
Pathways for Cardioprotection in Perspective: Focus on Remote Conditioning and Extracellular Vesicles



Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Ischemia/Reperfusion Injury
2.1. Pathophysiology of Myocardial Ischemia/Reperfusion Injury
2.1.1. Oxidative Stress
2.1.2. Calcium Overload
2.1.3. Nitric Oxide and Nitroxyl
2.1.4. Mitochondrial Permeability Transition Pore Opening
Events Triggering the mPTP Opening
Consequences of the mPTP Opening
Transient and Long-Lasting mPTP Opening
3. Cardioprotective Strategies
3.1. Target for Cardioprotective Therapies
3.2. Pre-Conditioning
3.3. Postconditioning
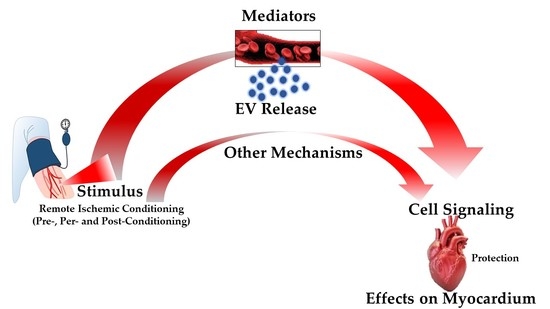
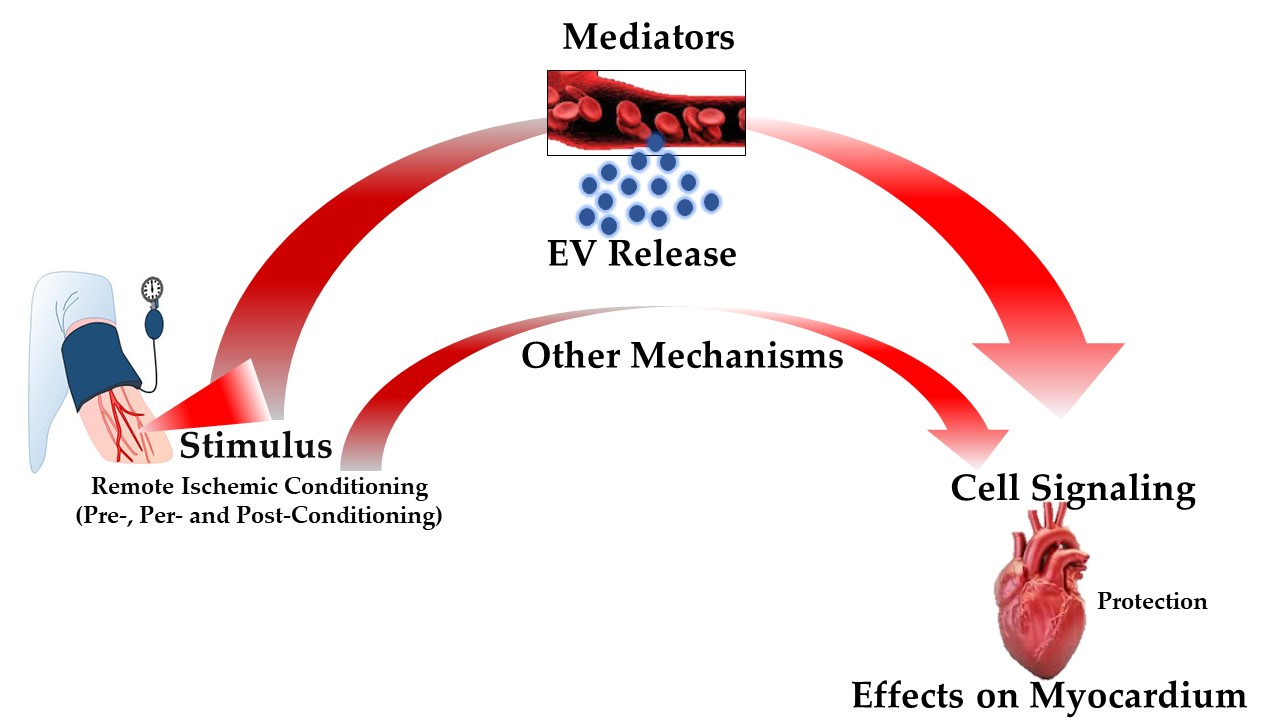
3.4. Remote Ischemic Conditioning
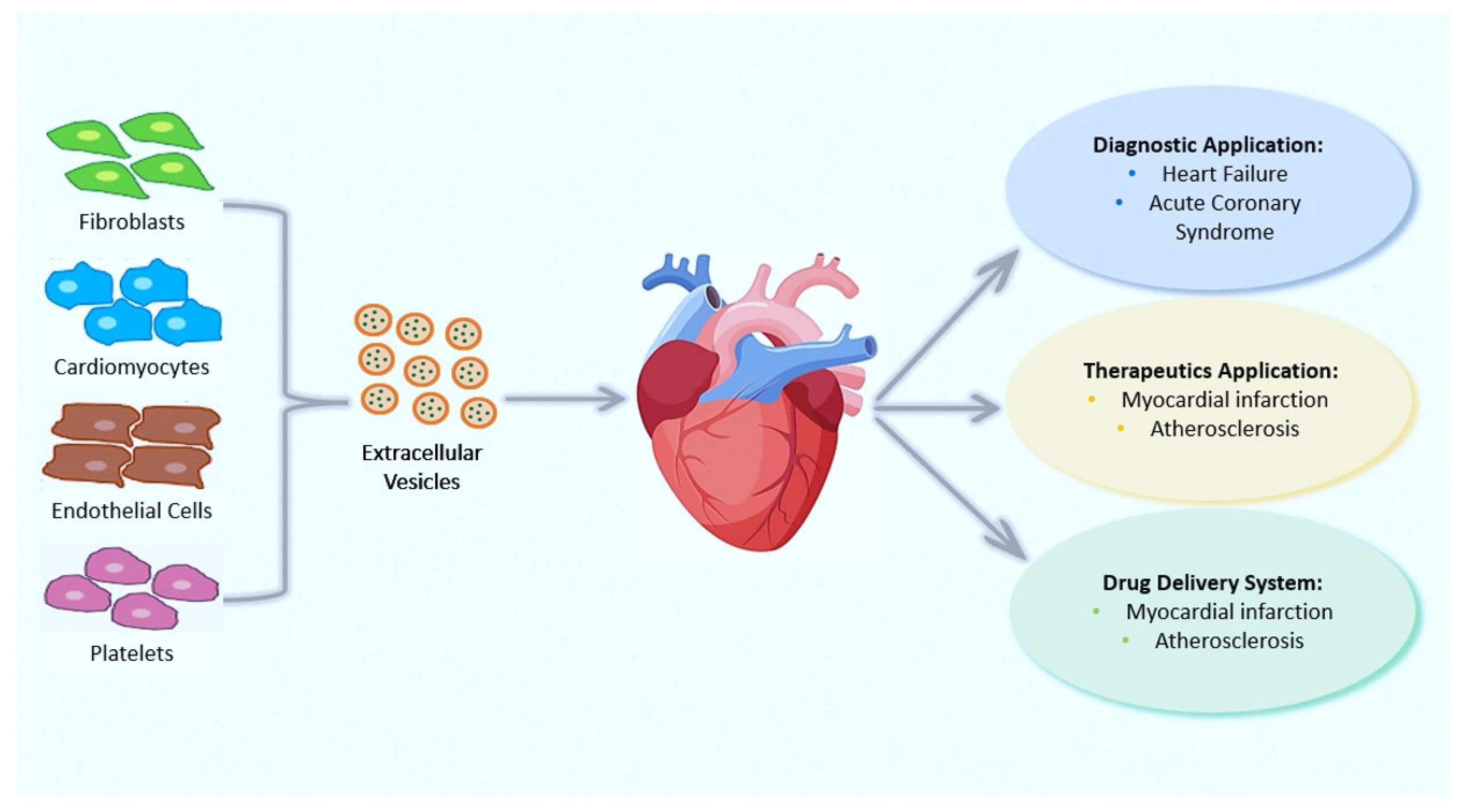
4. Extracellular Vesicles: Novel Communication Concept for Cardioprotective Maneuvers
Applications of EVs in Therapy and Their Roles as Prognostic and Diagnostic Biomarkers for CVDs
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mensah, G.A.; Roth, G.A.; Fuster, V. The Global Burden of Cardiovascular Diseases and Risk Factors: 2020 and Beyond. J. Am. Coll. Cardiol. 2019, 74, 2529–2532. [Google Scholar] [CrossRef] [PubMed]
- Geltman, E.M.; Ehsani, A.A.; Campbell, M.K.; Schechtman, K.; Roberts, R.; Sobel, B.E. The influence of location and extent of myocardial infarction on long-term ventricular dysrhythmia and mortality. Circulation 1979, 60, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Kelle, S.; Roes, S.D.; Klein, C.; Kokocinski, T.; de Roos, A.; Fleck, E.; Bax, J.J.; Nagel, E. Prognostic value of myocardial infarct size and contractile reserve using magnetic resonance imaging. J. Am. Coll. Cardiol. 2009, 54, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Stone, G.W.; Selker, H.P.; Thiele, H.; Patel, M.R.; Udelson, J.E.; Ohman, E.M.; Maehara, A.; Eitel, I.; Granger, C.B.; Jenkins, P.L.; et al. Relationship Between Infarct Size and Outcomes Following Primary PCI: Patient-Level Analysis From 10 Randomized Trials. J. Am. Coll. Cardiol. 2016, 67, 1674–1683. [Google Scholar] [CrossRef] [PubMed]
- Francone, M.; Bucciarelli-Ducci, C.; Carbone, I.; Canali, E.; Scardala, R.; Calabrese, F.A.; Sardella, G.; Mancone, M.; Catalano, C.; Fedele, F.; et al. Impact of primary coronary angioplasty delay on myocardial salvage, infarct size, and microvascular damage in patients with ST-segment elevation myocardial infarction: Insight from cardiovascular magnetic resonance. J. Am. Coll. Cardiol. 2009, 54, 2145–2153. [Google Scholar] [CrossRef]
- Ibáñez, B.; Heusch, G.; Ovize, M.; Van de Werf, F. Evolving therapies for myocardial ischemia/reperfusion injury. J. Am. Coll. Cardiol. 2015, 65, 1454–1471. [Google Scholar] [CrossRef]
- Boersma, H.; Califf, R.; Collins, R.; Deckers, J.W.; Simoons, M.L. Selection of reperfusion therapy for individual patients with evolving myocardial infarction. Eur. Heart J. 1997, 18, 1371–1381. [Google Scholar]
- Yellon, D.M.; Hausenloy, D.J. Myocardial Reperfusion Injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Efentakis, P.; Andreadou, I.; Iliodromitis, K.E.; Triposkiadis, F.; Ferdinandy, P.; Schulz, R.; Iliodromitis, E.K. Myocardial Protection and Current Cancer Therapy: Two Opposite Targets with Inevitable Cost. Int. J. Mol. Sci. 2022, 23, 14121. [Google Scholar] [CrossRef]
- Lecour, S.; Andreadou, I.; Bøtker, H.E.; Davidson, S.M.; Heusch, G.; Ruiz-Meana, M.; Schulz, R.; Zuurbier, C.J.; Ferdinandy, P.; Hausenloy, D.J. IMproving Preclinical Assessment of Cardioprotective Therapies (IMPACT) criteria: Guidelines of the EU-CARDIOPROTECTION COST Action. Basic Res. Cardiol. 2021, 116, 52. [Google Scholar] [CrossRef]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Bøtker, H.E.; Heusch, G.; Ibáñez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef]
- Alloatti, G.; Penna, C.; Comità, S.; Tullio, F.; Aragno, M.; Biasi, F.; Pagliaro, P. Aging, sex and NLRP3 inflammasome in cardiac ischaemic disease. Vascul. Pharmacol. 2022, 145, 107001. [Google Scholar] [CrossRef]
- Ferdinandy, P.; Hausenloy, D.J.; Heusch, G.; Baxter, G.F.; Schulz, R. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol. Rev. 2014, 66, 1142–1174. [Google Scholar] [CrossRef]
- Neely, J.R.; Morgan, H.E. Relationship Between Carbohydrate and Lipid Metabolism and the Energy Balance of Heart Muscle. Annu Rev Physiol. 2003, 36, 413–459. [Google Scholar] [CrossRef]
- Mastrocola, R.; Penna, C.; Tullio, F.; Femminò, S.; Nigro, D.; Chiazza, F.; Serpe, L.; Collotta, D.; Alloatti, G.; Cocco, M.; et al. Pharmacological Inhibition of NLRP3 Inflammasome Attenuates Myocardial Ischemia/Reperfusion Injury by Activation of RISK and Mitochondrial Pathways. Oxid. Med. Cell. Longev. 2016, 2016, 5271251. [Google Scholar] [CrossRef]
- Del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol. Rev. 2019, 99, 1765–1817. [Google Scholar] [CrossRef]
- Davidson, S.M.; Adameová, A.; Barile, L.; Cabrera-Fuentes, H.A.; Lazou, A.; Pagliaro, P.; Stensløkken, K.O.; Garcia-Dorado, D. EU-CARDIOPROTECTION COST Action (CA16225). Mitochondrial and mitochondrial-independent pathways of myocardial cell death during ischaemia and reperfusion injury. J. Cell. Mol. Med. 2020, 24, 3795. [Google Scholar] [CrossRef]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell. Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- Wang, H.B.; Yiang, G.T.; Ding, J.W.; Chen, L.H.; Li, S.; Liu, X.W.; Yang, C.J.; Fan, Z.X.; Yang, J. RNAi-Mediated Down-Regulation of CD47 Protects against Ischemia/Reperfusion-Induced Myocardial Damage via Activation of eNOS in a Rat Model. Cell. Physiol. Biochem. 2016, 40, 1163–1174. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investg. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Cadenas, S.; Aragonés, J.; Landázuri, M.O. Mitochondrial reprogramming through cardiac oxygen sensors in ischaemic heart disease. Cardiovasc. Res. 2010, 88, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Braunersreuther, V.; Jaquet, V. Reactive Oxygen Species in Myocardial Reperfusion Injury: From Physiopathology to Therapeutic Approaches. Curr. Pharm. Biotechnol. 2011, 13, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Tullio, F.; Angotti, C.; Perrelli, M.G.; Penna, C.; Pagliaro, P. Redox balance and cardioprotection. Basic Res. Cardiol. 2013, 108, 392. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, P.; Penna, C. Redox signalling and cardioprotection: Translatability and mechanism. Br. J. Pharmacol. 2015, 172, 1974. [Google Scholar] [CrossRef]
- Robin, E.; Guzy, R.D.; Loor, G.; Iwase, H.; Waypa, G.B.; Marks, J.D.; Hoek, T.L.; Schumacker, P.T. Oxidant Stress during Simulated Ischemia Primes Cardiomyocytes for Cell Death during Reperfusion. J. Biol. Chem. 2007, 282, 19133–19143. [Google Scholar] [CrossRef]
- Di Lisa, F.; Bernardi, P. Modulation of Mitochondrial Permeability Transition in Ischemia-Reperfusion Injury of the Heart. Advantages and Limitations. Curr. Med. Chem. 2015, 22, 2480–2487. [Google Scholar] [CrossRef]
- Ansari, M.; Kurian, G.A. Diabetic animal fed with high-fat diet prevents the protective effect of myocardial ischemic preconditioning effect in isolated rat heart perfusion model. J. Biochem. Mol. Toxicol. 2020, 34, e22457. [Google Scholar] [CrossRef]
- Davidson, S.M.; Andreadou, I.; Barile, L.; Birnbaum, Y.; Cabrera-Fuentes, H.A.; Cohen, M.V.; Downey, J.M.; Girao, H.; Pagliaro, P.; Penna, C. Circulating blood cells and extracellular vesicles in acute cardioprotection. Cardiovasc. Res. 2019, 115, 1156–1166. [Google Scholar] [CrossRef]
- Wider, J.; Undyala, V.V.R.; Whittaker, P.; Woods, J.; Chen, X.; Przyklenk, K. Remote ischemic preconditioning fails to reduce infarct size in the Zucker fatty rat model of type-2 diabetes: Role of defective humoral communication. Basic Res. Cardiol. 2018, 113, 16. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell Biology of Ischemia/Reperfusion Injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar]
- Correa, F.; Buelna-Chontal, M.; Chagoya, V.; García-Rivas, G.; Vigueras, R.M.; Pedraza-Chaverri, J.; García-Niño, W.R.; Hernández-Pando, R.; León-Contreras, J.C.; Zazueta, C. Inhibition of the nitric oxide/cyclic guanosine monophosphate pathway limited the cardioprotective effect of post-conditioning in hearts with apical myocardial infarction. Eur. J. Pharmacol. 2015, 765, 472–481. [Google Scholar] [CrossRef]
- Penna, C.; Cappello, S.; Mancardi, D.; Raimondo, S.; Rastaldo, R.; Gattullo, D.; Losano, G.; Pagliaro, P. Post-conditioning reduces infarct size in the isolated rat heart: Role of coronary flow and pressure and the nitric oxide/cGMP pathway. Basic Res. Cardiol. 2006, 101, 168–179. [Google Scholar] [CrossRef]
- Mancardi, D.; Pagliaro, P.; Ridnour, L.A.; Tocchetti, C.G.; Miranda, K.; Juhaszova, M.; Sollott, S.J.; Wink, D.A.; Paolocci, N. HNO Protects the Myocardium against Reperfusion Injury, Inhibiting the mPTP Opening via PKCε Activation. Antioxidants. 2022, 11, 382. [Google Scholar] [CrossRef]
- Chen, C.L.; Zhang, L.; Jin, Z.; Kasumov, T.; Chen, Y.R. Mitochondrial redox regulation and myocardial ischemia-reperfusion injury. Am. J. Physiol. Cell Physiol. 2022, 322, C12–C23. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Schulz, R.; Girao, H.; Kwak, B.R.; De Stefani, D.; Rizzuto, R.; Bernardi, P.; Di Lisa, F. Mitochondrial ion channels as targets for cardioprotection. J. Cell. Mol. Med. 2020, 24, 7102–7114. [Google Scholar] [CrossRef]
- Igbavboa, U.; Zwizinski, C.W.; Pfeiffer, D.R. Release of mitochondrial matrix proteins through a Ca2+-requiring, cyclosporin-sensitive pathway. Biochem. Biophys. Res. Commun. 1989, 161, 619–625. [Google Scholar] [CrossRef]
- Hurst, S.; Hoek, J.; Sheu, S.S. Mitochondrial Ca2+ and regulation of the permeability transition pore. J. Bioenerg. Biomembr. 2016, 49, 27–47. [Google Scholar] [CrossRef]
- Halestrap, A.P. The mitochondrial permeability transition: Its molecular mechanism and role in reperfusion injury. Biochem. Soc. Symp. 1999, 66, 181–203. [Google Scholar]
- Petronilli, V.; Nicolli, A.; Costantini, P.; Colonna, R.; Bernardi, P. Regulation of the permeability transition pore, a voltage-dependent mitochondrial channel inhibited by cyclosporin A. Biochim. Biophys. Acta 1994, 1187, 255–259. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Kerr, P.M.; Javadov, S.; Woodfield, K.Y. Elucidating the molecular mechanism of the permeability transition pore and its role in reperfusion injury of the heart. Biochim. Biophys. Acta 1998, 1366, 79–94. [Google Scholar] [CrossRef]
- Petronilli, V.; Penzo, D.; Scorrano, L.; Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J. Biol. Chem. 2001, 276, 12030–12034. [Google Scholar] [CrossRef]
- Hausenloy, D.; Wynne, A.; Duchen, M.; Yellon, D. Transient mitochondrial permeability transition pore opening mediates preconditioning-induced protection. Circulation 2004, 109, 1714–1717. [Google Scholar] [CrossRef] [PubMed]
- Juhaszova, M.; Kobrinsky, E.; Zorov, D.B.; Nuss, H.B.; Yaniv, Y.; Fishbein, K.W.; de Cabo, R.; Montoliu, L.; Gabelli, S.B.; Aon, M.A.; et al. ATP Synthase K+-and H+-Fluxes Drive ATP Synthesis and Enable Mitochondrial K+-”Uniporter” Function: I. Characterization of Ion Fluxes. Function 2021, 3, zqac001. [Google Scholar] [CrossRef] [PubMed]
- Juhaszova, M.; Kobrinsky, E.; Zorov, D.B.; Nuss, H.B.; Yaniv, Y.; Fishbein, K.W.; de Cabo, R.; Montoliu, L.; Gabelli, S.B.; Aon, M.A.; et al. ATP Synthase K+-and H+-fluxes Drive ATP Synthesis and Enable Mitochondrial K+-”Uniporter” Function: II. Ion and ATP Synthase Flux Regulation. Function 2022, 3, zqab065. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Garcia-Dorado, D.; Bøtker, H.E.; Davidson, S.M.; Downey, J.; Engel, F.B.; Jennings, R.; Lecour, S.; Leor, J.; Madonna, R.; et al. Novel targets and future strategies for acute cardioprotection: Position Paper of the European Society of Cardiology Working Group on Cellular Biology of the Heart. Cardiovasc. Res. 2017, 113, 564–585. [Google Scholar] [CrossRef]
- Abrial, M.; Da Silva, C.C.; Pillot, B.; Augeul, L.; Ivanes, F.; Teixeira, G.; Cartier, R.; Angoulvant, D.; Ovize, M.; Ferrera, R. Cardiac fibroblasts protect cardiomyocytes against lethal ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2014, 68, 56–65. [Google Scholar] [CrossRef]
- Lassen, T.R.; Just, J.; Hjortbak, M.V.; Jespersen, N.R.; Stenz, K.T.; Gu, T.; Yan, Y.; Su, J.; Hansen, J.; Bæk, R.; et al. Cardioprotection by remote ischemic conditioning is transferable by plasma and mediated by extracellular vesicles. Basic Res. Cardiol. 2021, 116, 16. [Google Scholar] [CrossRef]
- Penna, C.; Femminò, S.; Tapparo, M.; Lopatina, T.; Fladmark, K.E.; Ravera, F.; Comità, S.; Alloatti, G.; Giusti, I.; Dolo, V.; et al. The Inflammatory Cytokine IL-3 Hampers Cardioprotection Mediated by Endothelial Cell-Derived Extracellular Vesicles Possibly via Their Protein Cargo. Cells 2020, 10, 13. [Google Scholar] [CrossRef]
- D’Ascenzo, F.; Femminò, S.; Ravera, F.; Angelini, F.; Caccioppo, A.; Franchin, L.; Grosso, A.; Comità, S.; Cavallari, C.; Penna, C.; et al. Extracellular vesicles from patients with Acute Coronary Syndrome impact on ischemia-reperfusion injury. Pharmacol. Res. 2021, 170, 105715. [Google Scholar] [CrossRef]
- Femminò, S.; D’Ascenzo, F.; Ravera, F.; Comità, S.; Angelini, F.; Caccioppo, A.; Franchin, L.; Grosso, A.; Thairi, C.; Venturelli, E.; et al. Percutaneous Coronary Intervention (PCI) Reprograms Circulating Extracellular Vesicles from ACS Patients Impairing Their Cardio-Protective Properties. Int. J. Mol. Sci. 2021, 22, 10270. [Google Scholar] [CrossRef]
- Lionetti, V. The Role of Exosomes in Health and Disease. Int. J. Mol. Sci. 2022, 23, 11011. [Google Scholar] [CrossRef]
- Maroko, P.R.; Libby, P.; Covell, J.W.; Sobel, B.E.; Ross, J.; Braunwald, E. Precordial S-T segment elevation mapping: An atraumatic method for assessing alterations in the extent of myocardial ischemic injury. The effects of pharmacologic and hemodynamic interventions. Am. J. Cardiol. 1972, 29, 223–230. [Google Scholar] [CrossRef]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef]
- Liu, G.S.; Thornton, J.; Van Winkle, D.M.; Stanley, A.W.; Olsson, R.A.; Downey, J.M. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation 1991, 84, 350–356. [Google Scholar] [CrossRef]
- Cohen, M.V.; Yang, X.M.; Liu, G.S.; Heusch, G.; Downey, J.M. Acetylcholine, Bradykinin, Opioids, and Phenylephrine, but not Adenosine, Trigger Preconditioning by Generating Free Radicals and Opening Mitochondrial KATP Channels. Circ. Res. 2001, 89, 273–278. [Google Scholar] [CrossRef]
- Jones, S.P.; Tang, X.L.; Guo, Y.; Steenbergen, C.; Lefer, D.J.; Kukreja, R.C.; Kong, M.; Li, Q.; Bhushan, S.; Zhu, X.; et al. The NHLBI-Sponsored Consortium for preclinicAl assESsment of cARdioprotective Therapies (CAESAR): A new paradigm for rigorous, accurate, and reproducible evaluation of putative infarct-sparing interventions in mice, rabbits, and pigs. Circ. Res. 2015, 116, 572–586. [Google Scholar] [CrossRef]
- Bolli, R. CAESAR’s legacy: A new era of rigor in preclinical studies of cardioprotection. Basic Res. Cardiol. 2021, 116, 33. [Google Scholar] [CrossRef]
- Pagliaro, P.; Gattullo, D.; Rastaldo, R.; Losano, G. Ischemic preconditioning-From the first to the second window of protection. Life Sci. 2001, 69, 1–15. [Google Scholar] [CrossRef]
- Quarrie, R.; Cramer, B.M.; Lee, D.S.; Steinbaugh, G.E.; Erdahl, W.; Pfeiffer, D.R.; Zweier, J.L.; Crestanello, J.A. Ischemic preconditioning decreases mitochondrial proton leak and reactive oxygen species production in the postischemic heart. J. Surg. Res. 2011, 165, 5–14. [Google Scholar] [CrossRef]
- Dawn, B.; Bolli, R. Role of nitric oxide in myocardial preconditioning. Ann. N. Y. Acad. Sci. 2002, 962, 18–41. [Google Scholar] [CrossRef]
- Berȩsewicz, A.; Ma̧czewski, M.; Duda, M. Effect of classic preconditioning and diazoxide on endothelial function and O2− and NO generation in the post-ischemic guinea-pig heart. Cardiovasc. Res. 2004, 63, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, P.; Moro, F.; Tullio, F.; Perrelli, M.G.; Penna, C. Cardioprotective pathways during reperfusion: Focus on redox signaling and other modalities of cell signaling. Antioxid. Redox Signal. 2011, 14, 833–850. [Google Scholar] [CrossRef] [PubMed]
- Bolli, R. The Late Phase of Preconditioning. Circ. Res. 2000, 87, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Downey, J.M.; Davis, A.M.; Cohen, M.V. Signaling pathways in ischemic preconditioning. Heart Fail. Rev. 2007, 12, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D. Cardioprotection Techniques: Preconditioning, Postconditioning and Remote Con-ditioning (Basic Science). Curr. Pharm. Des. 2013, 19, 4544–4563. [Google Scholar] [CrossRef]
- Zhao, Z.Q.; Corvera, J.S.; Halkos, M.E.; Kerendi, F.; Wang, N.P.; Guyton, R.A.; Vinten-Johansen, J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: Comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003, 285, 579–588. [Google Scholar] [CrossRef]
- Skyschally, A.; Van Caster, P.; Iliodromitis, E.K.; Schulz, R.; Kremastinos, D.T.; Heusch, G. Ischemic postconditioning: Experimental models and protocol algorithms. Basic Res. Cardiol. 2009, 104, 469–483. [Google Scholar] [CrossRef]
- Przyklenk, K.; Bauer, B.; Ovize, M.; Kloner, R.A.; Whittaker, P. Regional ischemic ‘preconditioning’ protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 1993, 87, 893–899. [Google Scholar] [CrossRef]
- Heusch, G.; Bøtker, H.E.; Przyklenk, K.; Redington, A.; Yellon, D. Remote Ischemic Conditioning. J. Am. Coll. Cardiol. 2015, 65, 177–195. [Google Scholar] [CrossRef]
- Penna, C.; Sorge, M.; Tullio, F.; Comità, S.; Femminò, S.; Brancaccio, M.; Pagliaro, P. A TRICk to Improve the Effectiveness of RIC: Role of Limb Temperature in Enhancing the Effectiveness of Remote Ischemic Conditioning. Biology 2022, 11, 146. [Google Scholar] [CrossRef]
- Heusch, G. Cardioprotection: Chances and challenges of its translation to the clinic. Lancet 2013, 381, 166–175. [Google Scholar] [CrossRef]
- Shimizu, M.; Tropak, M.; Diaz, R.J.; Suto, F.; Surendra, H.; Kuzmin, E.; Li, J.; Gross, G.; Wilson, G.J.; Callahan, J.; et al. Transient limb ischaemia remotely preconditions through a humoral mechanism acting directly on the myocardium: Evidence suggesting cross-species protection. Clin. Sci. 2009, 117, 191–200. [Google Scholar] [CrossRef]
- Pedersen, C.M.; Cruden, N.L.; Schmidt, M.R.; Lau, C.; Bøtker, H.E.; Kharbanda, R.K.; Newby, D.E. Remote ischemic preconditioning prevents systemic platelet activation associated with ischemia-reperfusion injury in humans. J. Thromb. Haemost. 2011, 9, 404–407. [Google Scholar] [CrossRef]
- Kharbanda, R.K.; Mortensen, U.M.; White, P.A.; Kristiansen, S.B.; Schmidt, M.R.; Hoschtitzky, J.A.; Vogel, M.; Sorensen, K.; Redington, A.N.; MacAllister, R. Transient limb ischemia induces remote ischemic preconditioning in vivo. Circulation 2002, 106, 2881–2883. [Google Scholar] [CrossRef]
- Konstantinov, I.E.; Arab, S.; Kharbanda, R.K.; Li, J.; Cheung, M.M.; Cherepanov, V.; Downey, G.P.; Liu, P.P.; Cukerman, E.; Coles, J.G.; et al. The remote ischemic preconditioning stimulus modifies inflammatory gene expression in humans. Physiol. Genomics 2004, 19, 143–150. [Google Scholar] [CrossRef]
- Shimizu, M.; Saxena, P.; Konstantinov, I.E.; Cherepanov, V.; Cheung, M.M.; Wearden, P.; Zhangdong, H.; Schmidt, M.; Downey, G.P.; Redington, A.N. Remote ischemic preconditioning decreases adhesion and selectively modifies functional responses of human neutrophils. J. Surg. Res. 2010, 158, 155–161. [Google Scholar] [CrossRef]
- Loukogeorgakis, S.P.; Panagiotidou, A.T.; Broadhead, M.W.; Donald, A.; Deanfield, J.E.; MacAllister, R.J. Remote ischemic preconditioning provides early and late protection against endothelial ischemia-reperfusion injury in humans: Role of the autonomic nervous system. J. Am. Coll. Cardiol. 2005, 46, 450–456. [Google Scholar] [CrossRef]
- Chen, Y.; Shin, Y.K.; Bassham, D.C. YKT6 is a Core Constituent of Membrane Fusion Machineries at the Arabidopsis trans-Golgi Network. J. Mol. Biol. 2005, 350, 92–101. [Google Scholar] [CrossRef]
- Weinbrenner, C.; Schulze, F.; Sárváry, L.; Strasser, R.H. Remote preconditioning by infrarenal aortic occlusion is operative via δ1-opioid receptors and free radicals in vivo in the rat heart. Cardiovasc. Res. 2004, 61, 591–599. [Google Scholar] [CrossRef]
- Khanna, G.; Diwan, V.; Singh, M.; Singh, N.; Jaggi, A.S. Reduction of Ischemic, Pharmacological and Remote Preconditioning Effects by an Antioxidant N-Acetyl Cysteine Pretreatment in Isolated Rat Heart. Yakugaku Zasshi 2008, 128, 469–477. [Google Scholar] [CrossRef]
- Shahid, M.; Tauseef, M.; Sharma, K.K.; Fahim, M. Brief femoral artery ischaemia provides protection against myocardial ischaemia–reperfusion injury in rats: The possible mechanisms. Exp. Physiol. 2008, 93, 954–968. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.S.; Chien, C.T.; Ma, M.C.; Tseng, Y.Z.; Lin, F.Y.; Wang, S.S.; Chen, C.F. Protection ‘outside the box’ (skeletal remote preconditioning) in rat model is triggered by free radical pathway. J. Surg. Res. 2005, 126, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Mudaliar, H.; Rayner, B.; Billah, M.; Kapoor, N.; Lay, W.; Dona, A.; Bhindi, R. Remote ischemic preconditioning attenuates EGR-1 expression following myocardial ischemia reperfusion injury through activation of the JAK-STAT pathway. Int. J. Cardiol. 2017, 228, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Bromage, D.I.; Pickard, J.M.; Rossello, X.; Ziff, O.J.; Burke, N.; Yellon, D.M.; Davidson, S.M. Remote ischaemic conditioning reduces infarct size in animal in vivo models of ischaemia-reperfusion injury: A systematic review and meta-analysis. Cardiovasc. Res. 2017, 113, 288–297. [Google Scholar] [PubMed]
- Hausenloy, D.J.; Kharbanda, R.K.; Møller, U.K.; Ramlall, M.; Aarøe, J.; Butler, R.; Bulluck, H.; Clayton, T.; Dana, A.; Dodd, M.; et al. Effect of remote ischaemic conditioning on clinical outcomes in patients with acute myocardial infarction (CONDI-2/ERIC-PPCI): A single-blind randomised controlled trial. Lancet 2019, 394, 1415–1424. [Google Scholar] [CrossRef]
- Lieder, H.R.; Skyschally, A.; Sturek, M.; Heusch, G.; Kleinbongard, P. Remote ischemic conditioning in Ossabaw minipigs induces the release of humoral cardioprotective triggers, but the myocardium does not respond with reduced infarct size. Am. J. Physiol. Heart Circ. Physiol. 2022, 323, H1365–H1375. [Google Scholar] [CrossRef]
- Barani, B.; Rajasingh, S.; Rajasingh, J. Exosomes: Outlook for Future Cell-Free Cardiovascular Disease Therapy. Adv. Exp. Med. Biol. 2017, 998, 285–307. [Google Scholar]
- Ma, F.; Liu, H.; Shen, Y.; Zhang, Y.; Pan, S. Platelet-derived microvesicles are involved in cardio-protective effects of remote preconditioning. Int. J. Clin. Exp. Pathol. 2015, 8, 10832. [Google Scholar]
- Silva-Palacios, A.; Arroyo-Campuzano, M.; Flores-García, M.; Patlán, M.; Hernández-Díazcouder, A.; Alcántara, D.; Ramírez-Camacho, I.; Arana-Hidalgo, D.; Soria-Castro, E.; Sánchez, F.; et al. Citicoline Modifies the Expression of Specific miRNAs Related to Cardioprotection in Patients with ST-Segment Elevation Myocardial Infarction Subjected to Coronary Angioplasty. Pharmaceuticals. 2022, 15, 925. [Google Scholar] [CrossRef]
- Pitt, J.M.; Kroemer, G.; Zitvogel, L. Extracellular vesicles: Masters of intercellular communication and potential clinical interventions. J. Clin. Investg. 2016, 126, 1139–1143. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Hegyesi, H.; Pallinger, É.; Mecsei, S.; Hornyák, B.; Kovácsházi, C.; Brenner, G.B.; Giricz, Z.; Pálóczi, K.; Kittel, Á.; Tóvári, J.; et al. Circulating cardiomyocyte-derived extracellular vesicles reflect cardiac injury during systemic inflammatory response syndrome in mice. Cell. Mol. Life Sci. 2022, 79, 84. [Google Scholar] [CrossRef]
- Sluijter, J.P.G.; Davidson, S.M.; Boulanger, C.M.; Buzás, E.I.; de Kleijn, D.P.V.; Engel, F.B.; Giricz, Z.; Hausenloy, D.J.; Kishore, R.; Lecour, S.; et al. Extracellular vesicles in diagnostics and therapy of the ischaemic heart: Position Paper from the Working Group on Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc. Res. 2018, 114, 19–34. [Google Scholar] [CrossRef]
- Femminò, S.; Penna, C.; Margarita, S.; Comità, S.; Brizzi, M.F.; Pagliaro, P. Extracellular vesicles and cardiovascular system: Biomarkers and Cardioprotective Effectors. Vascul. Pharmacol. 2020, 135, 106790. [Google Scholar] [CrossRef]
- Terrasini, N.; Lionetti, V. Exosomes in Critical Illness. Crit. Care Med. 2017, 45, 1054–1060. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef]
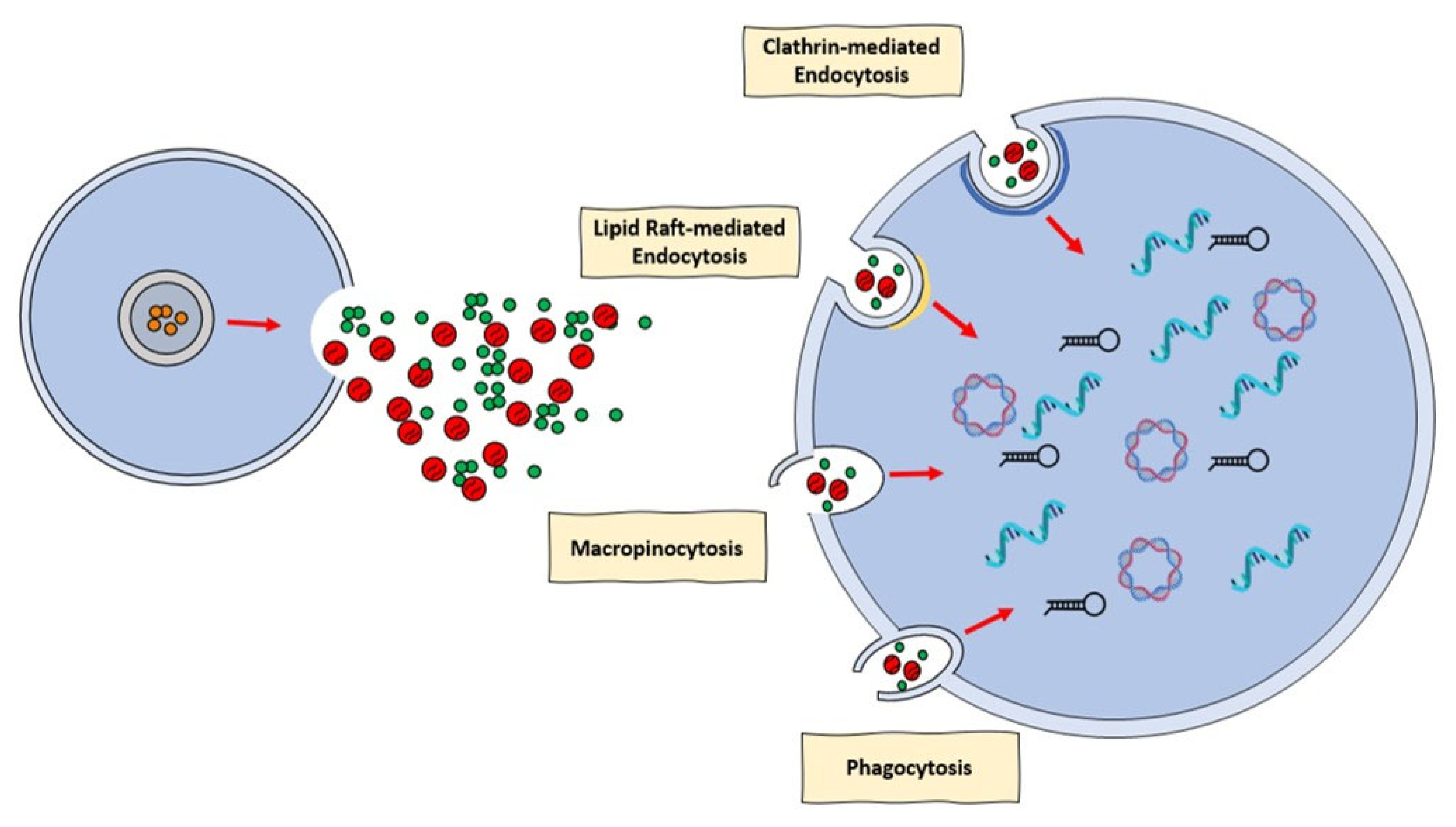
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 4, 3. [Google Scholar] [CrossRef]
- Record, M.; Carayon, K.; Poirot, M.; Silvente-Poirot, S. Exosomes as new vesicular lipid transporters involved in cell-cell communication and various pathophysiologies. Biochim. Biophys. Acta 2014, 1841, 108–120. [Google Scholar] [CrossRef]
- Chong, S.Y.; Lee, C.K.; Huang, C.; Ou, Y.H.; Charles, C.J.; Richards, A.M.; Neupane, Y.R.; Pavon, M.V.; Zharkova, O.; Pastorin, G.; et al. Extracellular Vesicles in Cardiovascular Diseases: Alternative Biomarker Sources, Therapeutic Agents, and Drug Delivery Carriers. Int. J. Mol. Sci. 2019, 20, 3272. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Hu, G.; Gao, L.; Hackfort, B.T.; Zucker, I.H. Extracellular vesicular MicroRNA-27a* contributes to cardiac hypertrophy in chronic heart failure. J. Mol. Cell. Cardiol. 2020, 143, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, P.; Kumari, R.; Goswami, S.K.; Li, J.; Pal, H.; Suleiman, Z.; Cheng, Z.; Krishnamurthy, P.; Kishore, R.; Verma, S.K. Myofibroblast-Derived Exosome Induce Cardiac Endothelial Cell Dysfunction. Front. Cardiovasc. Med. 2021, 8, 676267. [Google Scholar] [CrossRef] [PubMed]
- Shaihov-Teper, O.; Ram, E.; Ballan, N.; Brzezinski, R.Y.; Naftali-Shani, N.; Masoud, R.; Ziv, T.; Lewis, N.; Schary, Y.; Levin-Kotler, L.P.; et al. Extracellular Vesicles From Epicardial Fat Facilitate Atrial Fibrillation. Circulation 2021, 143, 2475–2493. [Google Scholar] [CrossRef]
- Caccioppo, A.; Franchin, L.; Grosso, A.; Angelini, F.; D’Ascenzo, F.; Brizzi, M.F. Ischemia Reperfusion Injury: Mechanisms of Damage/Protection and Novel Strategies for Cardiac Recovery/Regeneration. Int. J. Mol. Sci. 2019, 20, 5024. [Google Scholar] [CrossRef]
- Dignat-George, F.; Boulanger, C.M. The many faces of endothelial microparticles. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 27–33. [Google Scholar] [CrossRef]
- Li, C.; Pei, F.; Zhu, X.; Duan, D.D.; Zeng, C. Circulating microRNAs as novel and sensitive biomarkers of acute myocardial Infarction. Clin. Biochem. 2012, 45, 727–732. [Google Scholar] [CrossRef]
- Wang, K.; Gan, T.Y.; Li, N.; Liu, C.Y.; Zhou, L.Y.; Gao, J.N.; Chen, C.; Yan, K.W.; Ponnusamy, M.; Zhang, Y.H.; et al. Circular RNA mediates cardiomyocyte death via miRNA-dependent upregulation of MTP18 expression. Cell Death Differ. 2017, 24, 1111–1120. [Google Scholar] [CrossRef]
- Giricz, Z.; Varga, Z.V.; Baranyai, T.; Sipos, P.; Pálóczi, K.; Kittel, Á.; Buzás, E.I.; Ferdinandy, P. Cardioprotection by remote ischemic preconditioning of the rat heart is mediated by extracellular vesicles. J. Mol. Cell. Cardiol. 2014, 68, 75–78. [Google Scholar] [CrossRef]
- Xiong, F.; Mao, R.; Zhang, L.; Zhao, R.; Tan, K.; Liu, C.; Xu, J.; Du, G.; Zhang, T. CircNPHP4 in monocyte-derived small extracellular vesicles controls heterogeneous adhesion in coronary heart atherosclerotic disease. Cell Death Dis. 2021, 12, 948. [Google Scholar] [CrossRef]
- Irmscher, S.; Zipfel, S.L.H.; Halder, L.D.; Ivanov, L.; Gonzalez-Delgado, A.; Waldeyer, C.; Seiffert, M.; Brunner, F.J.; von der Heide, M.; Löschmann, I.; et al. Factor H-related protein 1 (FHR-1) is associated with atherosclerotic cardiovascular disease. Sci. Rep. 2021, 11, 22511. [Google Scholar] [CrossRef]
- Martins-Marques, T.; Ribeiro-Rodrigues, T.; de Jager, S.C.; Zuzarte, M.; Ferreira, C.; Cruz, P.; Reis, L.; Baptista, R.; Gonçalves, L.; Sluijter, J.P.; et al. Myocardial infarction affects Cx43 content of extracellular vesicles secreted by cardiomyocytes. Life Sci. Alliance 2020, 3, e202000821. [Google Scholar] [CrossRef]
- Ito, H. No-reflow phenomenon and prognosis in patients with acute myocardial infarction. Nat. Clin. Pract. Cardiovasc. Med. 2006, 3, 499–506. [Google Scholar] [CrossRef]
- Gough, D.J.; Koetz, L.; Levy, D.E. The MEK-ERK Pathway Is Necessary for Serine Phosphorylation of Mitochondrial STAT3 and Ras-Mediated Transformation. PLoS ONE 2013, 8, e83395. [Google Scholar] [CrossRef]
- Rossello, X.; Yellon, D.M. The RISK pathway and beyond. Basic Res. Cardiol. 2018, 113, 2. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Tsang, A.; Mocanu, M.M.; Yellon, D.M. Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am. J. Physiol. Hear. Circ. Physiol. 2005, 288, 971–976. [Google Scholar] [CrossRef]
- Ghaderi, S.; Alidadiani, N.; Dilaver, N.; Heidari, H.R.; Parvizi, R.; Rahbarghazi, R.; Soleimani-Rad, J.; Baradaran, B. Letter to the editor regarding article, ‘Role of glycogen synthase kinase following myocardial infarction and ischemia-reperfusion’. Apoptosis 2019, 24, 541. [Google Scholar] [CrossRef]
- Tyagi, S.; Singh, N.; Virdi, J.K.; Jaggi, A.S. Diabetes abolish cardioprotective effects of remote ischemic conditioning: Evidences and possible mechanisms. J. Physiol. Biochem. 2019, 75, 19–28. [Google Scholar] [CrossRef]
- Heusch, G.; Musiolik, J.; Kottenberg, E.; Peters, J.; Jakob, H.; Thielmann, M. STAT5 Activation and Cardioprotection by Remote Ischemic Preconditioning in Humans. Circ. Res. 2012, 110, 111–115. [Google Scholar] [CrossRef]
- Lecour, S. Multiple protective pathways against reperfusion injury: A SAFE path without Aktion? J. Mol. Cell. Cardiol. 2009, 46, 607–609. [Google Scholar] [CrossRef]
- O’Sullivan, K.E.; Breen, E.P.; Gallagher, H.C.; Buggy, D.J.; Hurley, J.P. Understanding STAT3 signaling in cardiac ischemia. Basic Res. Cardiol. 2016, 111, 27. [Google Scholar] [CrossRef] [PubMed]
- Comità, S.; Femminò, S.; Thairi, C.; Alloatti, G.; Boengler, K.; Pagliaro, P.; Penna, C. Regulation of STAT3 and its role in cardioprotection by conditioning: Focus on non-genomic roles targeting mitochondrial function. Basic Res. Cardiol. 2021, 116, 56. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Li, C.; Jia, X.; Xie, J.; Tang, Z.; Jin, M.; Chen, Q.; Sun, Y.; He, S.; Li, X.; et al. Extracellular vesicle-derived CircWhsc1 promotes cardiomyocyte proliferation and heart repair by activating TRIM59/STAT3/Cyclin B2 pathway. J. Adv. Res. 2022, 22, 00293-4. [Google Scholar] [CrossRef]
- Sánchez-Alonso, S.; Alcaraz-Serna, A.; Sánchez-Madrid, F.; Alfranca, A. Extracellular Vesicle-Mediated Immune Regulation of Tissue Remodeling and Angiogenesis After Myocardial Infarction. Front. Immunol. 2018, 9, 2799. [Google Scholar] [CrossRef] [PubMed]
- Rahmati, S.; Shojaei, F.; Shojaeian, A.; Rezakhani, L.; Dehkordi, M.B. An overview of current knowledge in biological functions and potential theragnostic applications of exosomes. Chem. Phys. Lipids 2020, 226, 104836. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhang, C.; Zhang, J.; Jiao, Z.; Dong, N.; Wang, G.; Wang, Z.; Wang, L. Localized injection of miRNA-21-enriched extracellular vesicles effectively restores cardiac function after myocardial infarction. Theranostics 2019, 9, 2346–2360. [Google Scholar] [CrossRef]
- Ciullo, A.; Biemmi, V.; Milano, G.; Bolis, S.; Cervio, E.; Fertig, E.T.; Gherghiceanu, M.; Moccetti, T.; Camici, G.G.; Vassalli, G.; et al. Exosomal Expression of CXCR4 Targets Cardioprotective Vesicles to Myocardial Infarction and Improves Outcome after Systemic Administration. Int. J. Mol. Sci. 2019, 20, 468. [Google Scholar] [CrossRef]
- Wang, X.; Chen, Y.; Zhao, Z.; Meng, Q.; Yu, Y.; Sun, J.; Yang, Z.; Chen, Y.; Li, J.; Ma, T.; et al. Engineered Exosomes With Ischemic Myocardium-Targeting Peptide for Targeted Therapy in Myocardial Infarction. J. Am. Heart Assoc. 2018, 7, e008737. [Google Scholar] [CrossRef]
- Heusch, G. Critical Issues for the Translation of Cardioprotection. Circ. Res. 2017, 120, 1477–1486. [Google Scholar] [CrossRef]
- Loyer, X.; Zlatanova, I.; Devue, C.; Yin, M.; Howangyin, K.Y.; Klaihmon, P.; Guerin, C.L.; Kheloufi, M.; Vilar, J.; Zannis, K.; et al. Intra-Cardiac Release of Extracellular Vesicles Shapes Inflammation Following Myocardial Infarction. Circ. Res. 2018, 123, 100–106. [Google Scholar] [CrossRef]
- Frey, U.H.; Klaassen, M.; Ochsenfarth, C.; Murke, F.; Thielmann, M.; Kottenberg, E.; Kleinbongard, P.; Klenke, S.; Engler, A.; Heusch, G.; et al. Remote ischaemic preconditioning increases serum extracellular vesicle concentrations with altered micro-RNA signature in CABG patients. Acta Anaesthesiol. Scand. 2019, 63, 483–492. [Google Scholar] [CrossRef]
- Abel, F.; Murke, F.; Gaida, M.; Garnier, N.; Ochsenfarth, C.; Theiss, C.; Thielmann, M.; Kleinbongard, P.; Giebel, B.; Peters, J.; et al. Extracellular vesicles isolated from patients undergoing remote ischemic preconditioning decrease hypoxia-evoked apoptosis of cardiomyoblasts after isoflurane but not propofol exposure. PLoS ONE 2020, 15, e0228948. [Google Scholar] [CrossRef]
- Haller, P.M.; Jäger, B.; Piackova, E.; Sztulman, L.; Wegberger, C.; Wojta, J.; Gyöngyösi, M.; Kiss, A.; Podesser, B.K.; Spittler, A.; et al. Changes in Circulating Extracellular Vesicles in Patients with ST-Elevation Myocardial Infarction and Potential Effects of Remote Ischemic Conditioning-A Randomized Controlled Trial. Biomedicines 2020, 8, 218. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Ntsekhe, M.; Yellon, D.M. A future for remote ischaemic conditioning in high-risk patients. Basic Res. Cardiol. 2020, 115, 35. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Comità, S.; Rubeo, C.; Giordano, M.; Penna, C.; Pagliaro, P. Pathways for Cardioprotection in Perspective: Focus on Remote Conditioning and Extracellular Vesicles. Biology 2023, 12, 308. https://doi.org/10.3390/biology12020308
Comità S, Rubeo C, Giordano M, Penna C, Pagliaro P. Pathways for Cardioprotection in Perspective: Focus on Remote Conditioning and Extracellular Vesicles. Biology. 2023; 12(2):308. https://doi.org/10.3390/biology12020308
Chicago/Turabian StyleComità, Stefano, Chiara Rubeo, Magalì Giordano, Claudia Penna, and Pasquale Pagliaro. 2023. "Pathways for Cardioprotection in Perspective: Focus on Remote Conditioning and Extracellular Vesicles" Biology 12, no. 2: 308. https://doi.org/10.3390/biology12020308
APA StyleComità, S., Rubeo, C., Giordano, M., Penna, C., & Pagliaro, P. (2023). Pathways for Cardioprotection in Perspective: Focus on Remote Conditioning and Extracellular Vesicles. Biology, 12(2), 308. https://doi.org/10.3390/biology12020308