
An Investigation of the Variations in Complete Mitochondrial Genomes of Lingula anatina in the Western Pacific Region

 ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
2. Results and Discussion
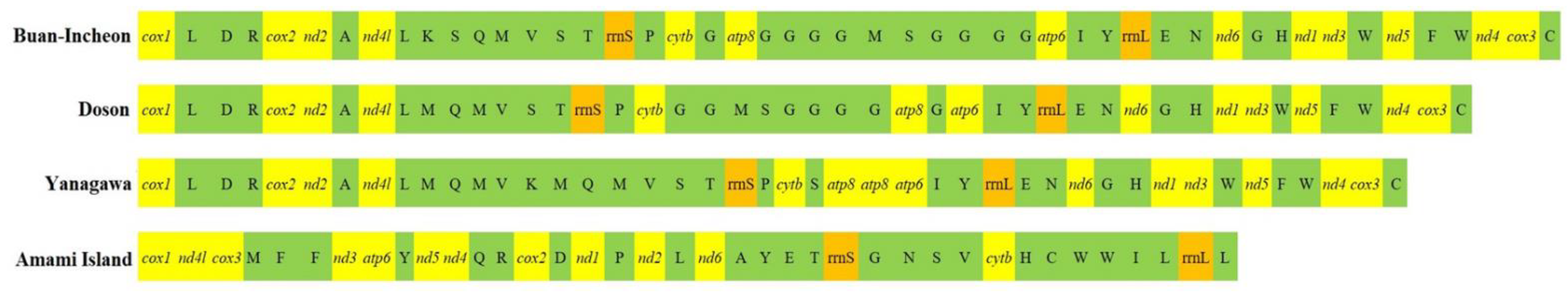
2.1. Mitogenome Structure
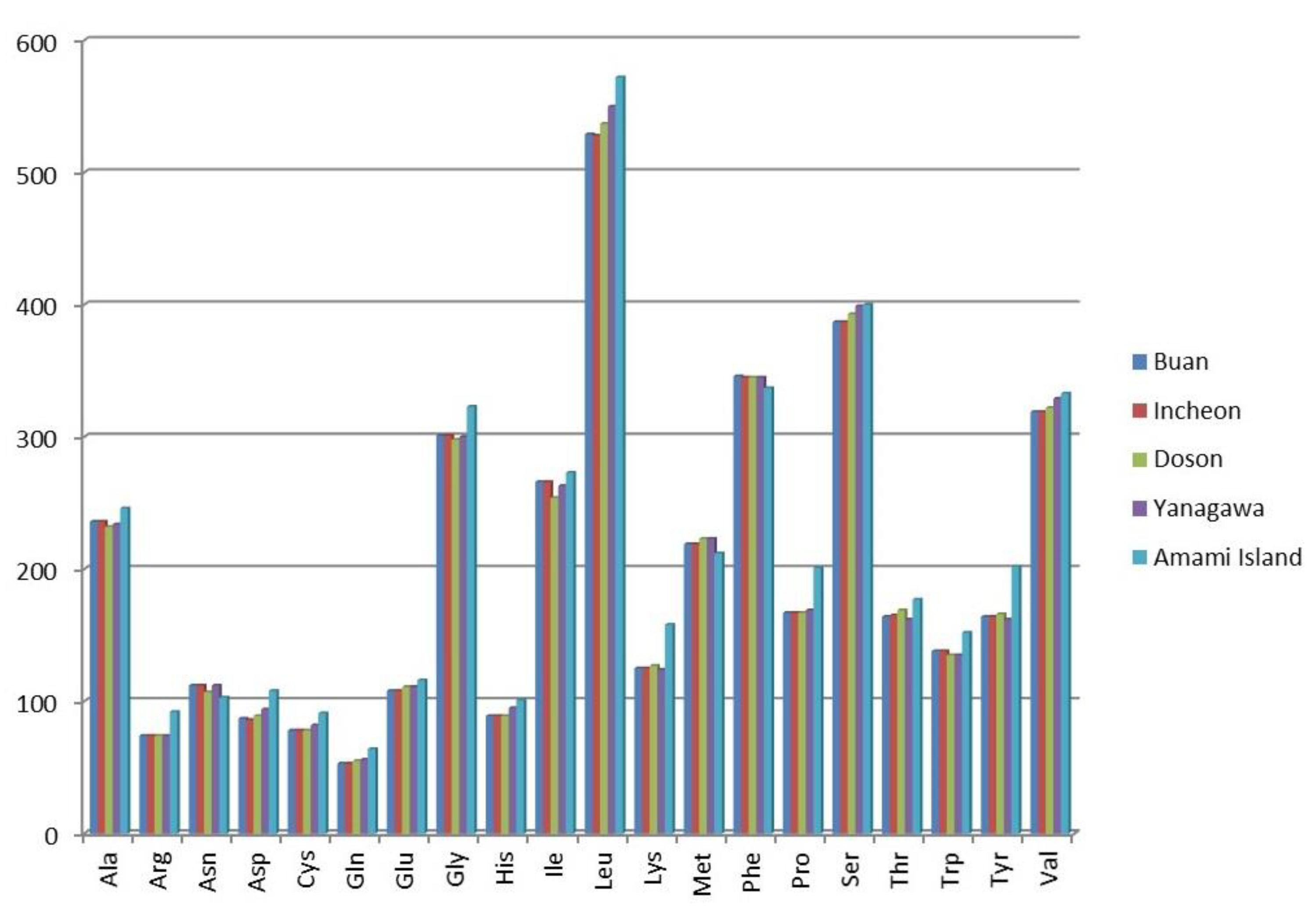
2.2. Protein-Coding Genes
2.3. Transfer RNAs and Ribosomal RNAs
2.4. Non-Coding Region
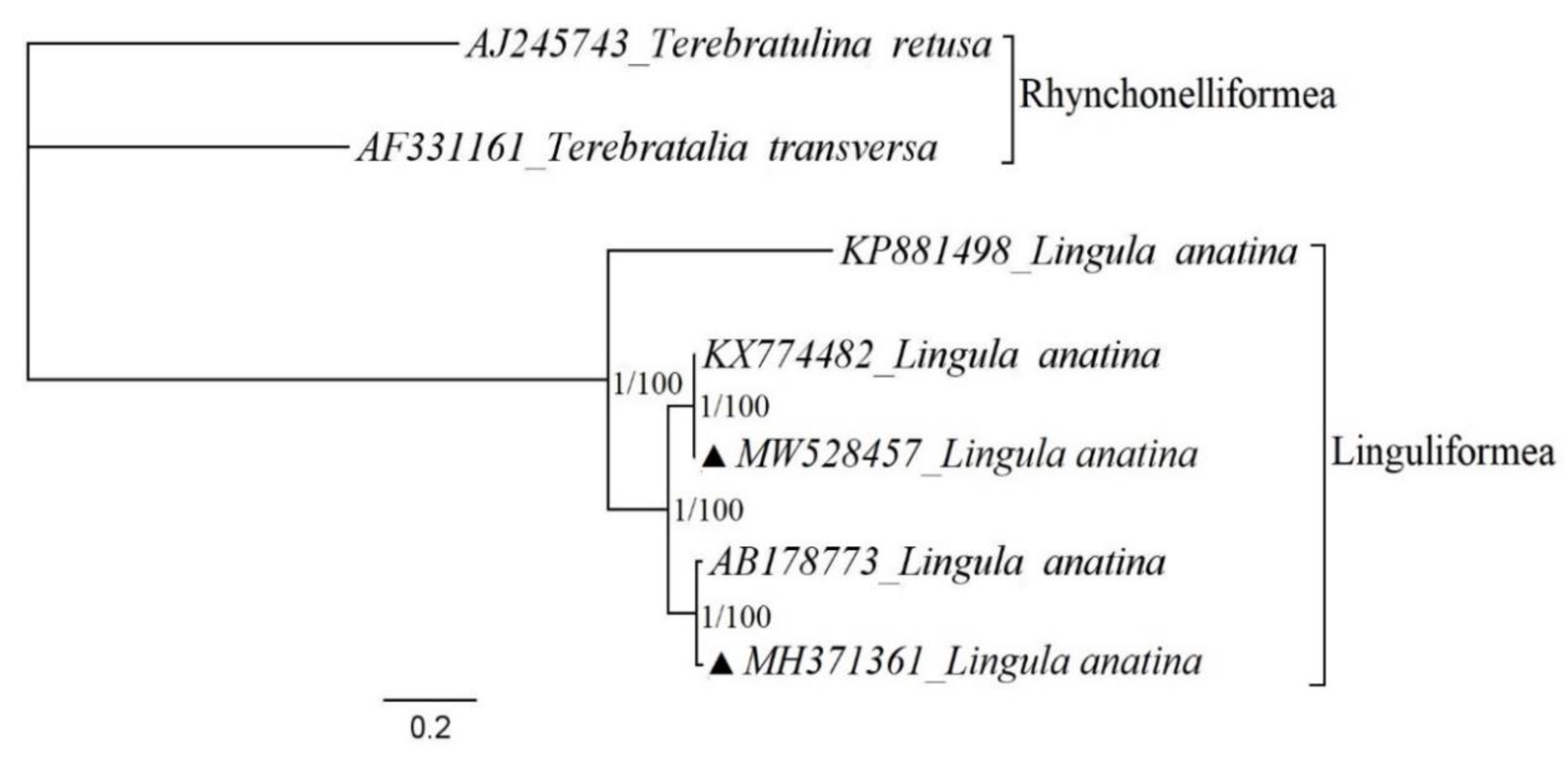
2.5. Phylogenetic Studies
3. Material and Methods
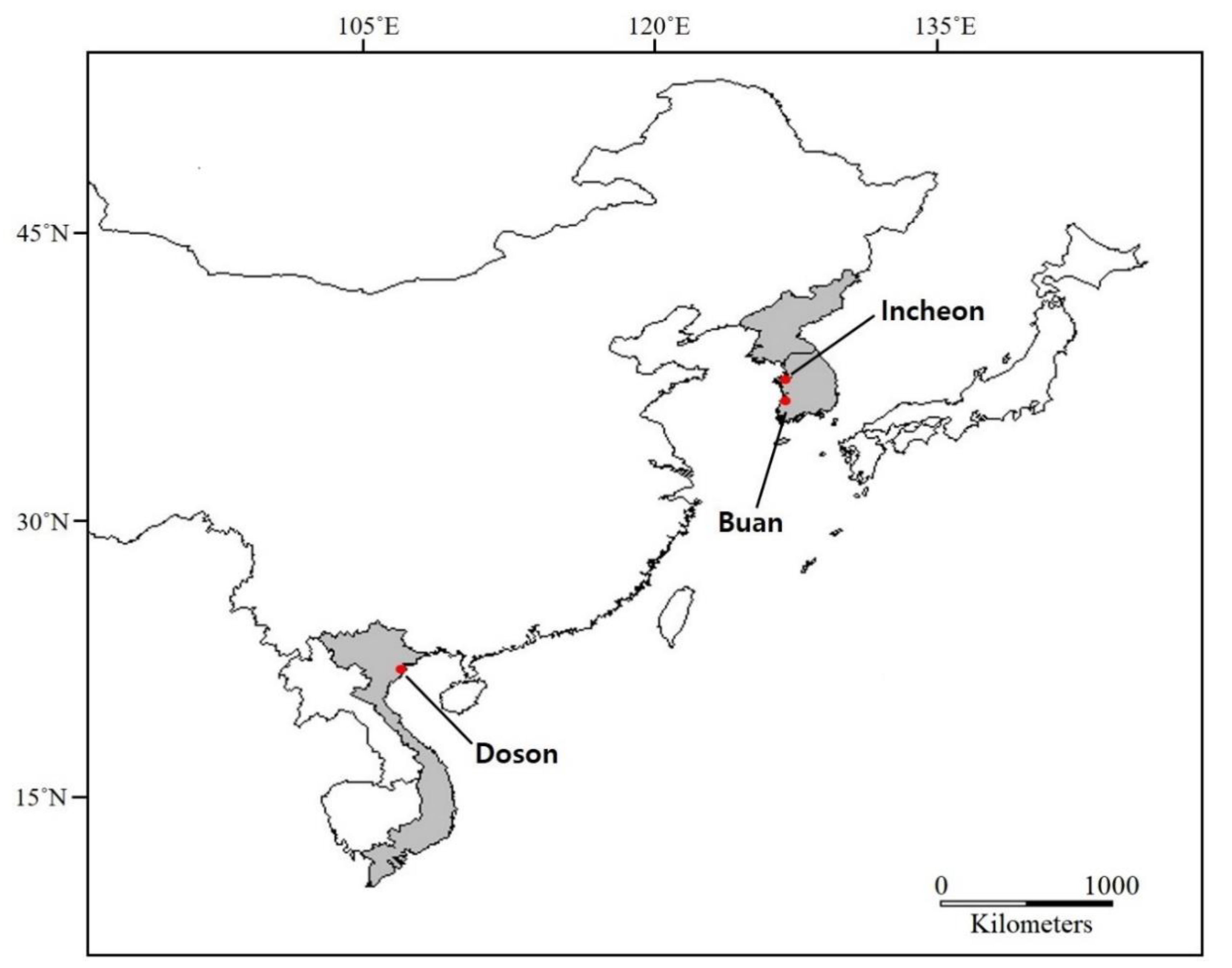
3.1. Sampling and DNA Sequencing
3.2. Analysis of Mitochondrial DNA
3.3. Phylogenetic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, D.X.; Hewitt, G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Curole, J.P.; Kocher, T.D. Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends Ecol. Evol. 1999, 14, 394–398. [Google Scholar] [CrossRef]
- Lin, C.P.; Danforth, B.N. How do insect nuclear and mitochondrial gene substitution patterns differ? Insights from Bayesian analyses of combined datasets. Mol. Phylogenet. Evol. 2004, 30, 686–702. [Google Scholar] [CrossRef]
- Avise, J.C. Mitochondrial DNA polymorphism and a connection between genetics and demography of relevance to conservation. Conser. Biol. 1995, 9, 686–690. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef]
- Luo, Y.J.; Satoh, N.; Endo, K. Mitochondrial gene order variation in the brachiopod Lingula anatina and its implications for mitochondrial evolution in lophotrochozoans. Mar. Genomics 2015, 1, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Emig, C.C.; Álvarez, F.; Bitner, M.A. Brachiopoda Database. Available online: http://paleopolis.rediris.es/brachiopoda_database/ (accessed on 1 February 2021).
- Harper, D.A.; Popov, L.E.; Holmer, L.E. Brachiopods: Origin and early history. Paleontology 2017, 60, 1–24. [Google Scholar] [CrossRef]
- Merkel, C.; Deuschle, J.; Griesshaber, E.; Enders, S.; Steinhauser, E.; Hochleitner, R.; Brand, U.; Schmahl, W.W. Mechanical properties of modern calcite-(Mergerlia truncata) and phosphate-shelled brachiopods (Discradisca stella and Lingula anatina) determined by nanoindentation. J. Struct. Biol. 2009, 168, 396–408. [Google Scholar] [CrossRef]
- Sperling, E.A.; Pisani, D.; Peterson, K.J. Molecular paleobiological insights into the origin of the Brachiopoda. Evol. Dev. 2011, 13, 290–303. [Google Scholar] [CrossRef]
- Yang, S.; Lai, X.; Sheng, G.; Wang, S. Deep genetic divergence within a “living fossil” brachiopod Lingula anatina. J. Paleontol. 2013, 87, 902–908. [Google Scholar] [CrossRef]
- Endo, K. The phylogenetic position of brachiopods inferred from mitochondrialgene orders. In Brachiopods: Past and Present. Systemic Association Special Volume; Brunton, H., Cocks, R., Long, S., Eds.; Taylor & Francis: London, UK, 2001; pp. 129–137. [Google Scholar]
- Emig, C.C. Proof that Lingula (Brachiopoda) is not a living fossil, and emended diagnoses of the family Linulidae. Carnets Geol. Noteb. Geol. 2003. [Google Scholar] [CrossRef]
- Kim, S.G.; Karagozlu, M.Z.; Kim, C.B. Phylogenetic investigations of Lingula anatina among some northwestern Pacific populations, based on mitochondrial DNA cytochrome c oxidase subunit I gene. J. Asia Pac. Biodivers 2017, 10, 162–166. [Google Scholar] [CrossRef]
- Nishizawa, A.; Sarashina, I.; Tsujimoto, Y.; Iijima, I. Artificial fertilization, early development and chromosome numbers in the brachiopod Lingula anatina. Palaeontology 2010, 84, 309–316. [Google Scholar]
- Endo, K.; Noguchi, Y.; Ueshima, R.; Jacobs, H.T. Novel repetitive structures, deviant protein-encoding sequences and unidentified ORFs in the mitochondrial genome of the brachiopod Lingula anatina. J. Mol. Evol. 2005, 61, 36–53. [Google Scholar] [CrossRef] [PubMed]
- Karagozlu, M.Z.; Kim, S.G.; Thinh, D.D.; Kim, C.B. Complete mitochondrial genome analysis of Lingula anatina from Korea (Brachiopoda, Lingulida, Lingulidae). Mitochondrial DNA B Resour. 2017, 2, 829–830. [Google Scholar] [CrossRef] [PubMed]
- Eo, S.H.; DeWoody, J.A. Evolutionary rates of mitochondrial genomes correspond to diversification rates and to contemporary species richness in birds and reptiles. Proc. Biol. Sci. 2010, 277, 3587–3592. [Google Scholar] [CrossRef]
- Wu, X.; Xu, X.; Yu, Z.; Wei, J.; Xia, J. The relationship between the rate of molecular evolution and the rate of genome rearrangement in animal mitochondrial genomes. J. Mol. Evol. 2006, 63, 375–392. [Google Scholar]
- Sun, Y.B.; Shen, Y.Y.; Irwin, D.M.; Zhang, Y.P. Evaluating the roles of energetic functional constraints on teleost mitochondrial-encoded protein evolution. Mol. Bio. Evol. 2011, 28, 39–44. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef]
- Yang, H.; Xia, J.; Zhang, J.E.; Yang, J.; Zhao, H.; Wang, Q.; Sun, J.; Xue, H.; Wu, Y.; Chen, J.; et al. Characterization of the complete mitochondrial genome sequences of three croakers (perciformes, sciaenidae) and novel insights into the phylogenetics. Int. J. Mol. Sci. 2018, 19, 1741. [Google Scholar] [CrossRef]
- Wolff, M.; Uribe, A.; Ortiz, A.; Duque, P. A preliminary study of forensic entomology in Medellin, Colombia. Forensic Sci. Int. 2001, 120, 53–59. [Google Scholar] [CrossRef]
- Yamauchi, M.M.; Miya, M.U.; Nishida, M. Complete mitochondrial DNA sequence of the swimming crab, Portunus trituberculatus (Crustacea: Decapoda: Brachyura). Gene 2003, 311, 129–135. [Google Scholar] [CrossRef]
- Ki, J.S.; Dahms, H.U.; Hwang, J.S.; Lee, J.S. The complete mitogenome of the hydrothermal vent crab Xenograpsus testudinatus (Decapoda, Brachyura) and comparison with brachyuran crabs. Comp. Biochem. Physiol. Part D Genom. Proteom. 2009, 4, 290–299. [Google Scholar] [CrossRef]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173. [Google Scholar] [PubMed]
- Ye, Y.; Li, J.; Wu, C. Genetic diversity and population connectivity of the Asian green mussel Perna viridis in South China Sea, inferred from mitochondria DNA markers. Biochem. Syst. Ecol. 2015, 61, 470–476. [Google Scholar] [CrossRef]
- Yan, S.; Catanese, G.; Brown, C.L.; Wang, M.; Yang, C.; Yang, T.B. Study on the chub mackerel (Scomber japonicus) in the northwestern Pacific indicates the late Pleistocene population isolation. Marine Ecol. Evol. Perspect. 2015, 36, 753–765. [Google Scholar] [CrossRef]
- Fujii, R.; Ueno, R.; Yamamoto, T. Breeding season and life history of Lingula anatina after settlement in Amami-Oshima Island, Kagoshima, Japan. Plankton Benthos Res. 2019, 14, 45–51. [Google Scholar] [CrossRef]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads-abaiting and iterative mapping approach. Nucleic Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef]
- Bernt, M.; Dnonath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucl. Aci. Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Raden, M.; Ali, S.M.; Alkhnbashi, O.S.; Busch, A.; Costa, F.; Davis, J.A.; Eggenhofer, F.; Gelhausen, R.; Georg, J.; Heyne, S. Freiburg RNA tools: A central online resource for RNA-focused research and teaching. Nucleic Acids Res. 2018, 46, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, J.; Zhao, X.Q.; Wang, J.; Wong, G.K.S.; Yu, J. Kaks_calculator: Calculating Ka and Ks through model selection and model averaging. Genom. Proteom. Bioinf. 2006, 4, 259–263. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML Version 8: A tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree v1.4.4. Available online: http://tree.bio.ed.ac.uk/softwar (accessed on 1 February 2021).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Position | Size (bp) | Initiation Codon | Termination Codon | Intergenic Sequence | Strand | |
|---|---|---|---|---|---|---|---|
| cox1 | 1 | 1722 | 1722 | GTG | TAG | 5 | + |
| tRNA-Leu | 1731 | 1798 | 68 | 8 | + | ||
| tRNA-Asp | 1802 | 1869 | 68 | 3 | + | ||
| tRNA-Arg | 1875 | 1942 | 68 | 5 | + | ||
| cox2 | 1944 | 2693 | 750 | ATG | TAA | 1 | + |
| nd2 | 2999 | 4036 | 1038 | ATG | TAA | 305 | + |
| tRNA-Ala | 4039 | 4104 | 66 | 2 | + | ||
| nd4l | 4109 | 4444 | 336 | ATG | TAG | 4 | + |
| tRNA-Leu | 4457 | 4523 | 67 | 12 | + | ||
| tRNA-Lys | 4808 | 4877 | 70 | 284 | + | ||
| tRNA-Ser | 4891 | 4960 | 70 | 13 | + | ||
| tRNA-Gln | 5036 | 5102 | 67 | 75 | + | ||
| tRNA-Met | 6736 | 6807 | 72 | 1633 | + | ||
| tRNA-Val | 6838 | 6904 | 67 | 30 | + | ||
| tRNA-Ser | 7425 | 7476 | 52 | 520 | |||
| tRNA-Thr | 7625 | 7693 | 69 | 148 | + | ||
| 12S rRNA | 7694 | 8964 | 1271 | 0 | + | ||
| tRNA-Pro | 8965 | 9033 | 69 | 0 | + | ||
| cytb | 9107 | 10,363 | 1257 | ATG | TAG | 73 | + |
| tRNA-Gly | 10,609 | 10,672 | 64 | 245 | + | ||
| atp8 | 10,675 | 10,851 | 177 | ATG | TAA | 2 | + |
| tRNA-Gly | 11,069 | 11,135 | 67 | 217 | + | ||
| tRNA-Gly | 11,627 | 11,693 | 67 | 491 | + | ||
| tRNA-Gly | 12,189 | 12,255 | 67 | 495 | + | ||
| tRNA-Gly | 12,733 | 12,799 | 67 | 477 | + | ||
| tRNA-Met | 13,171 | 13,236 | 66 | 371 | + | ||
| tRNA-Ser | 13,241 | 13,308 | 68 | 4 | + | ||
| tRNA-Gly | 13,328 | 13,395 | 68 | 19 | + | ||
| tRNA-Gly | 14,265 | 14,332 | 68 | 869 | + | ||
| tRNA-Gly | 14,880 | 14,947 | 68 | 547 | + | ||
| tRNA-Gly | 15,475 | 15,541 | 67 | 527 | + | ||
| atp6 | 15,547 | 16,275 | 729 | ATG | TAA | 5 | + |
| tRNA-Ile | 16,362 | 16,432 | 71 | 86 | + | ||
| tRNA-Tyr | 16,437 | 16,503 | 67 | 4 | + | ||
| 16S rRNA | 16,504 | 17,966 | 1463 | 0 | + | ||
| tRNA-Glu | 17,968 | 18,035 | 68 | 1 | + | ||
| tRNA-Asn | 18,050 | 18,119 | 70 | 14 | + | ||
| nd6 | 18,123 | 18,728 | 606 | ATG | TAG | 3 | + |
| tRNA-Gly | 18,734 | 18,800 | 67 | 5 | + | ||
| tRNA-His | 18,816 | 18,880 | 65 | 15 | + | ||
| nd1 | 18,931 | 19,839 | 909 | ATA | TAA | 50 | + |
| nd3 | 19,931 | 20,428 | 498 | ATG | TAG | 91 | + |
| tRNA-Trp | 20,503 | 20,571 | 69 | 74 | + | ||
| nd5 | 20,582 | 22,330 | 1749 | ATT | TAG | 10 | + |
| tRNA-Phe | 22,331 | 22,395 | 65 | 0 | + | ||
| tRNA-Trp | 22,412 | 22,480 | 69 | 16 | + | ||
| nd4 | 22,495 | 23,766 | 1272 | ATA | TAG | 14 | + |
| cox3 | 23,915 | 24,790 | 876 | GTG | TAA | 148 | + |
| tRNA-Cys | 24,803 | 24,870 | 68 | 12 | + | ||
| Gene | Position | Size (bp) | Initiation Codon | Termination Codon | Intergenic Sequence | Strand | |
|---|---|---|---|---|---|---|---|
| cox1 | 1 | 1725 | 1725 | ATA | TAA | 5 | + |
| tRNA-Leu | 1731 | 1798 | 68 | 5 | + | ||
| tRNA-Asp | 1802 | 1868 | 67 | 3 | + | ||
| tRNA-Arg | 1872 | 1939 | 68 | 3 | + | ||
| cox2 | 1941 | 2690 | 750 | ATG | TAG | 1 | + |
| nd2 | 2996 | 4033 | 1038 | ATG | TAA | 305 | + |
| tRNA-Ala | 4944 | 5009 | 66 | 910 | + | ||
| nd4l | 5013 | 5348 | 336 | GTG | TAG | 3 | + |
| tRNA-Leu | 5361 | 5427 | 67 | 12 | + | ||
| tRNA-Met | 5463 | 5534 | 72 | 35 | + | ||
| tRNA-Gln | 5542 | 5609 | 68 | 7 | + | ||
| tRNA-Met | 7238 | 7309 | 72 | 1628 | + | ||
| tRNA-Val | 7338 | 7404 | 67 | 28 | + | ||
| tRNA-Ser | 7925 | 7976 | 52 | 520 | + | ||
| tRNA-Thr | 8125 | 8193 | 69 | 148 | + | ||
| 12S rRNA | 8194 | 9463 | 1270 | 0 | + | ||
| tRNA-Pro | 9464 | 9532 | 69 | 0 | + | ||
| cytb | 9608 | 10,864 | 1257 | ATG | TAG | 75 | + |
| tRNA-Gly | 10,878 | 10,944 | 67 | 13 | + | ||
| tRNA-Gly | 11,455 | 11,521 | 67 | 510 | + | ||
| tRNA-Met | 11,900 | 11,965 | 66 | 378 | + | ||
| tRNA-Ser | 11,970 | 12,037 | 68 | 4 | + | ||
| tRNA-Gly | 12,057 | 12,123 | 67 | 19 | + | ||
| tRNA-Gly | 12,615 | 12,682 | 68 | 491 | + | ||
| tRNA-Gly | 14,048 | 14,115 | 68 | 1365 | + | ||
| tRNA-Gly | 15,471 | 15,538 | 68 | 1355 | + | ||
| atp8 | 15,543 | 15,719 | 177 | ATG | TAA | 4 | + |
| tRNA-Gly | 15,965 | 16,032 | 68 | 245 | + | ||
| atp6 | 16,038 | 16,766 | 729 | ATG | TAA | 5 | + |
| tRNA-Ile | 16,793 | 16,863 | 71 | 26 | + | ||
| tRNA-Tyr | 16,868 | 16,934 | 67 | 4 | + | ||
| 16S rRNA | 16,935 | 18,397 | 1463 | 0 | + | ||
| tRNA-Glu | 18,398 | 18,465 | 68 | 0 | + | ||
| tRNA-Asn | 18,480 | 18,549 | 70 | 14 | + | ||
| nd6 | 18,555 | 19,160 | 606 | ATG | TAG | 5 | + |
| tRNA-Gly | 19,166 | 19,232 | 67 | 5 | + | ||
| tRNA-His | 19,250 | 19,314 | 65 | 17 | + | ||
| nd1 | 19,335 | 20,273 | 939 | ATA | TAA | 20 | + |
| nd3 | 20,365 | 20,862 | 498 | ATG | TAG | 91 | + |
| tRNA-Trp | 20,937 | 21,005 | 69 | 74 | + | ||
| nd5 | 21,017 | 22,765 | 1749 | ATT | TAG | 11 | + |
| tRNA-Phe | 22,766 | 22,830 | 65 | 0 | + | ||
| tRNA-Trp | 22,845 | 22,913 | 69 | 14 | + | ||
| nd4 | 22,928 | 24,199 | 1272 | ATG | TAA | 14 | + |
| cox3 | 24,348 | 25,223 | 876 | GTG | TAA | 148 | + |
| tRNA-Cys | 25,232 | 25,300 | 69 | 8 | + | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karagozlu, M.Z.; Do, T.D.; Kim, J.-I.; Choi, T.-J.; Kim, S.-G.; Kim, C.-B. An Investigation of the Variations in Complete Mitochondrial Genomes of Lingula anatina in the Western Pacific Region. Biology 2021, 10, 367. https://doi.org/10.3390/biology10050367
Karagozlu MZ, Do TD, Kim J-I, Choi T-J, Kim S-G, Kim C-B. An Investigation of the Variations in Complete Mitochondrial Genomes of Lingula anatina in the Western Pacific Region. Biology. 2021; 10(5):367. https://doi.org/10.3390/biology10050367
Chicago/Turabian StyleKaragozlu, Mustafa Zafer, Thinh Dinh Do, Jung-Il Kim, Tae-June Choi, Seong-Geun Kim, and Chang-Bae Kim. 2021. "An Investigation of the Variations in Complete Mitochondrial Genomes of Lingula anatina in the Western Pacific Region" Biology 10, no. 5: 367. https://doi.org/10.3390/biology10050367
APA StyleKaragozlu, M. Z., Do, T. D., Kim, J.-I., Choi, T.-J., Kim, S.-G., & Kim, C.-B. (2021). An Investigation of the Variations in Complete Mitochondrial Genomes of Lingula anatina in the Western Pacific Region. Biology, 10(5), 367. https://doi.org/10.3390/biology10050367