
Validation of the Intermolecular Disulfide Bond in Caspase-2

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Crystal Structure Analysis
2.2. Molecular Cloning
2.3. A549 Cell Culture
2.4. Inducible Dimerization, Generation of Lysates, Treatment of Lysates
2.5. SDS–PAGE and Immunoblotting
2.6. Genotyping
2.7. Primary Cell Isolation and Culture
2.8. Cell Treatments and Assessment of Apoptosis via Annexin-V and PI
2.9. Preparation of Inoculum
2.10. In Vivo Infections
2.11. Plating for CFUs
2.12. Statistics
3. Results
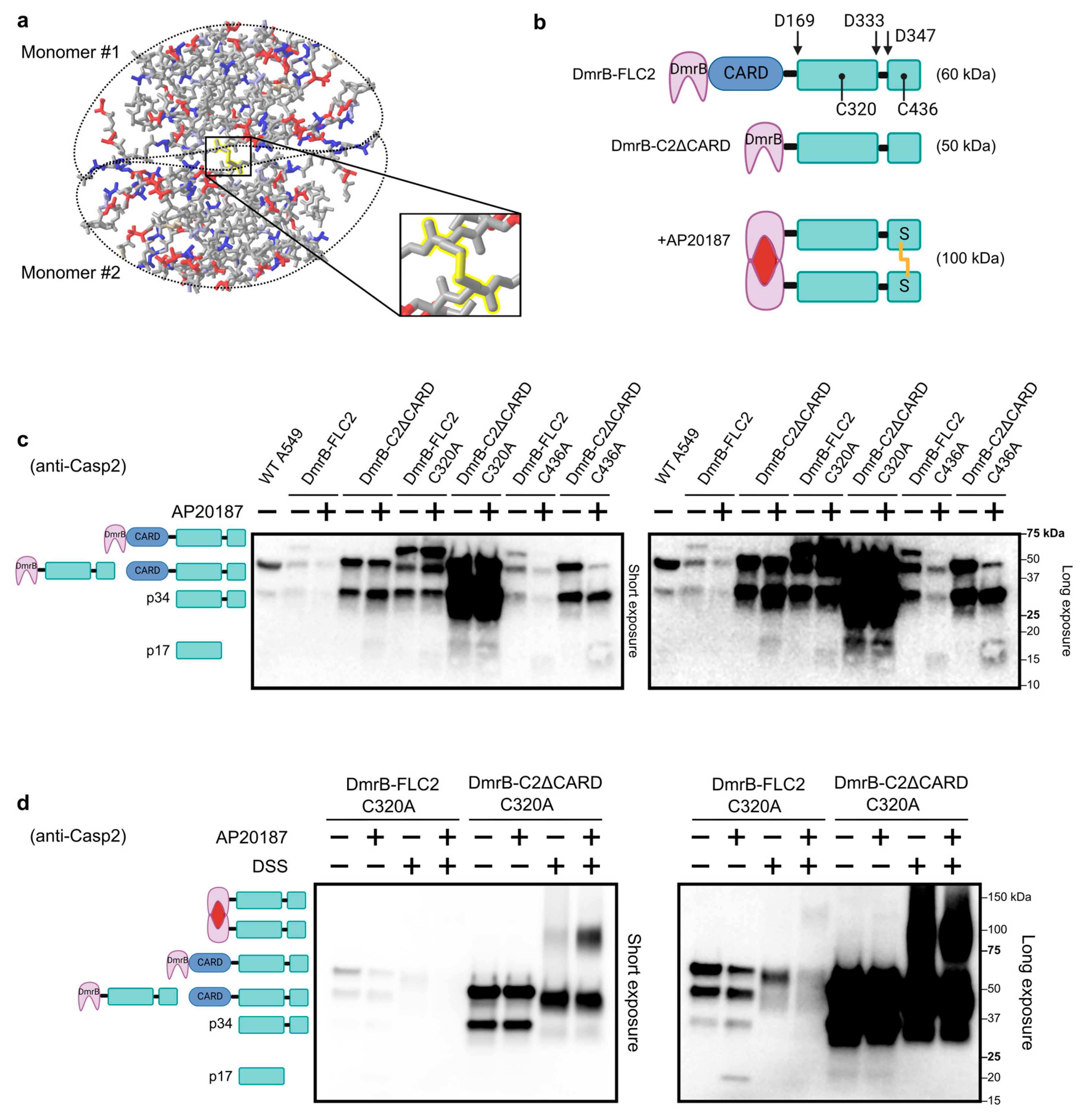
3.1. Inducible Dimerization of Caspase-2
3.2. Disulfide Bond Formation in Dimerized Caspase-2
3.3. Experimental Strategy to Study Cell Death Phenotypes in Caspase-2-Deficient Cells
3.4. DNA Damage and Cytoskeletal Disruption
3.5. Oxidative Stress
3.6. Ferroptosis
3.7. Heat Shock
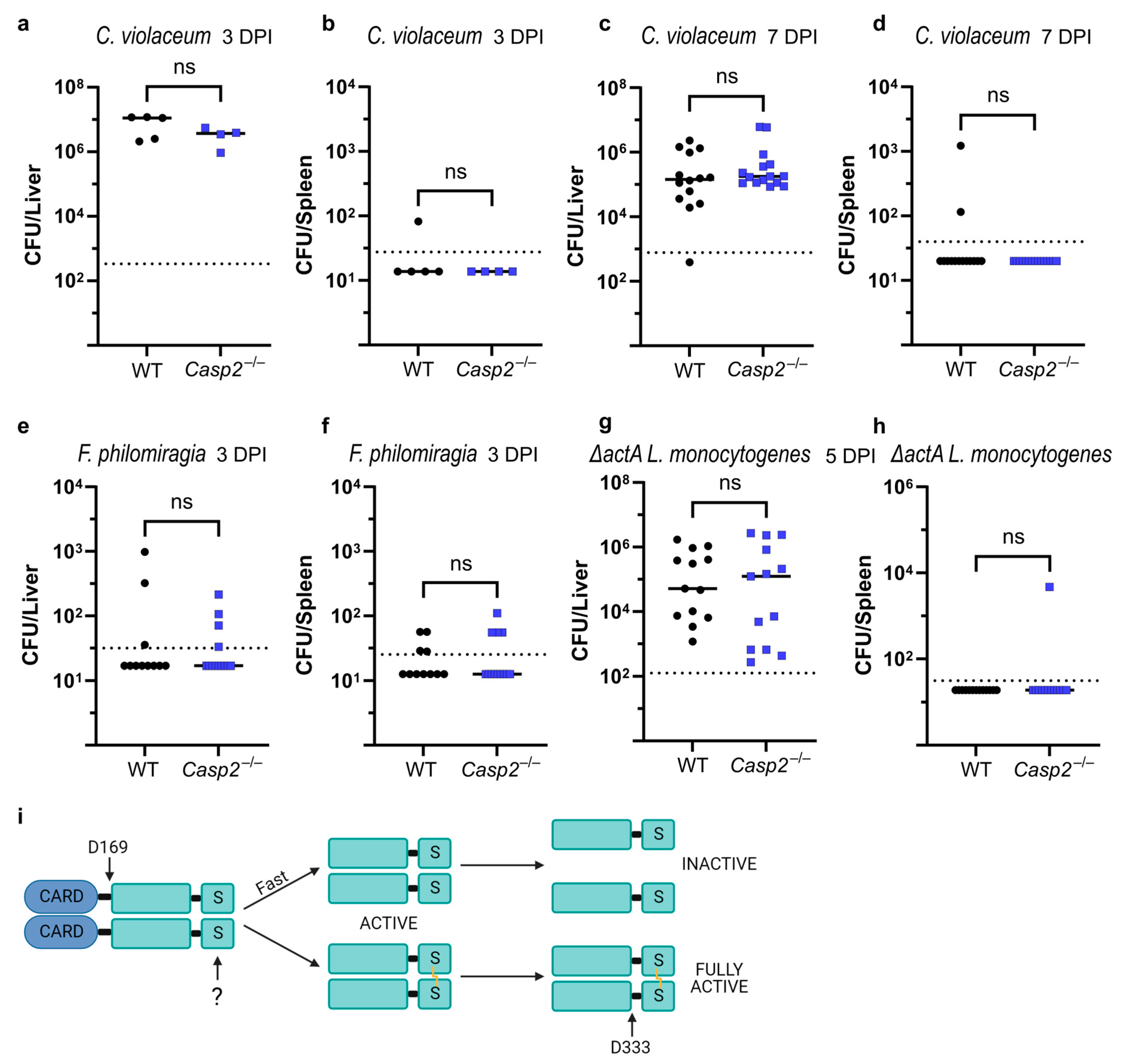
3.8. ROS and In Vivo Infections
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ellis, H.; Horvitz, H. Genetic control of programmed cell death in the nematode C. elegans. Cell 1986, 44, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Black, R.A.; Kronheim, S.R.; Sleath, P.R. Activation of interleukin- 1β by a co-induced protease. FEBS Lett. 1989, 247, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Alnemri, E.S.; Livingston, D.J.; Nicholson, D.W.; Salvesen, G.; Thornberry, N.A.; Wong, W.W.; Yuan, J. Human ICE/CED-3 protease nomenclature. Cell 1996, 87, 171. [Google Scholar] [CrossRef] [PubMed]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, I.; Zhang, Y.; Krantz, B.A.; Miao, E.A. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J. Exp. Med. 2016, 213, 2113–2128. [Google Scholar] [CrossRef] [PubMed]
- Lacey, C.A.; Miao, E.A. Programmed Cell Death in the Evolutionary Race against Bacterial Virulence Factors. Cold Spring Harb Perspect Biol. 2020, 12, a036459. [Google Scholar] [CrossRef] [PubMed]
- Cade, C.E.; Clark, A.C. Caspases—Key Players in Apoptosis. In Proteases in Apoptosis: Pathways, Protocols and Translational Advances; Bose, K., Ed.; Springer International Publishing: Berlin/Heidelberg, Germany, 2015; pp. 31–51. [Google Scholar] [CrossRef]
- Stennicke, H.; Salvesen, G. Catalytic properties of the caspases. Cell Death Differ. 1999, 6, 1054–1059. [Google Scholar] [CrossRef]
- Bao, Q.; Shi, Y. Apoptosome: A platform for the activation of initiator caspases. Cell Death Differ. 2007, 14, 56–65. [Google Scholar] [CrossRef]
- Tinel, A.; Tschopp, J. The PIDDosome, a Protein Complex Implicated in Activation of Caspase-2 in Response to Genotoxic Stress. Science 2004, 304, 843–846. [Google Scholar] [CrossRef]
- Sladky, V.; Schuler, F.; Fava, L.L.; Villunger, A. The resurrection of the PIDDosome—Emerging roles in the DNA-damage response and centrosome surveillance. J. Cell Sci. 2017, 130, 3779–3787. [Google Scholar] [CrossRef]
- Ribe, E.M.; Jean, Y.Y.; Goldstein, R.L.; Manzl, C.; Stefanis, L.; Villunger, A.; Troy, C.M. Neuronal caspase 2 activity and function requires RAIDD, but not PIDD. Biochem. J. 2012, 444, 591–599. [Google Scholar] [CrossRef]
- Berube, C.; Boucher, L.M.; Ma, W.; Wakeham, A.; Salmena, L.; Hakem, R.; Yeh, W.C.; Mak, T.W.; Benchimol, S. Apoptosis caused by p53-induced protein with death domain (PIDD) depends on the death adapter protein RAIDD. Proc. Natl. Acad. Sci. USA 2005, 102, 14314–14320. [Google Scholar] [CrossRef] [PubMed]
- Miles, M.A.; Kitevska-Ilioski, T.; Hawkins, C.J. Old and Novel Functions of Caspase-2. In International Review of Cell and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 332, pp. 155–212. [Google Scholar] [CrossRef]
- Brown-Suedel, A.N.; Bouchier-Hayes, L. Caspase-2 Substrates: To Apoptosis, Cell Cycle Control, and Beyond. Front. Cell Dev. Biol. 2020, 8, 610022. [Google Scholar] [CrossRef] [PubMed]
- Bouchier-Hayes, L. The role of caspase-2 in stress-induced apoptosis. J. Cell Mol. Med. 2010, 14, 1212–1224. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, L.; Perez, G.I.; Macdonald, G.; Shi, L.; Sun, Y.; Jurisicova, A.; Varmuza, S.; Latham, K.E.; Flaws, J.A.; Salter, J.C.M.; et al. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev. 1998, 12, 1304–1314. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Upton, J.W.; Long, A.B.; Livingston-Rosanoff, D.; Daley-Bauer, L.P.; Hakem, R.; Caspary, T.; Mocarski, E.S. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011, 471, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Kuida, K.; Haydar, T.F.; Kuan, C.Y.; Gu, Y.; Taya, C.; Karasuyama, H.; Su, M.S.S.; Rakic, P.; Flavell, R.A. Reduced Apoptosis and Cytochrome c–Mediated Caspase Activation in Mice Lacking Caspase 9. Cell 1998, 94, 325–337. [Google Scholar] [CrossRef]
- Kumar, S.; Kinoshita, M.; Noda, M.; Copeland, N.G.; Jenkins, N.A. Induction of apoptosis by the mouse Nedd2 gene, which encodes a protein similar to the product of the Caenorhabditis elegans cell death gene ced-3 and the mammalian IL-1 beta-converting enzyme. Genes Dev. 1994, 8, 1613–1626. [Google Scholar] [CrossRef]
- Schweizer, A.; Briand, C.; Grutter, M.G. Crystal structure of caspase-2, apical initiator of the intrinsic apoptotic pathway. J. Biol. Chem. 2003, 278, 42441–42447. [Google Scholar] [CrossRef]
- Baliga, B.C.; Read, S.H.; Kumar, S. The biochemical mechanism of caspase-2 activation. Cell Death Differ. 2004, 11, 1234–1241. [Google Scholar] [CrossRef]
- Bulleid, N.J.; Ellgaard, L. Multiple ways to make disulfides. Trends Biochem. Sci. 2011, 36, 485–492. [Google Scholar] [CrossRef]
- Bechtel, T.J.; Weerapana, E. From structure to redox: The diverse functional roles of disulfides and implications in disease. Proteomics 2017, 17, 1600391. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef]
- Bouchier-Hayes, L.; Oberst, A.; McStay, G.P.; Connell, S.; Tait, S.W.G.; Dillon, C.P.; Flanagan, J.M.; Beere, H.M.; Green, D.R. Characterization of Cytoplasmic Caspase-2 Activation by Induced Proximity. Mol. Cell. 2009, 35, 830–840. [Google Scholar] [CrossRef]
- Cremers, C.M.; Jakob, U. Oxidant Sensing by Reversible Disulfide Bond Formation. J. Biol. Chem. 2013, 288, 26489–26496. [Google Scholar] [CrossRef]
- Nagahara, N. Intermolecular disulfide bond to modulate protein function as a redox-sensing switch. Amino Acids. 2011, 41, 59–72. [Google Scholar] [CrossRef]
- Barford, D. The role of cysteine residues as redox-sensitive regulatory switches. Curr. Opin. Struct. Biol. 2004, 14, 679–686. [Google Scholar] [CrossRef]
- Le Gal, K.; Schmidt, E.E.; Sayin, V.I. Cellular Redox Homeostasis. Antioxidants 2021, 10, 1377. [Google Scholar] [CrossRef]
- Kohen, R.; Nyska, A. Invited Review: Oxidation of Biological Systems: Oxidative Stress Phenomena, Antioxidants, Redox Reactions, and Methods for Their Quantification. Toxicol. Pathol. 2002, 30, 620–650. [Google Scholar] [CrossRef]
- Dickinson, B.C.; Chang, C.J. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 2011, 7, 504–511. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Glutathione and modulation of cell apoptosis. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2012, 1823, 1767–1777. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Aspects Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Lushchak, V.I. Glutathione Homeostasis and Functions: Potential Targets for Medical Interventions. J. Amino Acids. 2012, 2012, 736837. [Google Scholar] [CrossRef]
- Zhang, H.; Forman, H.J. Glutathione synthesis and its role in redox signaling. Semin. Cell Dev. Biol. 2012, 23, 722–728. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J. Isolating, Cloning, and Sequencing DNA. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. Available online: https://www.ncbi.nlm.nih.gov/books/NBK26837/ (accessed on 12 December 2023).
- García-Nafría, J.; Watson, J.F.; Greger, I.H. IVA cloning: A single-tube universal cloning system exploiting bacterial In Vivo Assembly. Sci Rep. 2016, 6, 27459. [Google Scholar] [CrossRef]
- Swift, S.; Lorens, J.; Achacoso, P.; Nolan, G.P. Rapid Production of Retroviruses for Efficient Gene Delivery to Mammalian Cells Using 293T Cell–Based Systems. Curr. Protoc. Immunol. 1999, 31, 10–17. [Google Scholar] [CrossRef]
- Pear, W.S.; Nolan, G.P.; Scott, M.L.; Baltimore, D. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA 1993, 90, 8392–8396. [Google Scholar] [CrossRef]
- Gonçalves, R.; Mosser, D.M. The Isolation and Characterization of Murine Macrophages. Curr. Protoc. Immunol. 2015, 111. [Google Scholar] [CrossRef]
- Harvest, C.K.; Abele, T.J.; Yu, C.; Beatty, C.J.; Amason, M.E.; Billman, Z.P.; DePrizio, M.A.; Souza, F.W.; Lacey, C.A.; Maltez, V.I.; et al. An innate granuloma eradicates an environmental pathogen using Gsdmd and Nos2. Nat. Commun. 2023, 14, 6686. [Google Scholar] [CrossRef]
- Mahajan, I.M.; Chen, M.D.; Muro, I.; Robertson, J.D.; Wright, C.W.; Bratton, S.B. BH3-Only Protein BIM Mediates Heat Shock-Induced Apoptosis. PLoS ONE 2014, 9, e84388. [Google Scholar] [CrossRef] [PubMed]
- Fava, L.L.; Schuler, F.; Sladky, V.; Haschka, M.D.; Soratroi, C.; Eiterer, L.; Demetz, E.; Weiss, G.; Geley, S.; Nigg, E.A.; et al. The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev. 2017, 31, 34–45. [Google Scholar] [CrossRef]
- Rousculp, M.D.; Goldsmith, K.T.; Garver, R.I. Quantitative Evaluation of Retroviral Gene Transduction Efficiency in Human Lung Cancer Cells. Hum. Gene Ther. 1992, 3, 471–477. [Google Scholar] [CrossRef]
- Radke, J.R.; Siddiqui, Z.K.; Miura, T.A.; Routes, J.M.; Cook, J.L. E1A oncogene enhancement of caspase-2-mediated mitochondrial injury sensitizes cells to macrophage nitric oxide-induced apoptosis. J. Immunol. 2008, 180, 8272–8279. [Google Scholar] [CrossRef]
- Tang, J.; Frascaroli, G.; Lebbink, R.J.; Ostermann, E.; Brune, W. Human cytomegalovirus glycoprotein B variants affect viral entry, cell fusion, and genome stability. Proc. Natl. Acad. Sci. USA 2019, 116, 18021–18030. [Google Scholar] [CrossRef]
- Gargett, T.; Brown, M.P. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014, 5, 235. [Google Scholar] [CrossRef]
- Wolhuter, K.; Whitwell, H.J.; Switzer, C.H.; Burgoyne, J.R.; Timms, J.F.; Eaton, P. Evidence against Stable Protein S-Nitrosylation as a Widespread Mechanism of Post-translational Regulation. Mol. Cell. 2018, 69, 438–450.e5. [Google Scholar] [CrossRef]
- Andersen, J.L.; Johnson, C.E.; Freel, C.D.; Parrish, A.B.; Day, J.L.; Buchakjian, M.R.; Nutt, L.K.; Thompson, J.W.; Moseley, M.A.; Kornbluth, S. Restraint of apoptosis during mitosis through interdomain phosphorylation of caspase-2. EMBO J. 2009, 28, 3216–3227. [Google Scholar] [CrossRef]
- Dawar, S.; Shahrin, N.H.; Sladojevic, N.; D’Andrea, R.J.; Dorstyn, L.; Hiwase, D.K.; Kumar, S. Impaired haematopoietic stem cell differentiation and enhanced skewing towards myeloid progenitors in aged caspase-2-deficient mice. Cell Death Dis. 2016, 7, e2509. [Google Scholar] [CrossRef] [PubMed]
- Van Engeland, M.; Ramaekers, F.C.S.; Schutte, B.; Reutelingsperger, C.P.M. A novel assay to measure loss of plasma membrane asymmetry during apoptosis of adherent cells in culture. Cytometry 1996, 24, 131–139. [Google Scholar] [CrossRef]
- Logue, S.E.; Elgendy, M.; Martin, S.J. Expression, purification and use of recombinant annexin V for the detection of apoptotic cells. Nat. Protoc. 2009, 4, 1383–1395. [Google Scholar] [CrossRef] [PubMed]
- Cummings, B.S.; Schnellmann, R.G. Measurement of Cell Death in Mammalian Cells. Curr. Protoc. Pharmacol. 2004, 25, e210. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.; McStay, G.P.; Boucher, L.M.; Mak, T.; Beere, H.M.; Green, D.R. In situ trapping of activated initiator caspases reveals a role for caspase-2 in heat shock-induced apoptosis. Nat. Cell Biol. 2006, 8, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.P.; Austgen, K.; Nishino, M.; Coakley, K.M.; Hagen, A.; Han, D.; Papa, F.R.; Oakes, S.A. Caspase-2 Cleavage of BID Is a Critical Apoptotic Signal Downstream of Endoplasmic Reticulum Stress. Mol. Cell Biol. 2008, 28, 3943. [Google Scholar] [CrossRef] [PubMed]
- Paroni, G.; Henderson, C.; Schneider, C.; Brancolini, C. Caspase-2-induced apoptosis is dependent on caspase-9, but its processing during UV- or tumor necrosis factor-dependent cell death requires caspase-3. J. Biol. Chem. 2001, 276, 21907–21915. [Google Scholar] [CrossRef] [PubMed]
- Franklin, E.E.; Robertson, J.D. Requirement of Apaf-1 for mitochondrial events and the cleavage or activation of all procaspases during genotoxic stress-induced apoptosis. Biochem. J. 2007, 405, 115–122. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Robertson, J.D.; Enoksson, M.; Suomela, M.; Zhivotovsky, B.; Orrenius, S. Caspase-2 acts upstream of mitochondria to promote cytochrome c release during etoposide-induced apoptosis. J. Biol. Chem. 2002, 277, 29803–29809. [Google Scholar] [CrossRef] [PubMed]
- Lassus, P.; Opitz-Araya, X.; Lazebnik, Y. Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science 2002, 297, 1352–1354. [Google Scholar] [CrossRef]
- Ho, L.H.; Read, S.H.; Dorstyn, L.; Lambrusco, L.; Kumar, S. Caspase-2 is required for cell death induced by cytoskeletal disruption. Oncogene 2008, 27, 3393–3404. [Google Scholar] [CrossRef]
- Wagner, K.W.; Engels, I.H.; Deveraux, Q.L. Caspase-2 can function upstream of bid cleavage in the TRAIL apoptosis pathway. J. Biol. Chem. 2004, 279, 35047–35052. [Google Scholar] [CrossRef]
- Werner, A.B.; Tait, S.W.G.; de Vries, E.; Eldering, E.; Borst, J. Requirement for Aspartate-cleaved Bid in Apoptosis Signaling by DNA-damaging Anti-cancer Regimens*. J. Biol. Chem. 2004, 279, 28771–28780. [Google Scholar] [CrossRef]
- Bronner, D.N.; Abuaita, B.H.; Chen, X.; Fitzgerald, K.A.; Nuñez, G.; He, Y.; Yin, X.M.; O’Riordan, M.X.D. Endoplasmic Reticulum Stress Activates the Inflammasome via NLRP3- and Caspase-2-Driven Mitochondrial Damage. Immunity 2015, 43, 451–462. [Google Scholar] [CrossRef]
- Bronner, D.N.; O’Riordan, M.X.D.; He, Y. Caspase-2 mediates a Brucella abortus RB51-induced hybrid cell death having features of apoptosis and pyroptosis. Front. Cell Infect Microbiol. 2013, 3, 83. [Google Scholar] [CrossRef]
- Boice, A.G.; Lopez, K.E.; Pandita, R.K.; Parsons, M.J.; Charendoff, C.I.; Charaka, V.; Carisey, A.F.; Pandita, T.K.; Bouchier-Hayes, L. Caspase-2 regulates S-phase cell cycle events to protect from DNA damage accumulation independent of apoptosis. Oncogene 2022, 41, 204–219. [Google Scholar] [CrossRef]
- Ando, K.; Parsons, M.J.; Shah, R.B.; Charendoff, C.I.; Paris, S.L.; Liu, P.H.; Fassio, S.R.; Rohrman, B.A.; Thompson, R.; Oberst, A.; et al. NPM1 directs PIDDosome-dependent caspase-2 activation in the nucleolus. J. Cell Biol. 2017, 216, 1795–1810. [Google Scholar] [CrossRef]
- Bruni, E.; Reichle, A.; Scimeca, M.; Bonanno, E.; Ghibelli, L. Lowering Etoposide Doses Shifts Cell Demise from Caspase-Dependent to Differentiation and Caspase-3-Independent Apoptosis via DNA Damage Response, Inducing AML Culture Extinction. Front. Pharmacol. 2018, 9, 1307. Available online: https://www.frontiersin.org/articles/10.3389/fphar.2018.01307 (accessed on 7 April 2023). [CrossRef]
- Dawar, S.; Lim, Y.; Puccini, J.; White, M.; Thomas, P.; Bouchier-Hayes, L.; Green, D.R.; Dorstyn, L.; Kumar, S. Caspase-2-mediated cell death is required for deleting aneuploid cells. Oncogene 2017, 36, 2704–2714. [Google Scholar] [CrossRef]
- Nutt, L.K.; Margolis, S.S.; Jensen, M.; Herman, C.E.; Dunphy, W.G.; Rathmell, J.C.; Kornbluth, S. Metabolic Regulation of Oocyte Cell Death through the CaMKII-Mediated Phosphorylation of Caspase-2. Cell 2005, 123, 89–103. [Google Scholar] [CrossRef]
- Tiwari, M.; Sharma, L.K.; Vanegas, D.; Callaway, D.A.; Bai, Y.; Lechleiter, J.D.; Herman, B. A nonapoptotic role for CASP2/caspase 2. Autophagy 2014, 10, 1054–1070. [Google Scholar] [CrossRef]
- Sharma, R.; Callaway, D.; Vanegas, D.; Bendele, M.; Lopez-Cruzan, M.; Horn, D.; Guda, T.; Fajardo, R.; Abboud-Werner, S.; Herman, B. Caspase-2 Maintains Bone Homeostasis by Inducing Apoptosis of Oxidatively-Damaged Osteoclasts. PLoS ONE 2014, 9, e93696. [Google Scholar] [CrossRef] [PubMed]
- Tamm, C.; Zhivotovsky, B.; Ceccatelli, S. Caspase-2 activation in neural stem cells undergoing oxidative stress-induced apoptosis. Apoptosis 2008, 13, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Radke, J.R.; Figueroa, I.; Routes, J.M.; Cook, J.L. PIDD-dependent activation of caspase-2-mediated mitochondrial injury in E1A-induced cellular sensitivity to macrophage nitric oxide-induced apoptosis. Cell Death Discov. 2018, 4, 97. [Google Scholar] [CrossRef]
- Jimenez, P.T.; Frolova, A.I.; Chi, M.M.; Grindler, N.M.; Willcockson, A.R.; Reynolds, K.A.; Zhao, Q.; Moley, K.H. DHEA-Mediated Inhibition of the Pentose Phosphate Pathway Alters Oocyte Lipid Metabolism in Mice. Endocrinology 2013, 154, 4835–4844. [Google Scholar] [CrossRef]
- Heinz, S.; Freyberger, A.; Lawrenz, B.; Schladt, L.; Schmuck, G.; Ellinger-Ziegelbauer, H. Mechanistic Investigations of the Mitochondrial Complex I Inhibitor Rotenone in the Context of Pharmacological and Safety Evaluation. Sci. Rep. 2017, 7, 45465. [Google Scholar] [CrossRef]
- Srivastava, P.; Panda, D. Rotenone inhibits mammalian cell proliferation by inhibiting microtubule assembly through tubulin binding. FEBS J. 2007, 274, 4788–4801. [Google Scholar] [CrossRef]
- Shi, M.M.; Kugelman, A.; Iwamoto, T.; Tian, L.; Forman, H.J. Quinone-induced oxidative stress elevates glutathione and induces gamma-glutamylcysteine synthetase activity in rat lung epithelial L2 cells. J. Biol. Chem. 1994, 269, 26512–26517. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.X.; Berry, L.C.; Tanner, M.; Chang, M.; Myers, R.P.; Meyrick, B. Nitric oxide donors regulate nitric oxide synthase in bovine pulmonary artery endothelium. J. Cell Physiol. 2001, 186, 116–123. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Comish, P.B.; Tang, D.; Kang, R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021, 9, 637162. [Google Scholar] [CrossRef]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003, 3, 285–296. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef]
- Engel, S.; Doerflinger, M.; Lee, A.R.; Strasser, A.; Herold, M.J.; Bedoui, S.; Bachem, A. Caspase-2 does not play a critical role in cell death induction and bacterial clearance during Salmonella infection. Cell Death Differ. 2021, 28, 3371–3373. [Google Scholar] [CrossRef]
- Abuaita, B.H.; Sule, G.J.; Schultz, T.L.; Gao, F.; Knight, J.S.; O’Riordan, M.X. The IRE1α Stress Signaling Axis Is a Key Regulator of Neutrophil Antimicrobial Effector Function. J. Immunol. 2021, 207, 210–220. [Google Scholar] [CrossRef]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell Infect Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Song, C.H. Effect of Reactive Oxygen Species on the Endoplasmic Reticulum and Mitochondria during Intracellular Pathogen Infection of Mammalian Cells. Antioxidants 2021, 10, 872. [Google Scholar] [CrossRef] [PubMed]
- Macher, A.M. Chronic Granulomatous Disease of Childhood and Chromobacterium violaceum Infections in the Southeastern United States. Ann. Intern. Med. 1982, 97, 51. [Google Scholar] [CrossRef]
- Maltez, V.I.; Tubbs, A.L.; Cook, K.D.; Aachoui, Y.; Falcone, E.L.; Holland, S.M.; Whitmire, J.K.; Miao, E.A. Inflammasomes Coordinate Pyroptosis and Natural Killer Cell Cytotoxicity to Clear Infection by a Ubiquitous Environmental Bacterium. Immunity 2015, 43, 987–997. [Google Scholar] [CrossRef]
- Pizzolla, A.; Hultqvist, M.; Nilson, B.; Grimm, M.J.; Eneljung, T.; Jonsson, I.M.; Verdrengh, M.; Kelkka, T.; Gjertsson, I.; Segal, B.H.; et al. Reactive Oxygen Species Produced by the NADPH Oxidase 2 Complex in Monocytes Protect Mice from Bacterial Infections. J. Immunol. 2012, 188, 5003–5011. [Google Scholar] [CrossRef]
- Robles-Marhuenda, A.; Vaca, M.; Romero, P.; Ferreira, A.; López-Granados, E.; Arnalich, F. Francisella philomiragia: Think of Chronic Granulomatous Disease. J. Clin. Immunol. 2018, 38, 257–259. [Google Scholar] [CrossRef]
- Hoelzer, K.; Pouillot, R.; Dennis, S. Animal models of listeriosis: A comparative review of the current state of the art and lessons learned. Vet Res. 2012, 43, 18. [Google Scholar] [CrossRef]
- MacMicking, J.D.; Nathan, C.; Hom, G.; Chartrain, N.; Fletcher, D.S.; Trumbauer, M.; Stevens, K.; Xie, Q.-w.; Sokol, K.; Hutchinson, N.; et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell 1995, 81, 641–650. [Google Scholar] [CrossRef]
- Pasche, B.; Kalaydjiev, S.; Franz, T.J.; Kremmer, E.; Gailus-Durner, V.; Fuchs, H.; Hrabé De Angelis, M.; Lengeling, A.; Busch, D.H. Sex-Dependent Susceptibility to Listeria monocytogenes Infection Is Mediated by Differential Interleukin-10 Production. Infect Immun. 2005, 73, 5952–5960. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.; Marteyn, B.; Sansonetti, P.J.; Tang, C.M. Life on the inside: The intracellular lifestyle of cytosolic bacteria. Nat. Rev. Microbiol. 2009, 7, 333–340. [Google Scholar] [CrossRef]
- Pillich, H.; Puri, M.; Chakraborty, T. ActA of Listeria monocytogenes and Its Manifold Activities as an Important Listerial Virulence Factor. In The Actin Cytoskeleton and Bacterial Infection; Mannherz, H.G., Ed.; Current Topics in Microbiology and Immunology; Springer International Publishing: Berlin/Heidelberg, Germany, 2016; Volume 399, pp. 113–132. [Google Scholar] [CrossRef]
- Reniere, M.L.; Whiteley, A.T.; Hamilton, K.L.; John, S.M.; Lauer, P.; Brennan, R.G.; Portnoy, D.A. Glutathione activates virulence gene expression of an intracellular pathogen. Nature 2015, 517, 170–173. [Google Scholar] [CrossRef]
- Deponte, M.; Horst Lillig, C. Enzymatic control of cysteinyl thiol switches in proteins. Biol. Chem. 2015, 396, 401–413. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Orchestrating Redox Signaling Networks through Regulatory Cysteine Switches. ACS Chem. Biol. 2010, 5, 47–62. [Google Scholar] [CrossRef]
- Forsberg, J.; Zhivotovsky, B.; Olsson, M. Caspase-2: An orphan enzyme out of the shadows. Oncogene 2017, 36, 5441–5444. [Google Scholar] [CrossRef]
- Li, J.; Billiar, T.R.; Talanian, R.V.; Kim, Y.M. Nitric Oxide Reversibly Inhibits Seven Members of the Caspase Family via S-Nitrosylation. Biochem. Biophys. Res. Commun. 1997, 240, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Rayamajhi, M.; Miao, E.A. Just say NO to NLRP3. Nat. Immunol. 2013, 14, 12–14. [Google Scholar] [CrossRef]
- Lopez-Cruzan, M.; Sharma, R.; Tiwari, M.; Karbach, S.; Holstein, D.; Martin, C.R.; Lechleiter, J.D.; Herman, B. Caspase-2 resides in the mitochondria and mediates apoptosis directly from the mitochondrial compartment. Cell Death Discov. 2016, 2, 16005. [Google Scholar] [CrossRef]
- Kim, J.Y.; Garcia-Carbonell, R.; Yamachika, S.; Zhao, P.; Dhar, D.; Loomba, R.; Kaufman, R.J.; Saltiel, A.R.; Karin, M. ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 2018, 175, 133–145.e15. [Google Scholar] [CrossRef]
- Colussi, P.A.; Harvey, N.L.; Kumar, S. Prodomain-dependent Nuclear Localization of the Caspase-2 (Nedd2) Precursor. J. Biol. Chem. 1998, 273, 24535–24542. [Google Scholar] [CrossRef]
- Baliga, B.C.; Colussi, P.A.; Read, S.H.; Dias, M.M.; Jans, D.A.; Kumar, S. Role of prodomain in importin-mediated nuclear localization and activation of caspase-2. J. Biol. Chem. 2003, 278, 4899–4905. [Google Scholar] [CrossRef]
- Mancini, M.; Machamer, C.E.; Roy, S.; Nicholson, D.W.; Thornberry, N.A.; Casciola-Rosen, L.A.; Rosen, A. Caspase-2 is localized at the Golgi complex and cleaves golgin-160 during apoptosis. J. Cell Biol. 2000, 149, 603–612. [Google Scholar] [CrossRef]
- Adams, L.; Franco, M.C.; Estevez, A.G. Reactive nitrogen species in cellular signaling. Exp. Biol. Med. 2015, 240, 711–717. [Google Scholar] [CrossRef]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 2018, 215, 827–840. [Google Scholar] [CrossRef]
- Go, Y.M.; Jones, D.P. Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta BBA—Gen. Subj. 2008, 1780, 1273–1290. [Google Scholar] [CrossRef]
- Pallardó, F.V.; Markovic, J.; García, J.L.; Viña, J. Role of nuclear glutathione as a key regulator of cell proliferation. Mol. Aspects Med. 2009, 30, 77–85. [Google Scholar] [CrossRef]
- Ahuja, D.; Sáenz-Robles, M.T.; Pipas, J.M. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene 2005, 24, 7729–7745. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef]
- Baptiste-Okoh, N.; Barsotti, A.M.; Prives, C. A role for caspase 2 and PIDD in the process of p53-mediated apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 1937–1942. [Google Scholar] [CrossRef]
- Heinke, L. Mitochondrial ROS drive cell cycle progression. Nat. Rev. Mol. Cell Biol. 2022, 23, 581. [Google Scholar] [CrossRef]
- Gagliardi, L.J.; Shain, D.H. Is intracellular pH a clock for mitosis? Theor. Biol. Med. Model. 2013, 10, 8. [Google Scholar] [CrossRef]
- Hecht, F.; Zocchi, M.; Alimohammadi, F.; Harris, I.S. Regulation of antioxidants in cancer. Mol. Cell. 2023, 84, 23–33. [Google Scholar] [CrossRef]
- Navarro, J.; Obrador, E.; Carretero, J.; Petschen, I.; Aviñó, J.; Perez, P.; Estrela, J.M. Changes in glutathione status and the antioxidant system in blood and in cancer cells associate with tumour growth in vivo. Free Radic. Biol. Med. 1999, 26, 410–418. [Google Scholar] [CrossRef]
- Wilson, C.H.; Shalini, S.; Filipovska, A.; Richman, T.R.; Davies, S.; Martin, S.D.; McGee, S.L.; Puccini, J.; Nikolic, A.; Dorstyn, L.; et al. Age-related proteostasis and metabolic alterations in Caspase-2-deficient mice. Cell Death Dis. 2015, 6, e1615. [Google Scholar] [CrossRef]
- Wilson, C.H.; Nikolic, A.; Kentish, S.J.; Keller, M.; Hatzinikolas, G.; Dorstyn, L.; Page, A.J.; Kumar, S. Caspase-2 deficiency enhances whole-body carbohydrate utilisation and prevents high-fat diet-induced obesity. Cell Death Dis. 2017, 8, e3136. [Google Scholar] [CrossRef]
- Nutt, L.K.; Buchakjian, M.R.; Gan, E.; Darbandi, R.; Yoon, S.Y.; Wu, J.Q.; Miyamoto, Y.J.; Gibbon, J.A.; Andersen, J.L.; Freel, C.D.; et al. Metabolic Control of Oocyte Apoptosis Mediated by 14-3-3ζ-Regulated Dephosphorylation of Caspase-2. Dev. Cell. 2009, 16, 856–866. [Google Scholar] [CrossRef]
- Shi, H.; Wang, Y.; Li, X.; Zhan, X.; Tang, M.; Fina, M.; Su, L.; Pratt, D.; Bu, C.H.; Hildebrand, S.; et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2016, 17, 250–258. [Google Scholar] [CrossRef]
- Slauch, J.M. How does the oxidative burst of macrophages kill bacteria? Still an open question. Mol. Microbiol. 2011, 80, 580–583. [Google Scholar] [CrossRef]
- Fang, F.C. Antimicrobial Actions of Reactive Oxygen Species. mBio 2011, 2, e00141-11. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Itoh, K.; Wakabayashi, J.; Motohashi, H.; Noda, S.; Takahashi, S.; Imakado, S.; Kotsuji, T.; Otsuka, F.; Roop, D.R.; et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003, 35, 238–245. [Google Scholar] [CrossRef]
- Andersen, J.L.; Kornbluth, S. The tangled circuitry of metabolism and apoptosis. Mol. Cell. 2013, 49, 399–410. [Google Scholar] [CrossRef]
- Buchakjian, M.R.; Kornbluth, S. The engine driving the ship: Metabolic steering of cell proliferation and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 715–727. [Google Scholar] [CrossRef]
- Rivière, I.; Brose, K.; Mulligan, R.C. Effects of retroviral vector design on expression of human adenosine deaminase in murine bone marrow transplant recipients engrafted with genetically modified cells. Proc. Natl. Acad. Sci. USA 1995, 92, 6733–6737. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stimulus | Previously Tested Cell Type | Cellular Effect | References |
|---|---|---|---|
| TNF/CHX | HeLa; Jurkat; MEF; IMR90E1A; MCF7 | Death receptor | [26,55,56,57] |
| Etoposide | Jurkat; MEF *; IMR90E1A; HeLa; HCT116; BMM *; U2OS *; U937 | DNA damage | [26,55,58,59,60,61,62,63,64,65,66,67,68] |
| Taxol | HeLa; Jurkat; MEF *; Splenocytes * | Cytoskeletal disruption | [26,55,61,67,69] |
| DHEA | HeLa; Xenopus | Oxidative stress | [26,70] |
| Rotenone | MEF *; osteoclasts * | Oxidative stress | [71,72] |
| DMNQ | Neural stem cells | Oxidative stress | [73] |
| Heat shock | HeLa; Jurkat; Splenocytes * +; MEF * | Heat shock | [26,43,55,71] |
| NONOate | 3T3; H4; BMK | Oxidative stress | [46,74] |
| SNAP | ― | Oxidative stress | This work |
| Erastin | ― | Ferroptosis | This work |
| RSL3 | ― | Ferroptosis | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amason, M.E.; Li, L.; Harvest, C.K.; Lacey, C.A.; Miao, E.A. Validation of the Intermolecular Disulfide Bond in Caspase-2. Biology 2024, 13, 49. https://doi.org/10.3390/biology13010049
Amason ME, Li L, Harvest CK, Lacey CA, Miao EA. Validation of the Intermolecular Disulfide Bond in Caspase-2. Biology. 2024; 13(1):49. https://doi.org/10.3390/biology13010049
Chicago/Turabian StyleAmason, Megan E., Lupeng Li, Carissa K. Harvest, Carolyn A. Lacey, and Edward A. Miao. 2024. "Validation of the Intermolecular Disulfide Bond in Caspase-2" Biology 13, no. 1: 49. https://doi.org/10.3390/biology13010049
APA StyleAmason, M. E., Li, L., Harvest, C. K., Lacey, C. A., & Miao, E. A. (2024). Validation of the Intermolecular Disulfide Bond in Caspase-2. Biology, 13(1), 49. https://doi.org/10.3390/biology13010049