyproline is used as a parameter of collagen catabolism [37]. Increased ROS formation is also known to accelerate collagen degradation [37].
Overall, our findings indicate that the hypertrophy of cardiomyocytes in carriers of HCM variants is regulated not only by factors predisposing cardiac hypertrophy, but also by factors that attempt to counteract hypertrophy in response to hemodynamic loading (Figure 1). In the carriers of DCM variants, we found a metabolite profile compatible with vascular endothelial dysfunction, a decrease in antioxidant precursors, an increase in ROS generation, an increase in cardiac remodeling, and a high protein turnover in cardiomyocytes, indicating a disturbed collagen metabolism in carriers of DCM variants (Figure 2).
The strength of our study is a large size of our population-based study cohort and detailed analyses of genetic variants and 1098 metabolites. Additionally, we identified several novel metabolites associated with pathogenic genetic variants for HCM and DCM. A major limitation of our study is that both HCM and DCM are rare diseases, and therefore, the prevalence of these diseases is small, which makes it difficult to obtain statistically significant results. To improve the power of our study, we selected five controls for each case of HCM or DCM. Other limitations of our study are that only middle-aged and elderly Finnish men were included in the study. We do not know if the results are valid for women, all age groups, and other ethnic and racial groups. Therefore, our findings need to be replicated in other studies. Finally, our study is an association study that does not allow us to make causal conclusions from our results.
4. Materials and Methods
4.1. Subjects
The METSIM study is a randomly selected, population-based study comprising of 10,197 men recruited from Kuopio and surrounding communities in Eastern Finland, aged from 45 to 73 years at baseline [15]. The METSIM study was approved by the Ethics Committee of the University of Eastern Finland and Kuopio University Hospital and was conducted in accordance with the Declaration of Helsinki. Written informed consent was given by all participants.
Patients with HCM had either echocardiographic or Magnetic Resonance Imaging measurement of a left ventricular wall thickness. All of them had a left ventricular wall thickness at least 15 mm, and they did not have systemic hypertension. All patients with DCM had left ventricular or biventricular systolic dysfunction and dilatation that are not explained by abnormal loading conditions or coronary artery disease [4].
4.2. Whole Exome Sequencing and Classification of Genetic Variants for Cardiomyopathies
A total of 9928 participants of the METSIM study had whole exome sequencing results. The methods of sequencing have been previously described in detail [50]. We identified pathogenic and likely pathogenic genetic variants from known genes causing HCM or DCM and classified these variants according to the ACMG/AMP 2015 guidelines [51]. We validated the results by Sanger sequencing using a BigDye Terminator v1.1 Cycle Sequencing Kit and analysis using 3500XL Genetic Analyzer (Thermo Fisher Scientific Inc., Waltham, MA, USA). TaqMan Allelic Discrimination Assay (Applied Biosystems QuantStudio 5 Real-Time PCR System, Thermo Fisher Scientific Inc., Waltham, MA, USA) was performed to identify the Gln1061Ter variant of MYBPC3.
We included in our analyses only the variants located in the protein coding regions or in canonical splice sites.
4.3. Metabolomics
Non-targeted metabolomics profiling was performed at Metabolon, Inc. (Morrisville, NC, USA) on EDTA plasma samples obtained after an overnight fast from 8679 participants, as previously described in detail [51,52,53]. The Metabolon DiscoveryHD4 platform was applied to assay named metabolites. All samples were processed together for peak quantification and data scaling. We quantified raw mass spectrometry peaks for each metabolite using the area under the curve and evaluated overall process variability by the median relative standard deviation for endogenous metabolites present in all 20 technical replicates in each batch. We adjusted for variation caused by day-to-day instrument tuning differences and columns used for biochemical extraction by scaling the raw peak quantifications to the median for each metabolite by the Metabolon batch. We included 1098 metabolites in the statistical analysis.
4.4. Statistical Analyses
We conducted statistical analyses using SPSS (version 27, IBM Corp., Armonk, NY, USA). Metabolite levels were log-transformed and standardized to a mean of 0 and a standard deviation of 1. We also log-transformed all other variables having a skewed distribution for statistical analyses and present the data for continuous variables as mean ± standard deviation (SD). All metabolite concentrations were compared between the variant carriers and controls by one-way analysis of variance (ANOVA). p < 0.05 was considered statistically significant. Each carrier of a pathogenic or likely pathogenic genetic variant for HCM or DCM was matched with five controls (without a known HCM/DCM variant and disease phenotype) based on age and body mass index (BMI). The fuzz relaxation was 0.25 to the standard deviation (SD) of age (SD = 1.75) and BMI (SD = 1). The iteration was repeated until the desired number of controls were found (the case-to-control ratio 1:5).
5. Conclusions
In conclusion, we identified several novel metabolites associated with the pathogenic or likely pathogenic sarcomere genetic variants for the development of HCM and DCM.
Author Contributions
Formal analysis, writing—original draft, R.R.; formal analysis, writing—original draft, visualization, L.F.S.; formal analysis, writing—review and editing, J.V., M.M., J.R. and S.H.; conceptualization, project administration, writing—review and editing, M.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by grants from the Academy of Finland (321428), the National Institutes of Health (NIH no. 2U01DK062370-15, NIH no. 5R01DK093757-10), the Sigrid Juselius Foundation, the Finnish Foundation for Cardiovascular Research, Kuopio University Hospital, and the Centre of Excellence of Cardiovascular and Metabolic Diseases, supported by the Academy of Finland, no. 2285733 (to M.L.). Academy of Finland, PROFI5, no. 325 022 (to S.H.). GenomMed Doctoral Programme, co-funded by Horizon 2020 Framework Programme of the European Union, Marie Skłodowska-Curie grant agreement no. 740264 (to R.R.).
Institutional Review Board Statement
The study was approved by the Ethics Committee of the Kuopio University Hospital (number: 174/2004; approval: 29 November 2004), and it was conducted in accordance with the Helsinki Declaration.
Informed Consent Statement
All study participants provided written informed consent.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to preserving the confidentiality of the participants.
Conflicts of Interest
The authors declare no conflict of interest.
References
- McKenna, W.J.; Judge, D.P. Epidemiology of the inherited cardiomyopathies. Nat. Rev. Cardiol. 2021, 18, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Semsarian, C.; Ingles, J.; Maron, M.S.; Maron, B.J. New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll Cardiol. 2015, 65, 1249–1254. [Google Scholar] [CrossRef]
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B.; American Heart Association; et al. Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Function. Circulation 2006, 113, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kühl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society of Cardiology Working Group on myocardial and pericardial diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Reichart, D.; Magnussen, C.; Zeller, T.; Blankenberg, S. Dilated cardiomyopathy: From epidemiologic to genetic phenotypes: A translational review of current literature. J. Intern. Med. 2019, 286, 362–372. [Google Scholar] [CrossRef]
- Yotti, R.; Seidman, C.E.; Seidman, J.G. Advances in the genetic basis and pathogenesis of sarcomere cardiomyopathies. Ann. Rev. Genomic. Hum. Genet. 2019, 20, 129–153. [Google Scholar] [CrossRef]
- Jääskeläinen, P.; Vangipurapu, J.; Raivo, J.; Kuulasmaa, T.; Heliö, T.; Aalto-Setälä, K.; Kaartinen, M.; Ilveskoski, E.; Vanninen, S.; Hämäläinen, L.; et al. Genetic basis and outcome in a nationwide study of Finnish patients with hypertrophic cardiomyopathy. ESC Hearth Fail. 2019, 6, 436–445. [Google Scholar] [CrossRef]
- Mestroni, L.; Rocco, C.; Gregori, D.; Sinagra, G.; Di Lenarda, A.; Miocic, S.; Vatta, M.; Pinamonti, B.; Muntoni, F.; Caforio, A.L.; et al. Familial dilated cardiomyopathy: Evidence for genetic and phenotypic heterogeneity. J. Am. Coll Cardiol. 1999, 34, 181–190. [Google Scholar] [CrossRef]
- Stege, N.M.; de Boer, R.A.; van den Berg, M.P.; Silljé, H.H.W. The time has come to explore plasma biomarkers in genetic cardiomyopathies. Int. J. Mol. Sci. 2021, 22, 2955. [Google Scholar] [CrossRef]
- Greenwell, A.A.; Gopal, K.; Ussher, J.R. Myocardial energy metabolism in non-ischemic cardiomyopathy. Front. Physiol. 2020, 11, 570421. [Google Scholar] [CrossRef]
- Wolf, C.M. Hypertrophic cardiomyopathy: Genetics and cellular perspectives. Cardiovasc. Diagn. Ther. 2019, 9, S388–S415. [Google Scholar] [CrossRef] [PubMed]
- Ranjbarvaziri, S.; Kooiker, K.B.; Ellenberger, M.; Fajardo, G.; Zhao, M.; Vander Roest, A.S.; Woldeyes, R.A.; Koyano, T.T.; Fong, R.; Ma, N.; et al. Altered cardiac energetics and mitochondrial dysfunction in hypertrophic cardiomyopathy. Circulation 2021, 144, 1714–1731. [Google Scholar] [CrossRef] [PubMed]
- Nagana Gowda, G.A.; Djukovic, D. Overview of mass spectrometry-based metabolomics: Opportunities and challenges. Method Mol. Biol. 2014, 1198, 3–12. [Google Scholar]
- Mueller-Hennessen, M.; Sigl, J.; Fuhrmann, J.C.; Witt, H.; Reszka, R.; Schmitz, O.; Kastler, J.; Fischer, J.J.; Müller, O.J.; Giannitsis, E.; et al. Metabolic profiles in heart failure due to non-ischemic cardiomyopathy at rest and under exercise. ESC Heart Fail. 2017, 4, 178–189. [Google Scholar] [CrossRef]
- Laakso, M.; Kuusisto, J.; Stančáková, A.; Kuulasmaa, T.; Pajukanta, P.; Lusis, A.J.; Collins, F.S.; Mohlke, K.L.; Boehnke, M. The Metabolic Syndrome in Men study: A resource for studies of metabolic and cardiovascular diseases. J. Lipid Res. 2017, 58, 481–493. [Google Scholar] [CrossRef]
- Ingles, J.; Goldstein, J.; Thaxton, C.; Caleshu, C.; Corty, E.W.; Crowley, S.B.; Dougherty, K.; Harrison, S.M.; McGlaughon, J.; Milko, L.V.; et al. Evaluating the clinical validity of hypertrophic cardiomyopathy genes. Circ. Genom. Precis. Med. 2019, 12, e002460. [Google Scholar] [CrossRef]
- Jordan, E.; Peterson, L.; Ai, T.; Asatryan, B.; Bronicki, L.; Brown, E.; Celeghin, R.; Edwards, M.; Fan, J.; Ingles, J.; et al. Evidence-based assessment of genes in dilated cardiomyopathy. Circulation 2021, 144, 7–19. [Google Scholar] [CrossRef]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef]
- Vikhorev, P.G.; Vikhoreva, N.N.; Yeung, W.; Li, A.; Lal, S.; Dos Remedios, C.G.; Blair, C.A.; Guglin, M.; Campbell, K.S.; Yacoub, M.H. Titin-truncating mutations associated with dilated cardiomyopathy alter length-dependent activation and its modulation via phosphorylation. Cardiovasc. Res. 2022, 118, 241–253. [Google Scholar] [CrossRef]
- Deidda, M.; Noto, A.; Pasqualucci, D.; Fattuoni, C.; Barberini, L.; Piras, C.; Bassareo, P.P.; Porcu, M.; Mercuro, G.; Dessalvi, C.C. The echocardiographic parameters of systolic function are associated with specific metabolomic fingerprints in obstructive and non-obstructive hypertrophic cardiomyopathy. Metabolites 2021, 11, 787. [Google Scholar] [CrossRef]
- Schuldt, M.; van Driel, B.; Algül, S.; Parbhudayal, R.Y.; Barge-Schaapveld, D.; Güçlü, A.; Jansen, M.; Michels, M.; Baas, A.F.; van de Wiel, M.A.; et al. Distinct metabolomic signatures in preclinical and obstructive hypertrophic cardiomyopathy. Cells 2021, 10, 2950. [Google Scholar] [CrossRef] [PubMed]
- Jørgenrud, B.; Jalanko, M.; Heliö, T.; Jääskeläinen, P.; Laine, M.; Hilvo, M.; Nieminen, M.S.; Laakso, M.; Hyötyläinen, T.; Orešič, M.; et al. The metabolome in Finnish carriers of the MYBPC3-Q1061X mutation for hypertrophic cardiomyopathy. PLoS ONE 2015, 10, e0134184. [Google Scholar] [CrossRef] [PubMed]
- Simpson, P.B.; Woollacott, A.J.; Hill, R.G.; Seabrook, G.R. Functional characterization of bradykinin analogues on recombinant human bradykinin B(1) and B(2) receptors. Eur. J. Pharmacol. 2000, 392, 1–9. [Google Scholar] [CrossRef]
- Feldman, D.S.; Carnes, C.A.; Abraham, W.T.; Bristow, M.R. Mechanisms of disease: β-adrenergic receptors—Alterations in signal transduction and pharmacogenomics in heart failure. Nat. Clin. Pract. Cardiovasc. Med. 2005, 2, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.J.; Engelhardt, S.; Eschenhagen, T. What is the role of β-adrenergic signaling in heart failure? Circ. Res. 2003, 93, 896–906. [Google Scholar] [CrossRef] [PubMed]
- De Lucia, C.; Eguchi, A.; Koch, W.J. New Insights in Cardiac β-Adrenergic Signaling During Heart Failure and Aging. Front. Pharmacol. 2018, 9, 904. [Google Scholar] [CrossRef]
- Kamide, T.; Okumura, S.; Ghosh, S.; Shinoda, Y.; Mototani, Y.; Ohnuki, Y.; Jin, H.; Cai, W.; Suita, K.; Sato, I. Oscillation of cAMP and Ca2+ in cardiac myocytes: A systems biology approach. J. Physiol. Sci. 2015, 65, 195–200. [Google Scholar] [CrossRef]
- Lowes, B.D.; Gilbert, E.M.; Abraham, W.T.; Minobe, W.A.; Larrabee, P.; Ferguson, D.; Wolfel, E.E.; Lindenfeld, J.; Tsvetkova, T.; Robertson, A.D.; et al. Myocardial gene expression in dilated cardiomyopathy treated with β-blocking agents. N. Engl. J. Med. 2002, 346, 1357–1365. [Google Scholar] [CrossRef]
- Frey, N.; McKinsey, T.A.; Olson, E.N. Decoding calcium signals involved in cardiac growth and function. Nat. Med. 2000, 6, 1221–1227. [Google Scholar] [CrossRef]
- Abraham, W.T.; Gilbert, E.M.; Lowes, B.D.; Minobe, W.A.; Larrabee, P.; Roden, R.L.; Dutcher, D.; Sederberg, J.; Lindenfeld, J.A.; Wolfel, E.E.; et al. Coordinate changes in myosin heavy chain isoform gene expression are selectively associated with alterations in dilated cardiomyopathy phenotype. Mol. Med. 2002, 8, 750–760. [Google Scholar] [CrossRef]
- Goetze, J.P.; Bruneau, B.G.; Ramos, H.R.; Ogawa, T.; de Bold, M.K.; de Bold, A.J. Cardiac natriuretic peptides. Nat. Rev. Cardiol. 2020, 17, 698–717. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; He, F.J.; Dong, Y.; Huang, Y.; Harshfield, G.A.; Zhu, H. Sodium reduction, metabolomic profiling, and cardiovascular disease risk in untreated black hypertensives. Hypertension 2019, 74, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Kubli, D.A.; Gustafsson, A.B. Mitochondria and mitophagy: The yin and yang of cell death control. Circ. Res. 2012, 111, 1208–1221. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, A.S.; Appling, D.R. Compartmentalization of mammalian folate-mediated one-carbon metabolism. Ann. Rev. Nutr. 2010, 30, 57–81. [Google Scholar] [CrossRef] [PubMed]
- Katayama, A.; Funamoto, M.; Shimizu, K.; Gempei, M.; Sunagawa, Y.; Wada, H.; Hasegawa, K. Analysis of the effects of EPA and DHA on cardiomyocyte hypertrophy. Eur. Cardiol. 2018, 13, 121. [Google Scholar] [CrossRef]
- Shimojo, N.; Jesmin, S.; Sakai, S.; Maeda, S.; Miyauchi, T.; Mizutani, T.; Aonuma, K.; Kawano, S. Fish oil constituent eicosapentaenoic acid inhibits endothelin-induced cardiomyocyte hypertrophy via PPAR-α. Life Sci. 2014, 118, 173–178. [Google Scholar] [CrossRef]
- Wishart, D.S.; Guo, A.C.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Gautam, V.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef]
- Han, J.H.; Cao, C.; Kim, S.M.; Piao, F.L.; Kim, S.H. Attenuation of lysophosphatidylcholine-induced suppression of ANP release from hypertrophied atria. Hypertension 2004, 43, 243–248. [Google Scholar] [CrossRef][Green Version]
- Ronkainen, J.J.; Vuolteenaho, O.; Tavi, P. Calcium-calmodulin kinase II is the common factor in calcium-dependent cardiac expression and secretion of A- and B-type natriuretic peptides. Endocrinology 2007, 148, 2815–2820. [Google Scholar] [CrossRef][Green Version]
- Coppini, R.; Ferrantini, C.; Yao, L.; Fan, P.; Del Lungo, M.; Stillitano, F.; Sartiani, L.; Tosi, B.; Suffredini, S.; Tesi, C.; et al. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation 2013, 127, 575–584. [Google Scholar] [CrossRef]
- Torimoto, K.; Okada, Y.; Mori, H.; Tanaka, Y. Low levels of 1,5-anhydro-D-glucitol are associated with vascular endothelial dysfunction in type 2 diabetes. Cardiovasc. Diabetol. 2014, 13, 99. [Google Scholar] [CrossRef] [PubMed]
- Roura, S.; Bayes-Genis, A. Vascular dysfunction in idiopathic dilated cardiomyopathy. Nat. Rev. Cardiol. 2009, 6, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.; Lombardi, R.; Rodriguez, G.; Mitchell, M.M.; Marian, A.J. Metabolomic distinction and insights into the pathogenesis of human primary dilated cardiomyopathy. Eur. J. Clin. Investig. 2011, 41, 527–538. [Google Scholar] [CrossRef]
- Zhou, T.; Chen, X.; Li, Z.; Li, L. Relationship between Serum Bilirubin and Left Ventricular Hypertrophy in Patients with Essential Hypertension. PLoS ONE 2015, 10, e0125275. [Google Scholar]
- Zheng, H.; Li, Y.; Xie, N. Association of serum total bilirubin levels with diastolic dysfunction in heart failure with preserved ejection fraction. Biol. Res. 2014, 47, 7. [Google Scholar] [CrossRef][Green Version]
- Bahls, M.; Atzler, D.; Markus, M.R.P.; Friedrich, N.; Böger, R.H.; Völzke, H.; Felix, S.B.; Schwedhelm, E.; Dörr, M. Low-circulating homoarginine is associated with dilatation and decreased function of the left ventricle in the general population. Biomolecules 2018, 8, 63. [Google Scholar] [CrossRef]
- Faller, K.; Atzler, D.; McAndrew, D.J.; Zervou, S.; Whittington, H.J.; Simon, J.N.; Aksentijevic, D.; Ten Hove, M.; Choe, C.U.; Isbrandt, D.; et al. Impaired cardiac contractile function in arginine:glycine amidinotransferase knockout mice devoid of creatine is rescued by homoarginine but not creatine. Cardiovasc. Res. 2018, 114, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Ruszczycky, M.W.; Liu, H.W. Biochemistry: The surprising history of an antioxidant. Nature 2017, 551, 37–38. [Google Scholar] [CrossRef]
- Matsuda, Y.; Ozawa, N.; Shinozaki, T.; Wakabayashi, K.-I.; Suzuki, K.; Kawano, Y.; Ohtsu, I.; Tatebayashi, Y. Ergothioneine, a metabolite of the gut bacterium Lactobacillus reuteri, protects against stress-induced sleep disturbances. Transl. Psychiatry 2020, 10, 170. [Google Scholar] [CrossRef]
- Locke, A.E.; Steinberg, K.M.; Chiang, C.; Service, S.K.; Havulinna, A.S.; Stell, L.; Pirinen, M.; Abel, H.J.; Chiang, C.C.; Fulton, R.S.; et al. Exome sequencing of Finnish isolates enhances rare-variant association power. Nature 2019, 572, 323–328. [Google Scholar] [CrossRef]
- Fernandes Silva, L.; Vangipurapu, J.; Kuulasmaa, T.; Laakso, M. An intronic variant in the GCKR gene is associated with multiple lipids. Sci. Rep. 2019, 9, 10240. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Chan, L.S.; Bose, D.; Jackson, A.U.; VandeHaar, P.; Locke, A.E.; Fuchsberger, C.; Stringham, H.M.; Welch, R.; Boehnke, M.; et al. Genome-wide association studies of metabolites in Finnish men identify disease-relevant loci. Nat. Commun. 2022, 13, 1644. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).