
Respiratory Parameters for the Classification of Dysfunctional Insulin Secretion by Pancreatic Islets


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
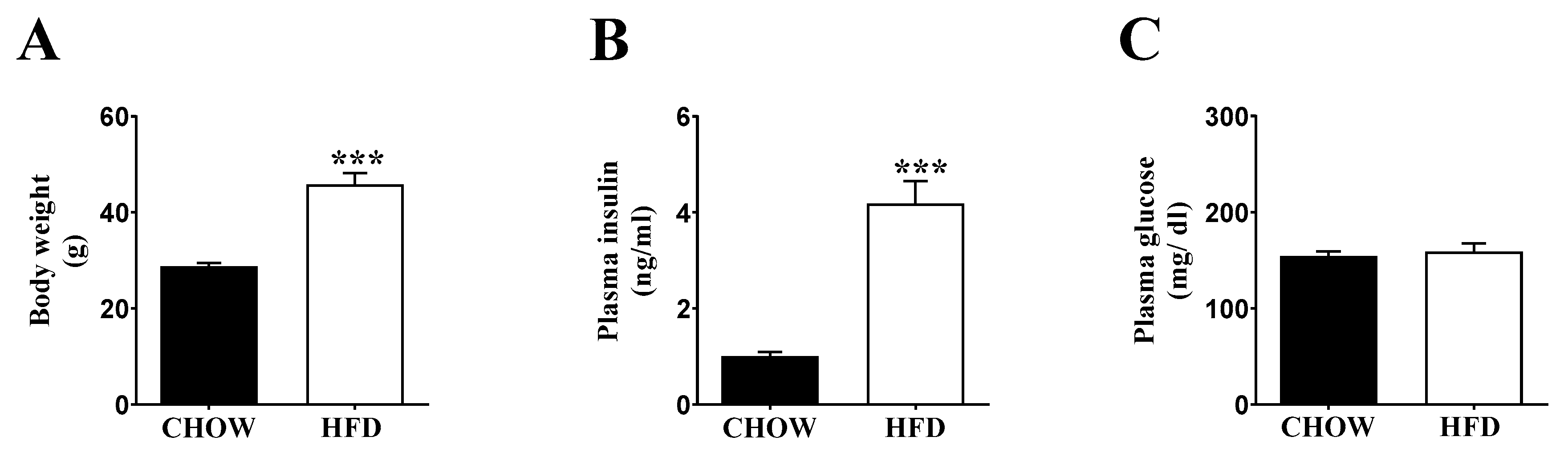
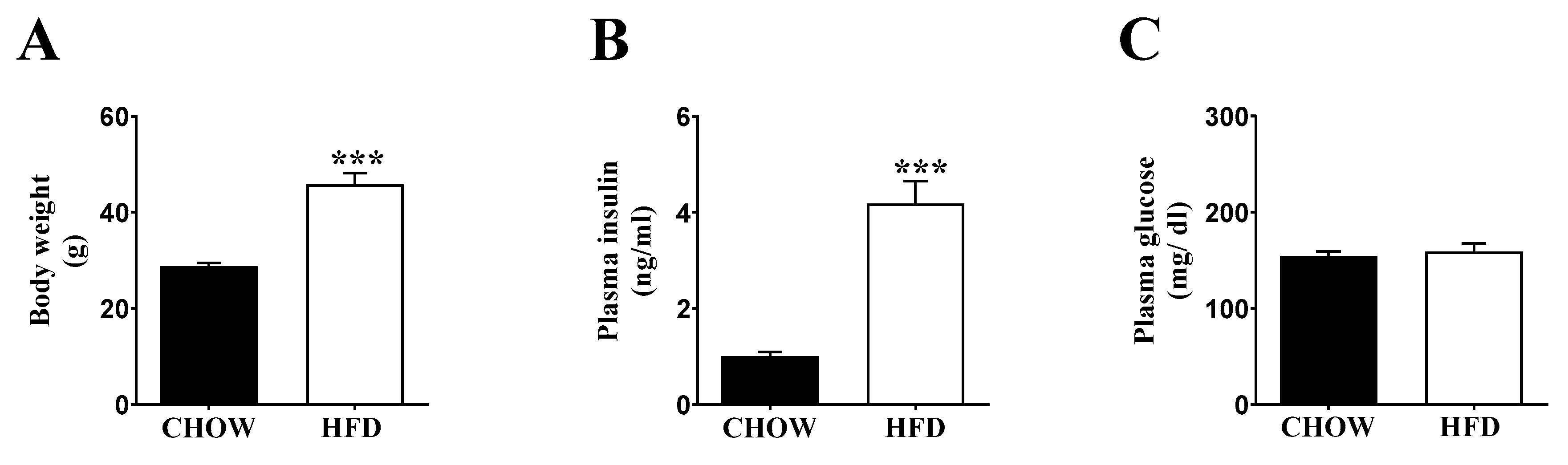
2.1. Inducing Metabolic Stress in Mice
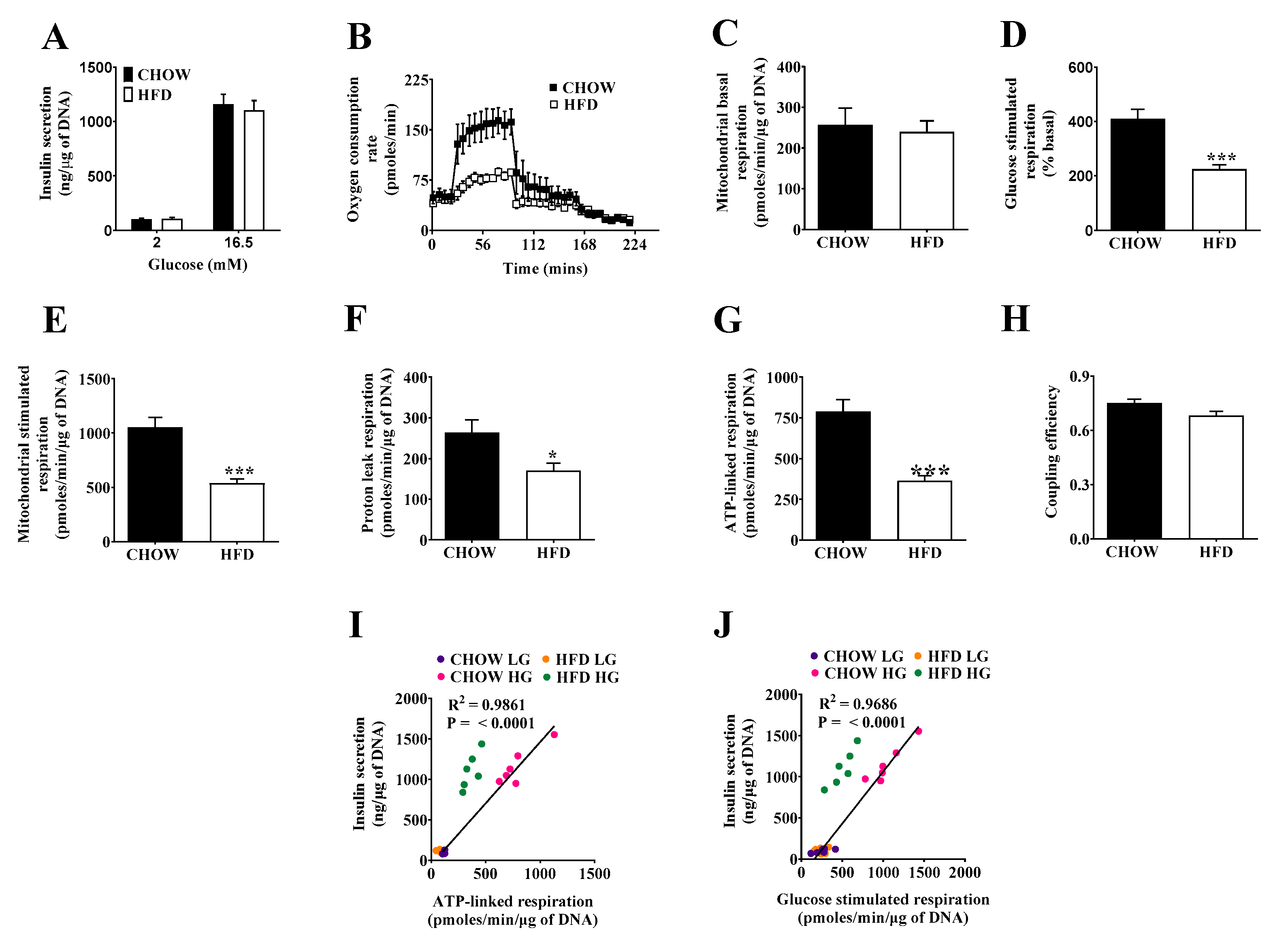
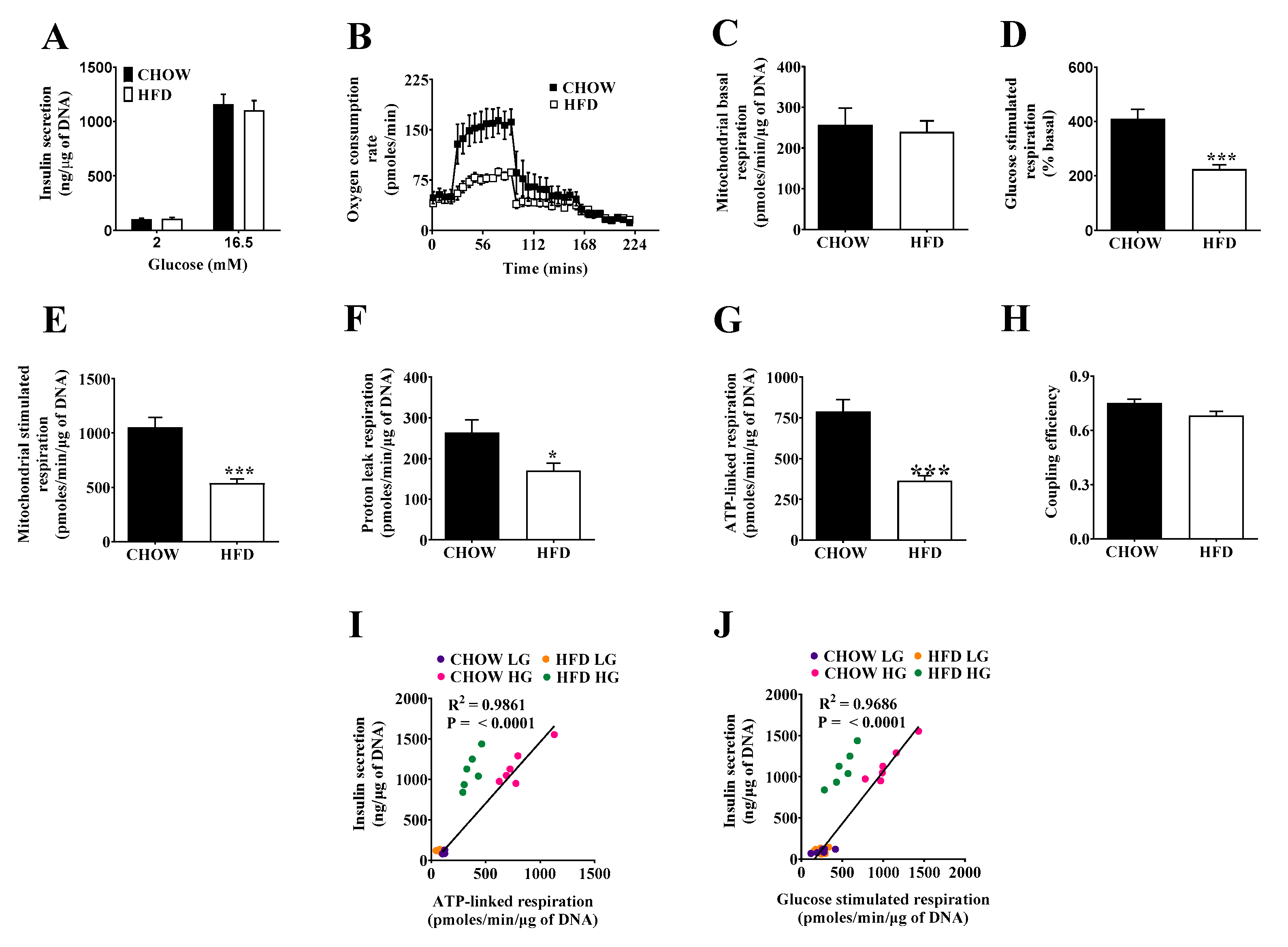
2.2. Respiratory Activity but Not Insulin Secretion Is Impaired in Islets of DIO Mice
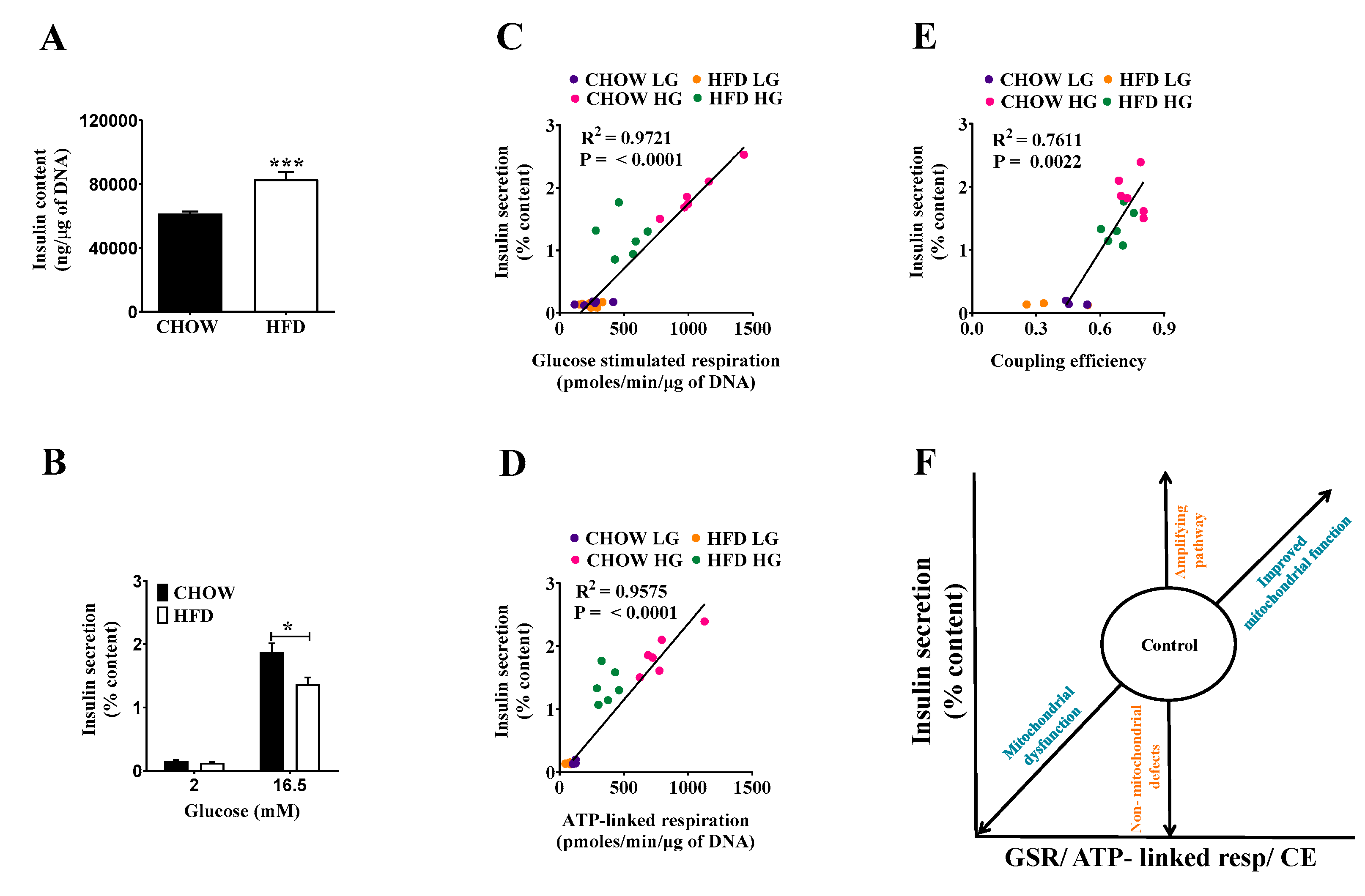
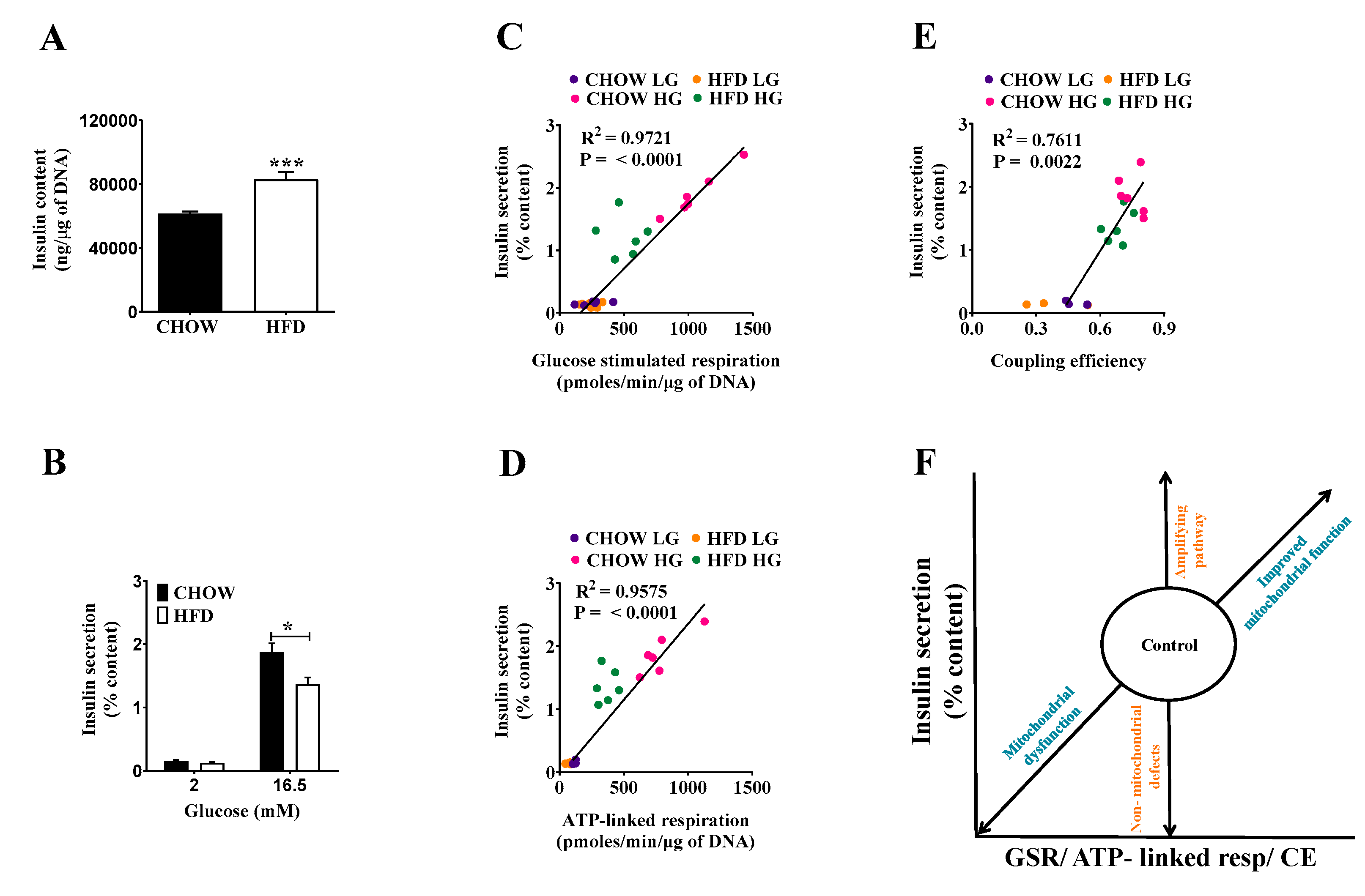
2.3. Compensating Increase of Insulin Content in DIO Islets Masks Defects of the Triggering Pathway
2.4. Classifying Defects in Insulin Secretion
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rutter, G.A.; Pullen, T.J.; Hodson, D.J.; Martinez-Sanchez, A. Pancreatic beta-cell identity, glucose sensing and the control of insulin secretion. Biochem. J. 2015, 466, 203–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henquin, J.C. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 2000, 49, 1751–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashcroft, F.M. ATP-sensitive potassium channelopathies: Focus on insulin secretion. J. Clin. Investig. 2005, 115, 2047–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergsten, P.; Lin, J.; Westerlund, J. Pulsatile insulin release: Role of cytoplasmic Ca2+ oscillations. Diabetes Metab. 1998, 24, 41–45. [Google Scholar] [PubMed]
- Lewandowski, S.L.; Cardone, R.L.; Foster, H.R.; Ho, T.; Potapenko, E.; Poudel, C.; VanDeusen, H.R.; Sdao, S.M.; Alves, T.C.; Zhao, X.; et al. Pyruvate kinase controls signal strength in the insulin secretory pathway. Cell Metab. 2020, 32, 736–750. [Google Scholar] [CrossRef] [PubMed]
- Gembal, M.; Detimary, P.; Gilon, P.; Gao, Z.Y.; Henquin, J.C. Mechanisms by which glucose can control insulin release independently from its action on adenosine triphosphate-sensitive K+ channels in mouse B cells. J. Clin. Investig. 1993, 91, 871–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maechler, P.; Li, N.; Casimir, M.; Vetterli, L.; Frigerio, F.; Brun, T. Role of mitochondria in beta-cell function and dysfunction. In The Islets of Langerhans. Advances in Experimental Medicine and Biology; Springer: Dordrecht, The Netherlands, 2010; Volume 654, pp. 193–216. [Google Scholar] [CrossRef]
- Affourtit, C.; Brand, M.D. Measuring mitochondrial bioenergetics in INS-1E insulinoma cells. In Methods in Enzymology; William, S.A., Anne, N.M., Eds.; Elsevier: San Diego, CA, USA, 2009; Volume 457, pp. 405–424. [Google Scholar] [CrossRef]
- Gerencser, A.A.; Mookerjee, S.A.; Jastroch, M.; Brand, M.D. Measurement of the Absolute Magnitude and Time Courses of Mitochondrial Membrane Potential in Primary and Clonal Pancreatic Beta-Cells. PLoS ONE 2016, 11, e0159199. [Google Scholar] [CrossRef] [PubMed]
- Gerencer, A.A.; Chinopoulos, C.; Birket, M.J.; Jastroch, M.; Vitelli, C.; Nicholls, D.G.; Brand, M.D. Quantitative measurement of mitochondrial membrane potential in cultured cells: Calcium-induced de-and hyperpolarization of neuronal mitochondria. J. Physiol. 2012, 590, 2845–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mookerjee, S.A.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 2017, 292, 7189–7207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Affourtit, C.; Jastroch, M.; Brand, M.D. Uncoupling protein-2 attenuates glucose-stimulated insulin secretion in INS-1E insulinoma cells by lowering mitochondrial reactive oxygen species. Free Radic. Biol. Med. 2011, 50, 609–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, C.; Raun, K.; Yan, F.F.; Larsen, M.O.; Tang-Christensen, M. Laboratory animals as surrogate models of human obesity. Acta Pharmacol. Sin. 2012, 33, 173–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalwat, M.A.; Cobb, M.H. Mechanisms of the amplifying pathway of insulin secretion in the β cell. Pharmacol. Ther. 2017, 179, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Mosser, R.E.; Maulis, M.F.; Moullé, V.S.; Dunn, J.C.; Carboneau, B.A.; Arasi, K.; Pappan, K.; Poitout, V.; Gannon, M. High-fat diet-induced β-cell proliferation occurs prior to insulin resistance in C57Bl/6J male mice. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E573–E582. [Google Scholar] [CrossRef] [PubMed]
- Kabra, U.D.; Pfuhlmann, K.; Migliorini, A.; Keipert, S.; Lamp, D.; Korsgren, O.; Gegg, M.; Woods, S.C.; Pfluger, P.T.; Lickert, H.; et al. Direct substrate delivery into mitochondrial fission–deficient pancreatic islets rescue insulin secretion. Diabetes 2017, 66, 1247–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlow, J.; Solomon, T.P.J.; Affourtit, C. Pro-inflammatory cytokines attenuate glucose-stimulated insulin secretion from INS-1E insulinoma cells by restricting mitochondrial pyruvate oxidation capacity—Novel mechanistic insight from real-time analysis of oxidative phosphorylation. PLoS ONE 2018, 13, e0199505. [Google Scholar] [CrossRef] [PubMed]
- Schulz, N.; Kluth, O.; Jastroch, M.; Schürmann, A. Minor role of mitochondrial respiration for fatty-acid induced insulin secretion. Int. J. Mol. Sci. 2013, 14, 18989–18998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kabra, U.D.; Affourtit, C.; Jastroch, M. Respiratory Parameters for the Classification of Dysfunctional Insulin Secretion by Pancreatic Islets. Metabolites 2021, 11, 405. https://doi.org/10.3390/metabo11060405
Kabra UD, Affourtit C, Jastroch M. Respiratory Parameters for the Classification of Dysfunctional Insulin Secretion by Pancreatic Islets. Metabolites. 2021; 11(6):405. https://doi.org/10.3390/metabo11060405
Chicago/Turabian StyleKabra, Uma D., Charles Affourtit, and Martin Jastroch. 2021. "Respiratory Parameters for the Classification of Dysfunctional Insulin Secretion by Pancreatic Islets" Metabolites 11, no. 6: 405. https://doi.org/10.3390/metabo11060405
APA StyleKabra, U. D., Affourtit, C., & Jastroch, M. (2021). Respiratory Parameters for the Classification of Dysfunctional Insulin Secretion by Pancreatic Islets. Metabolites, 11(6), 405. https://doi.org/10.3390/metabo11060405