1. Introduction
Since its discovery in the early eighties AIDS, the clinical result of infection by HIV has caused over 25 million deaths worldwide [
1]. The global reach and fatal prognosis of human immunodeficiency virus (HIV) infection have emphasised the need for effective antiretroviral therapies. While several effective treatment regimens have been devised, involving inhibitors that target multiple viral proteins [
2], the emergence of mutations in these proteins is a contributing factor to the eventual failure of treatment. Understanding the interactions of inhibitory drugs and viral proteins may in the future allow us to tailor treatment to individual patients and develop more effective therapies.
The essential role played by reverse transcriptase (RT) in the replicative cycle of HIV has made it an attractive target for HIV/AIDS therapy. The enzyme uses the single stranded RNA viral genome as a template to create a single strand of DNA which is in turn used as a template to create a double stranded DNA (
dsDNA) copy of the genome. The
dsDNA copy is then suitable for integration into the chromosomes of human host cells. HIV-1 RT is a multifunctional enzyme with distinct polymerase and RNaseH active sites. At the polymerase active site incoming nucleotides matching the template RNA or DNA are incorporated into the growing complementary DNA chain. The RNaseH active site catalyses the breakdown of the RNA genome, freeing the DNA copy to act as a template for the creation of the final double stranded DNA genome. Inhibition of either enzymatic activity compromises viral replication. Two classes of inhibitor are currently in clinical use, called nucleotide analogue RT inhibitors (NRTIs) and non-nucleoside analogue RT inhibitors (NNRTIs). The former mimic the natural deoxynucleotide triphosphate (dNTP) substrate, binding to nascent DNA chains and acting as chain terminators preventing further elongation. NNRTIs operate allosterically, binding approximately 10 Å from the polymerase active site in a pocket which does not exist in the apoenzyme (see
Figure 1).
HIV-1 RT is a heterodimeric protein consisting of 440 and 560 amino acid subunits (known as p51 and p66 respectively due to their molecular weights). Both subunits are derived from the same
gag-pol polyprotein with the p51 subunit lacking the C-terminal domain (containing the RNaseH active site), which is cleaved from it by the HIV-1 protease. The PDB [
3] contains many different crystal structures of HIV-1 RT in a range of states including those bound to both NRTIs [
4,
5] and NNRTIs [
6,
7,
8], natural DNA and RNA substrates [
9,
10,
11,
12] in addition to the apoenzyme [
13]. The supposed resemblance of the p66 to a right hand has led to the shared subdomains being known as the fingers, palm, thumb and connection [
14]. The reported crystal structures of the unliganded enzyme show the tips of the p66 fingers and thumb domains in close proximity, with a separation of less than 15 Å (one structure does not exhibit this feature but was created by soaking out an NNRTI), in what is known as the closed conformation. Despite deriving from the same sequence the subunits have very different forms once folded. All template/primer bound systems show the enzyme in a more open configuration with the template/primer substrate fitting between the thumb and fingers domains [
15]. A similar conformation, but with greater separation between the p66 fingers and thumb, is seen in all NNRTI bound structures.
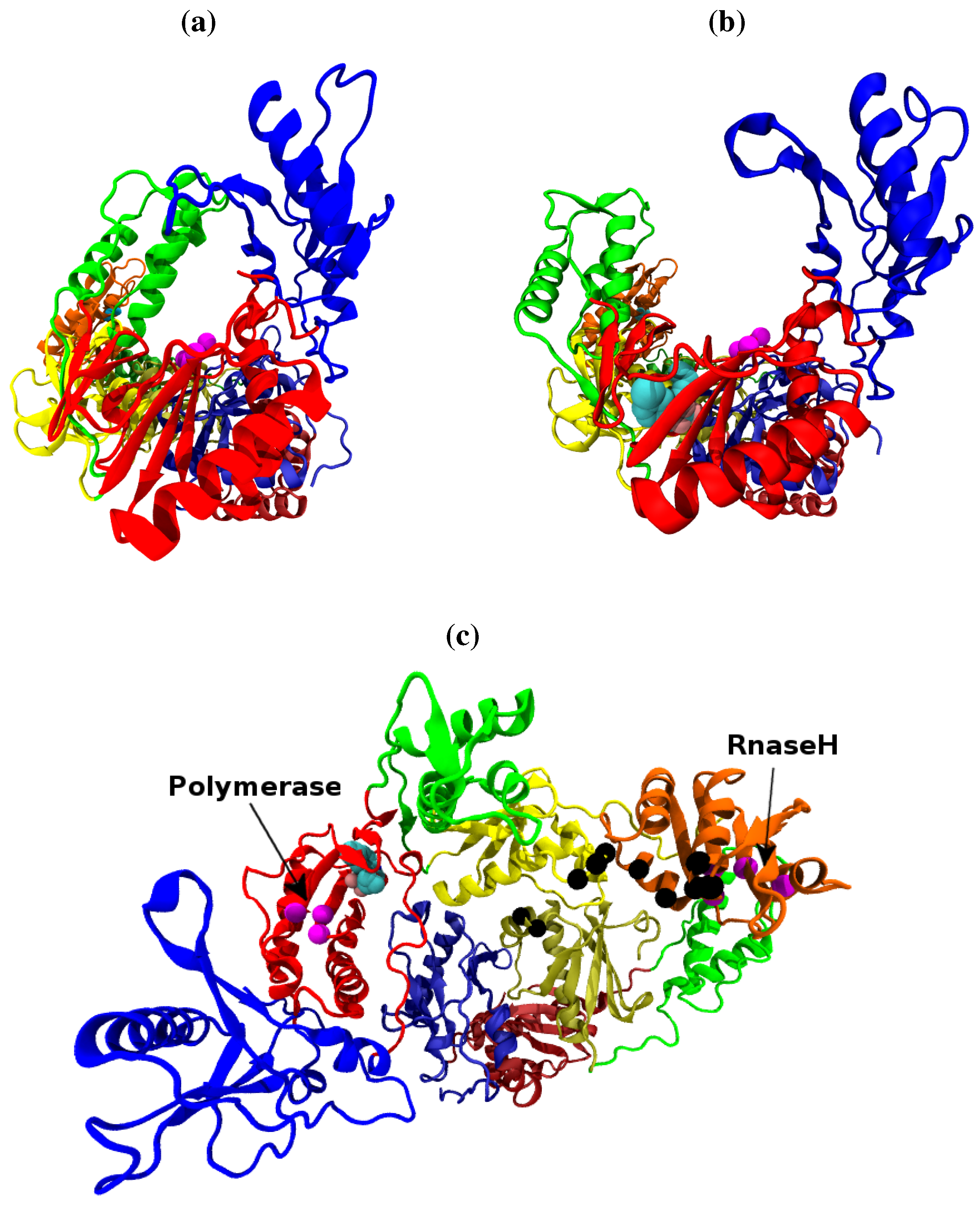
Figure 1 shows both the closed apo and open NNRTI bound conformations of the enzyme. Both the polymerase and RNase H catalytic sites are located within the p66 subunit. The polymerase active site (formed by Asp110, Asp185 and Asp186) is found in the palm subdomain, the RNaseH active site being located at the opposite end of the enzyme with a distance of approximately 30 Å separating them along the central template/primer binding cleft. When bound to a RNA/DNA hybrid template/primer approximately 19 base pairs are contained between the two catalytic sites [
12]. The template/primer is positioned at the two active sites by contacts with regions known as the polymerase primer grip (p66 residues 227 to 235) and the RNaseH primer grip (p66 residues 359, 360, 361, 473, 475, 476, 501 and 505, and p51 residues 395 and 396) [
12].
Figure 1.
The structure of the HIV-1 RT shown along the template/primer binding cleft in the (a) closed apo (based on the 1DLO crystal structure) and (b) open NNRTI bound conformation (based on the EFV bound 1IKW structure, the van der Waals surface of the drug is shown). In this view the p66 subunit is most prominent, in (c) viewed from above the template/primer binding cleft both the p66 and p51 monomers are clearly visible. The subunits are named after the structures supposed likeness to a right hand. It is in fact, the folding of the p66 seen in (a) and (b) which results in the likeness. However the subdomains retain their name in p51 as seen in (b). The subdomains are known as the (F)ingers (blue), (P)alm (red), (T)humb (green), (C)onnection (yellow) and (R)NaseH (orange). The first 4 subdomains form the polymerase domain and are common to both subunits. In all figures the residues of the polymerase and RNaseH active sites are shown as magenta beads (the former is located in the palm). The residues of the RNaseH primer grip are indicated by black beads.
Figure 1.
The structure of the HIV-1 RT shown along the template/primer binding cleft in the (a) closed apo (based on the 1DLO crystal structure) and (b) open NNRTI bound conformation (based on the EFV bound 1IKW structure, the van der Waals surface of the drug is shown). In this view the p66 subunit is most prominent, in (c) viewed from above the template/primer binding cleft both the p66 and p51 monomers are clearly visible. The subunits are named after the structures supposed likeness to a right hand. It is in fact, the folding of the p66 seen in (a) and (b) which results in the likeness. However the subdomains retain their name in p51 as seen in (b). The subdomains are known as the (F)ingers (blue), (P)alm (red), (T)humb (green), (C)onnection (yellow) and (R)NaseH (orange). The first 4 subdomains form the polymerase domain and are common to both subunits. In all figures the residues of the polymerase and RNaseH active sites are shown as magenta beads (the former is located in the palm). The residues of the RNaseH primer grip are indicated by black beads.
![Biology 01 00222 g001]()
Several mechanisms by which NNRTIs may inhibit reverse transcriptase have been proposed. Two of the most widely described are that NNRTI binding disrupts the geometry of the polymerase active site [
16], or that the conformation or dynamics of the polymerase primer grip are perturbed preventing catalytically competent positioning of the 3
'-end of the primer. Another common model is the “arthritic thumb” model in which NNRTI binding is said to restrict the movement of the p66 thumb [
17,
18,
19]. The reduction of thumb motility is thought to prevent the translocation of the template/primer complex during nucleic acid polymerisation. Recently, the need to reconcile data indicating that NNRTIs can influence the rate of RNaseH processing [
20,
21] and the clinical observation of resistance mutations distal from the NNRTI binding pocket [
22,
23,
24] have led to the realisation that a more subtle picture accounting for bidirectional coupling between the two active sites is necessary for a full understanding of NNRTI function.
In contrast to the wealth of structural data comparatively little experimental evidence is available describing the conformation and dynamics of the entire enzyme in solution that could help to reach this understanding. Spin labelling experiments have shown that in solution the enzyme exists in an equilibrium between this state and a more open conformation [
25], and more recently hydrogen exchange mass spectrometry (HXMS) has been used to provide information on the dynamics of the peptide backbone [
26,
27]. Similarly the relatively large size of HIV-1 RT has limited the number and scope of computational studies. Recently, studies have begun to emerge that probe NNRTI binding [
28,
29,
30] and its impact on enzyme dynamics at the atomistic level. In particular molecular dynamics (MD) simulations by Ivetac and McCammon [
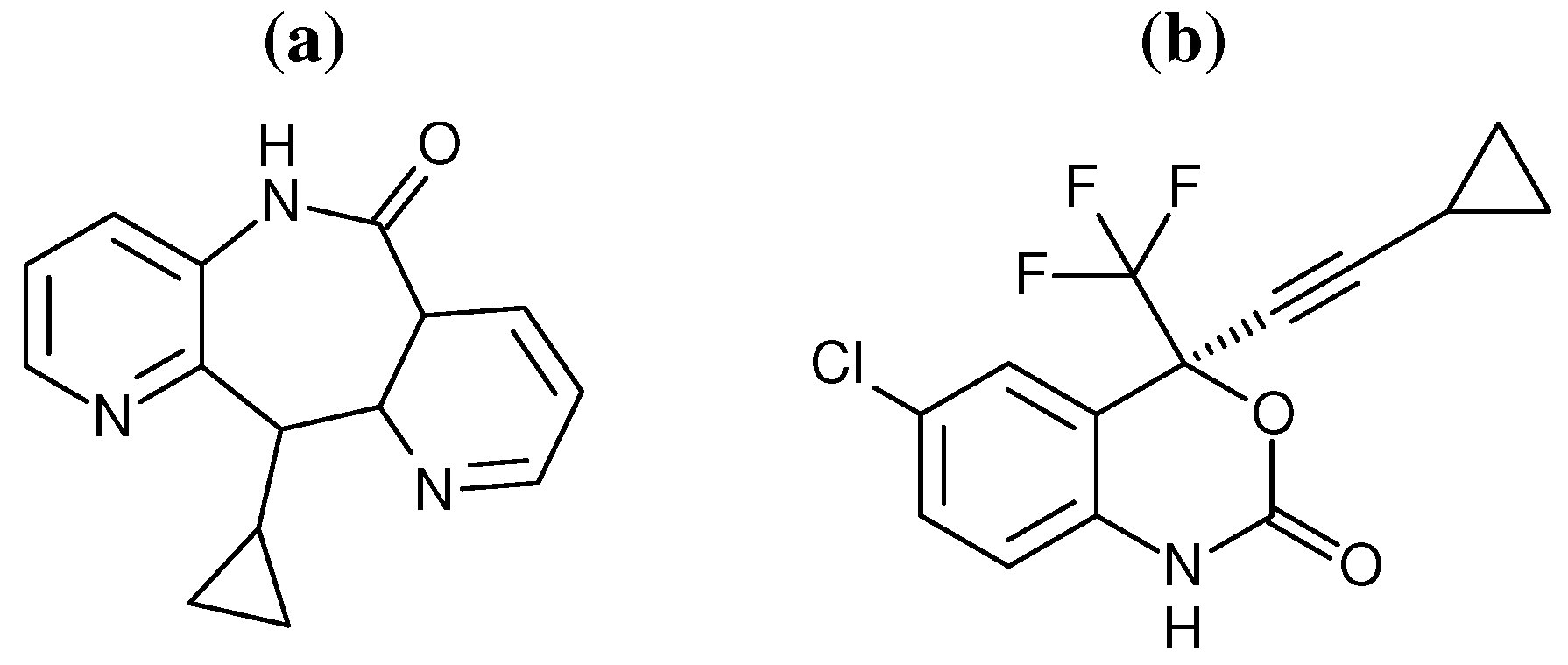
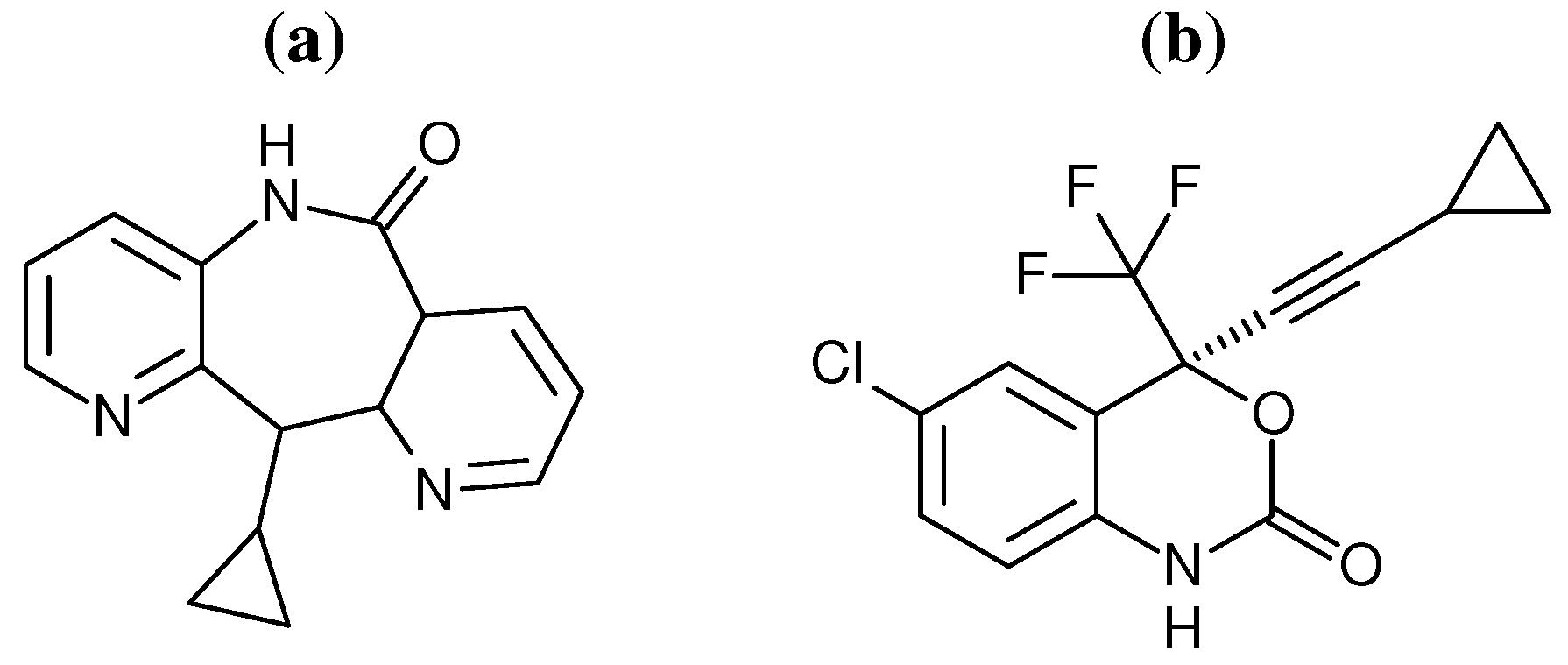
29] have provided evidence to support the “arthritic thumb” hypothesis. However, coarse grained network model analysis, based on crystal structures, has indicated that different NNRTIs alter domain movements in distinctive ways. Here we use a combination of network models and atomistic MD to investigate the impact of the binding of two different NNRTIs on the dynamics of distant regions of the HIV-1 RT structure. The inhibitors efavirenz (EFZ) and nevirapine (NVP) were chosen to represent the NNRTI class of drugs (the chemical structures of both drugs are shown in
Figure 2).
Figure 2.
Chemical structures of the two NNRTIs used in this study: (a) nevirapine (NVP) and (b) efavirenz (EFV).
Figure 2.
Chemical structures of the two NNRTIs used in this study: (a) nevirapine (NVP) and (b) efavirenz (EFV).
3. Results: Coarse Grained Models of HIV-1 RT Dynamics
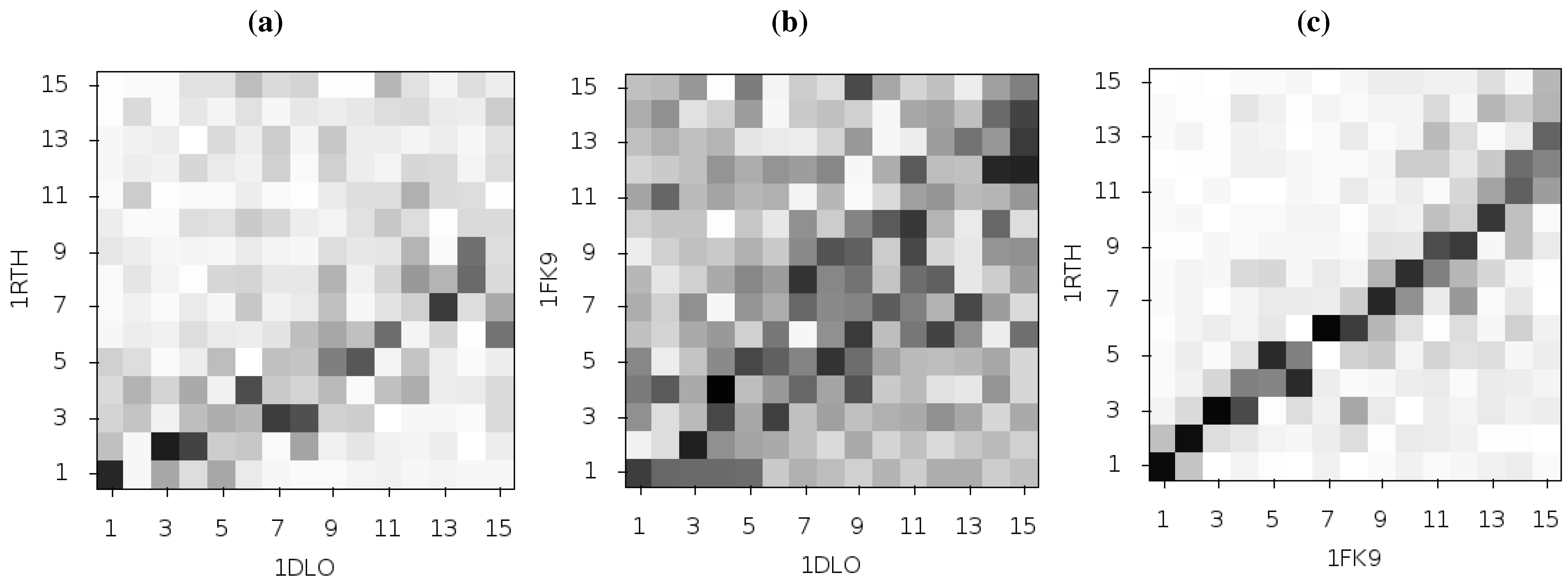
Temiz and Bahar [
33] suggest that differences in the second ANM mode calculated for NPV and EFV bound HIV-1 RT may be linked to the differing efficacy of the two drugs. This was based on analysis of the 1RTH NVP bound and 1FK9 EFV bound PDB crystal structures. The modes for both structures were observed to differ noticeably from those computed for the apo 1DLO structure.
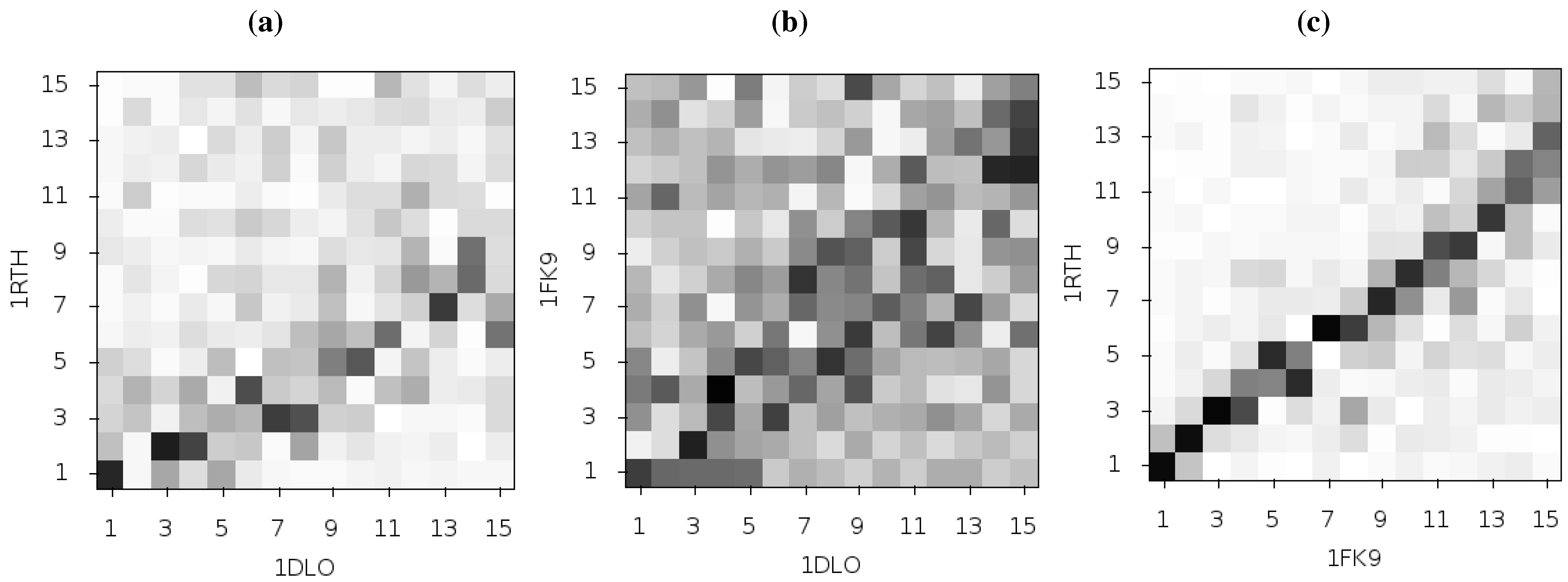
Figure 3 shows the pairwise correlation between the first 15 modes calculated for all three of these structures. In each comparison darker squares indicate higher magnitude of correlation between two individual modes and hence that the shape of the motions represented is similar (there is no physical meaning to the direction of the mode and consequently no meaning attached to a whether a pair of modes are correlated or anti-correlated). A dark diagonal in a heat map is indicative that not only are the modes generated for both structures similar but that their rank ordering is also the same. Clear differences between the two NNRTI bound structures and the apo structure are seen (in
Figure 2a,b), with only a clear correlation between mode 1 of both classes of structure and between mode 3 of the apo and mode 2 of the drug bound structures apparent. In contrast, there are strong correlations close to the diagonal in
Figure 2c indicating many of the modes generated for 1RTH and 1FK9 are very similar, and are similarly ordered. The second mode describes anti-correlated motions of the fingers and thumb in both cases (see
Figure 4), suggestive of the possibility that the NNRTI bound HIV-1 RT can adopt a closed conformation despite this not having been observed crystallographically. Despite the qualitative similarities the comparison of this mode between the two systems exhibits some level of correlation with a range of other modes, reflecting the differences observed by Temiz and Bahar [
33]. There are three principle sources of these differences which are illustrated in
Figure 5; these are reduced motions of the p66 fingers (residues 1 to 84 and 120 to 150) and thumb (residues 244 to 322), along with additional motions in the p51 domain (between residues 170 and 260) of the EFV bound structure relative to that bound to NVP.
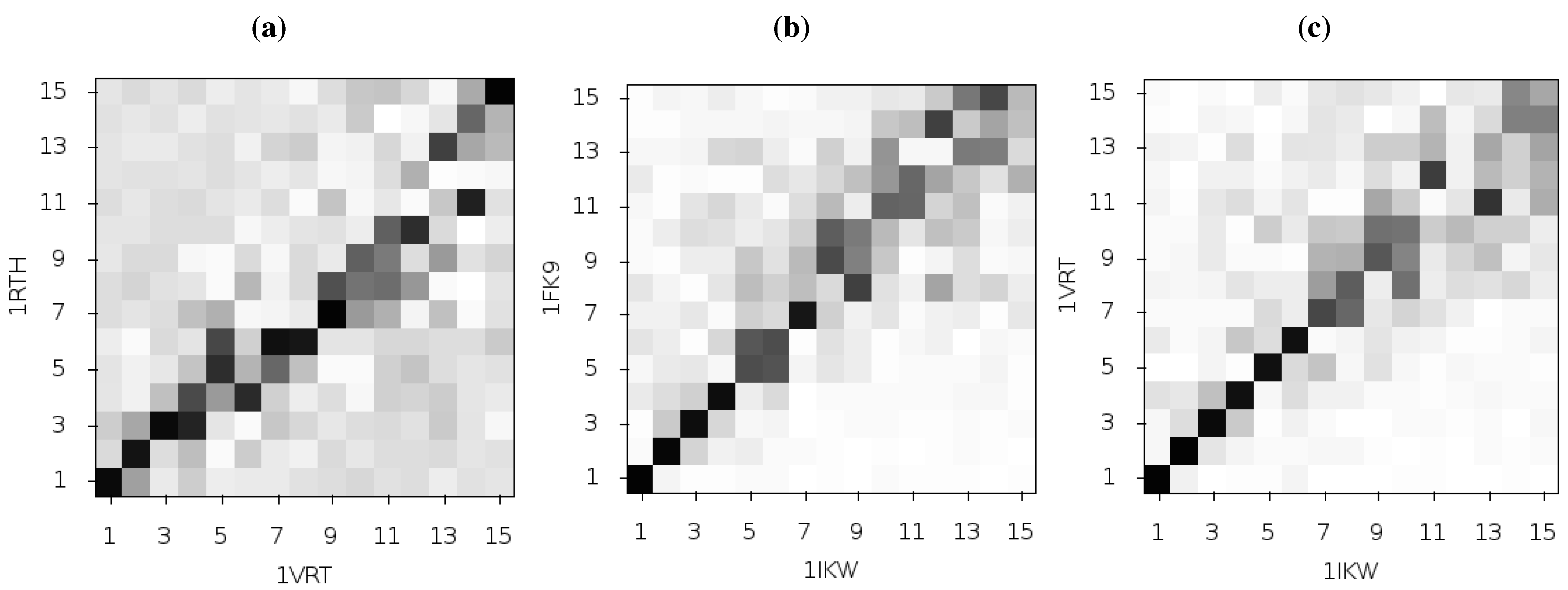
Figure 3.
Comparison of the first 15 ANM modes generated from the apo HIV-1 RT structure and those of the NVP and EFV bound enzyme using the 1DLO, 1RTH and 1FK9 crystal structures respectively. The matrices show the pairwise correlation of modes, the darker the square the higher the magnitude of the correlation. (a) The apo to NVP bound and (b) apo to EFV bound comparisons show similar patterns with high correlations between the first and third modes and lower correlation in most other modes. (c) shows that the modes for the EFV and NVP bound structures are much more similar. The correlation of mode 2 for both structures with other modes indicates that this mode is not identical between the two structures.
Figure 3.
Comparison of the first 15 ANM modes generated from the apo HIV-1 RT structure and those of the NVP and EFV bound enzyme using the 1DLO, 1RTH and 1FK9 crystal structures respectively. The matrices show the pairwise correlation of modes, the darker the square the higher the magnitude of the correlation. (a) The apo to NVP bound and (b) apo to EFV bound comparisons show similar patterns with high correlations between the first and third modes and lower correlation in most other modes. (c) shows that the modes for the EFV and NVP bound structures are much more similar. The correlation of mode 2 for both structures with other modes indicates that this mode is not identical between the two structures.
3.1. Influence of Initial Crystal Structure Choice
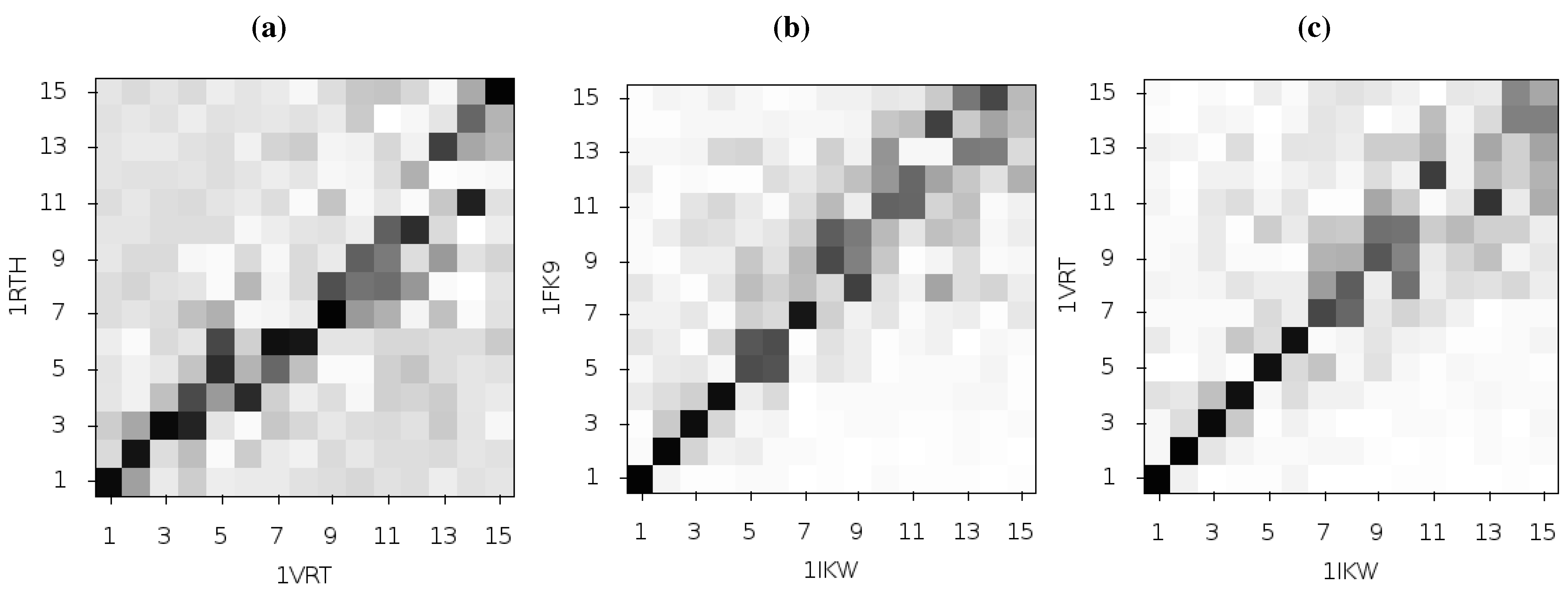
If the differences observed between the modes seen for 1FK9 and 1RTH is caused by differences in the bound inhibitor, then similar differences should be observed using different EFV and NVP bound structures. We used all available EFV and NVP crystal structures as inputs to ANMs. We found that whilst all structures showed similar second modes, there was no correlation between the particular drug bound and the predictions of the flexibility of the p66 thumb and fingers, or the presence or absence of fluctuations in the p51 subunit. A comparison between the results for the 1IKW and 1VRT crystal structures (EFV and NVP bound, respectively) and those of 1FK9 and 1RTH (EFV and NVP bound, respectively) is shown in
Figure 6. The NVP bound 1VRT and 1RTH structures in particular show dissimilarities across a range of modes. Comparison of the fluctuations described by mode 2 for 1IKW and 1VRT shown in
Figure 7 indicates that not only are the motions more restrained in these models but both exhibit fluctuations in p51, removing the clear distinction seen between 1FK9 and 1RTH. Furthermore, the inclusion or exclusion of a node representing the drug, placed at the drug centre of mass as in [
33], is not found to impact results (see
Supporting Information Supplementary Figure S2).
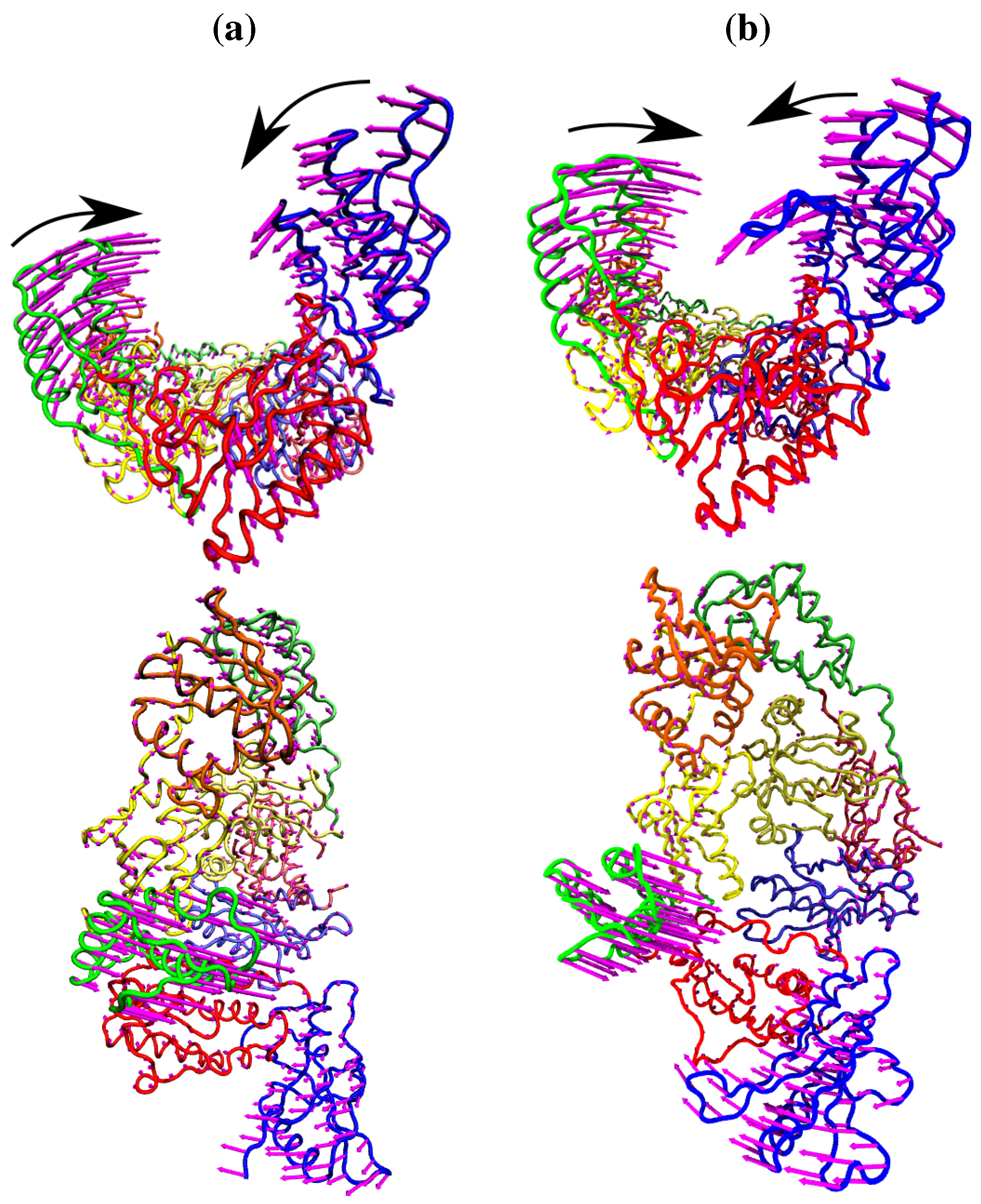
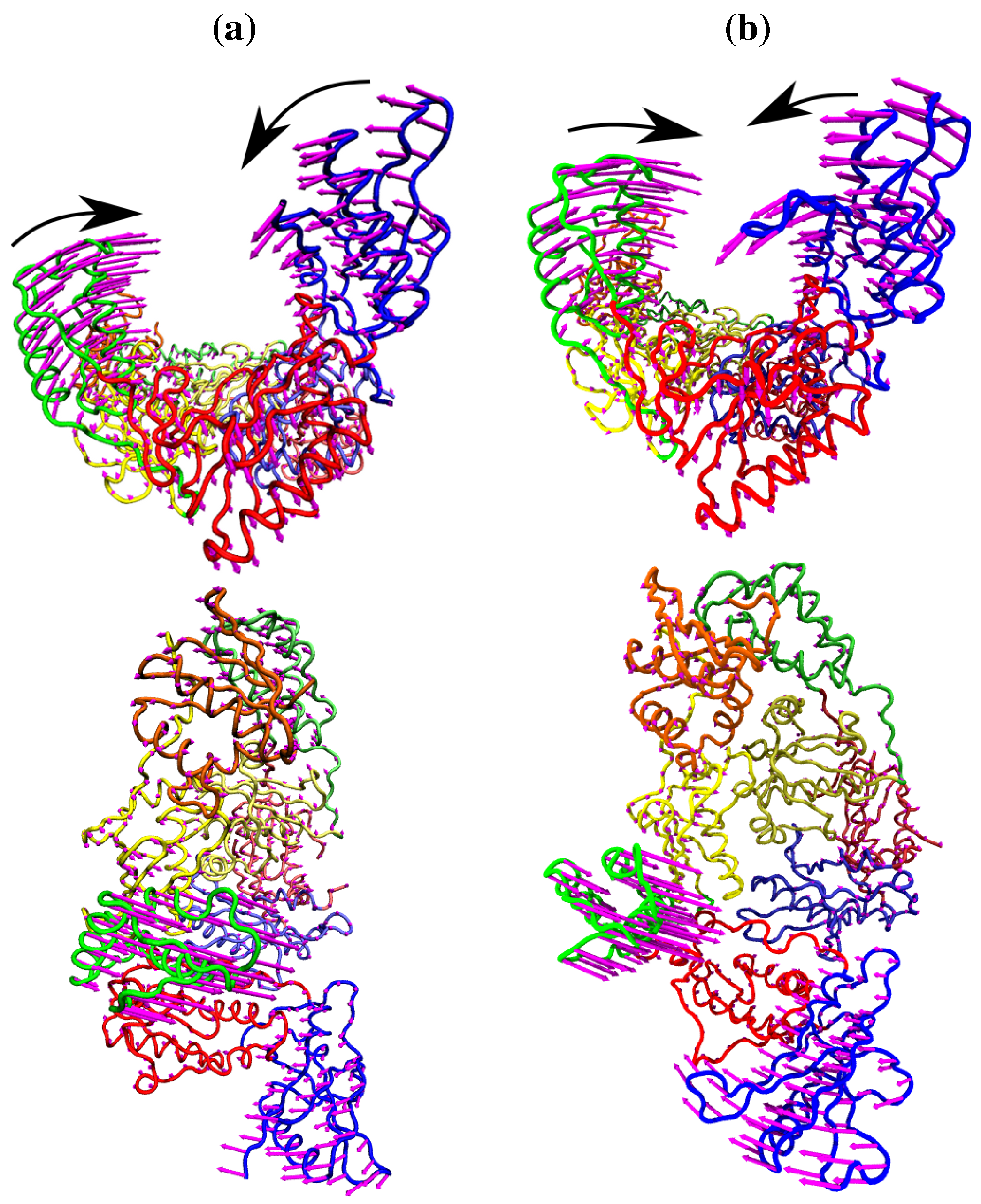
Figure 4.
The fluctuations of the NNRTI bound HIV-1 RT described by the second ANM mode for (a) the EFV bound 1FK9 and (b) NVP bound 1RTH structures. The fluctuations of each residue are shown as magenta arrows. In both cases two views are shown, one above the other; along the DNA binding cleft and looking down into it. In both cases the motions are dominated by anti-correlated movements of the p66 thumb and fingers (as highlighted by the black arrows).
Figure 4.
The fluctuations of the NNRTI bound HIV-1 RT described by the second ANM mode for (a) the EFV bound 1FK9 and (b) NVP bound 1RTH structures. The fluctuations of each residue are shown as magenta arrows. In both cases two views are shown, one above the other; along the DNA binding cleft and looking down into it. In both cases the motions are dominated by anti-correlated movements of the p66 thumb and fingers (as highlighted by the black arrows).
Figure 5.
Comparison of the squared fluctuations predicted by ANM mode 2 of the NVP bound 1RTH (red) and EFV bound 1FK9 structures (black).
Figure 5.
Comparison of the squared fluctuations predicted by ANM mode 2 of the NVP bound 1RTH (red) and EFV bound 1FK9 structures (black).
Figure 6.
Comparison of the first 15 ANM modes generated from different HIV-1 RT structures bound to NVP and EFV, using the 1RTH and 1VRT NVP bound structures and 1FK9 and 1IKW EFV bound structures. The matrices show the magnitude of the pairwise correlation of the modes, the darker the square the higher the correlation. (
a) and (
b) show comparisons of the modes generated from different crystal structures but with RT bound to the same inhibitor, NVP and EFV respectively. In (
c) the 1IKW (EFV) and 1VRT (NVP) bound structures are compared, more of the low modes show high levels of similarity than in the plot for 1FK9 and 1RTH shown in
Figure 2c.
Figure 6.
Comparison of the first 15 ANM modes generated from different HIV-1 RT structures bound to NVP and EFV, using the 1RTH and 1VRT NVP bound structures and 1FK9 and 1IKW EFV bound structures. The matrices show the magnitude of the pairwise correlation of the modes, the darker the square the higher the correlation. (
a) and (
b) show comparisons of the modes generated from different crystal structures but with RT bound to the same inhibitor, NVP and EFV respectively. In (
c) the 1IKW (EFV) and 1VRT (NVP) bound structures are compared, more of the low modes show high levels of similarity than in the plot for 1FK9 and 1RTH shown in
Figure 2c.
Figure 7.
Comparison of the squared fluctuations predicted by ANM mode 2 of the NVP bound 1VRT (red) and EFV bound 1IKW structures (black). The main difference between the two structures is in the extent of the predicted mobility of the p66 thumb (residues 244 to 322); in both cases this is much than that observed for 1RTH and 1FK9 (see
Figure 5).
Figure 7.
Comparison of the squared fluctuations predicted by ANM mode 2 of the NVP bound 1VRT (red) and EFV bound 1IKW structures (black). The main difference between the two structures is in the extent of the predicted mobility of the p66 thumb (residues 244 to 322); in both cases this is much than that observed for 1RTH and 1FK9 (see
Figure 5).
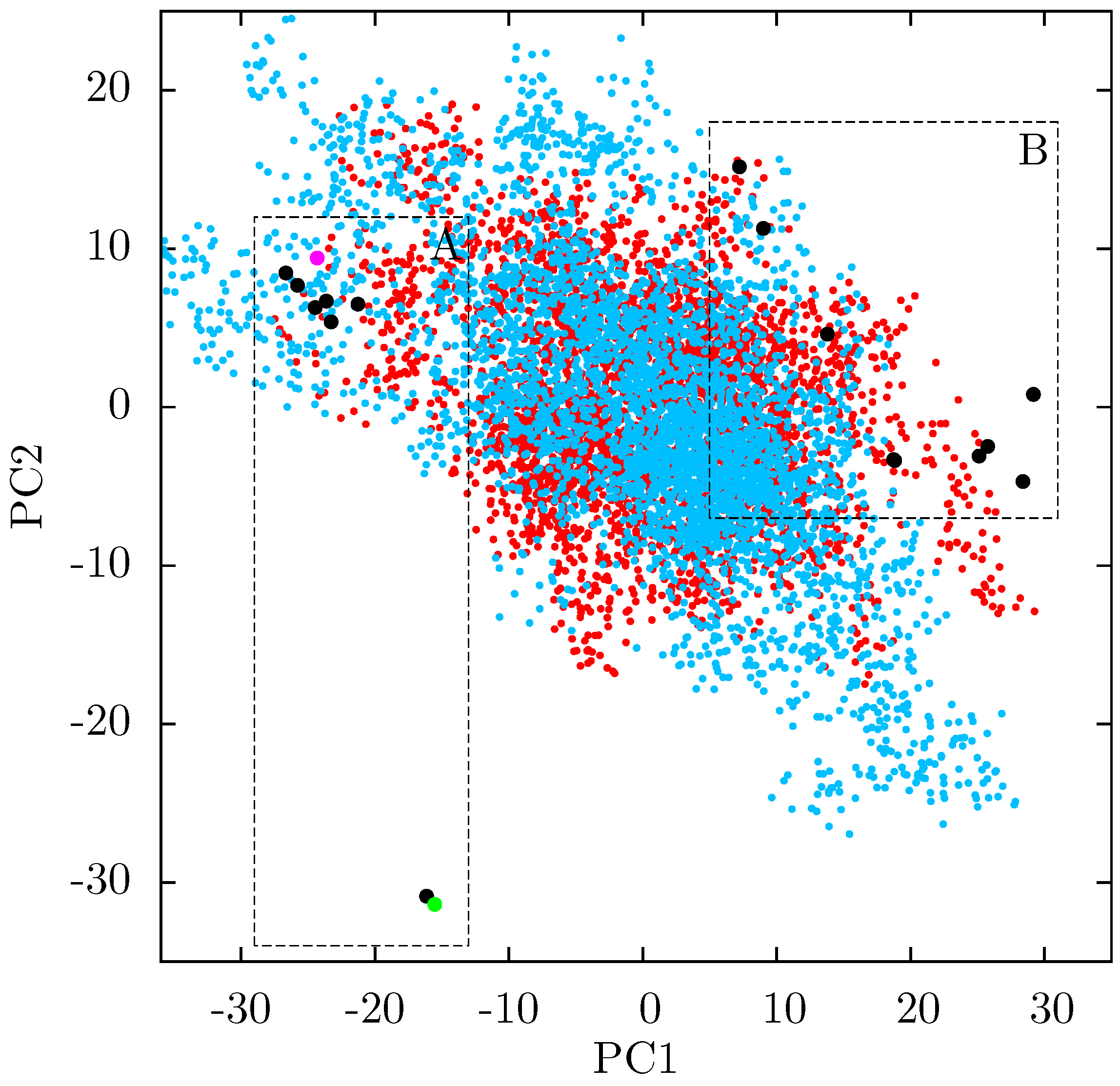
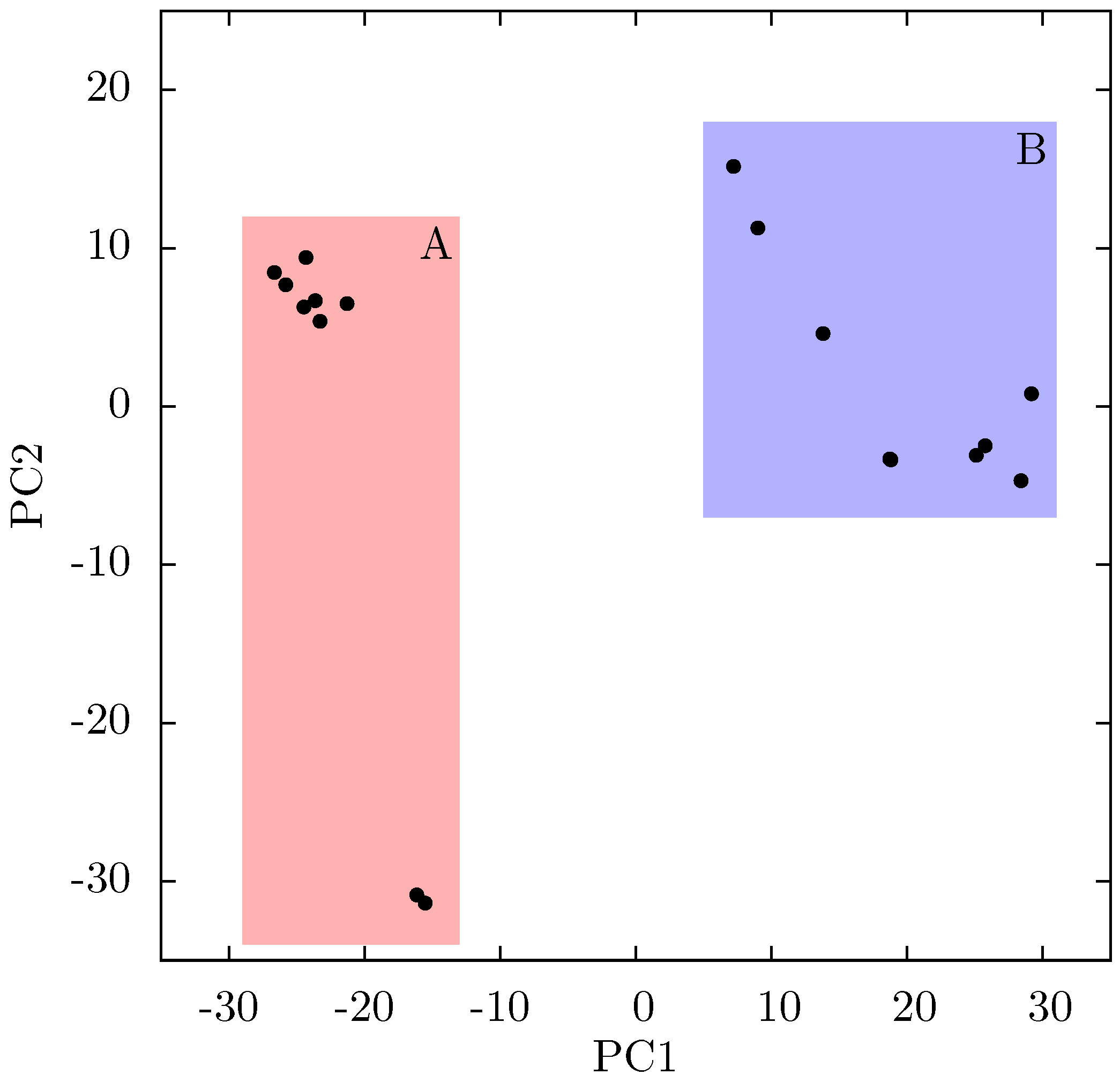
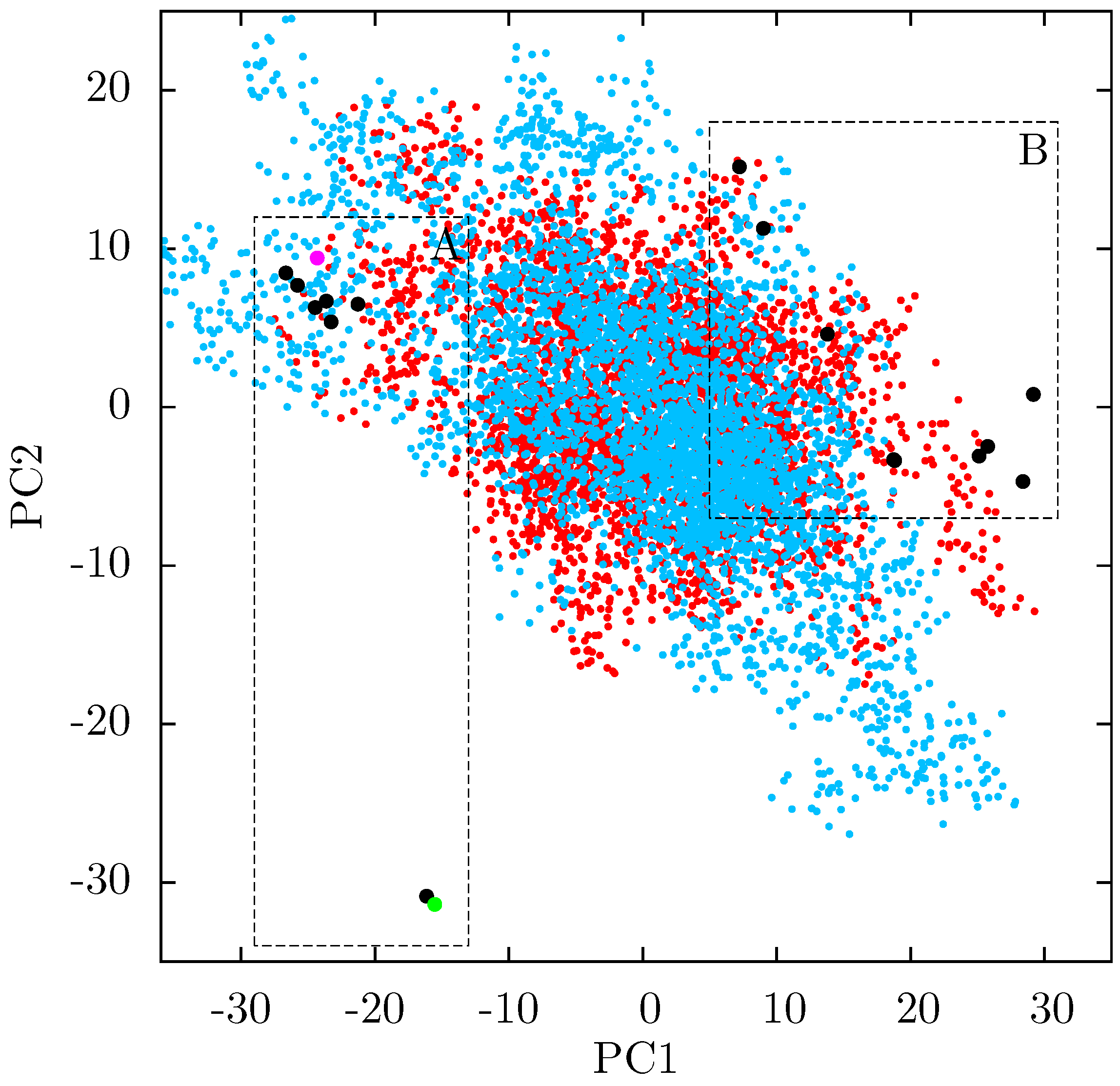
If there is no link between the specific inhibitor bound and the differences seen in the ANM modes of NNRTI bound HIV-1 RT, the obvious question is which features of the structures are causing the observed variation. In order to assess these differences observed in the ANMs we used PCA to analyse the differences in the available EFV and NVP bound structures. Analysis of all available crystal structures revealed one prominent principal component (PC), but this only identified differences between the 3HVT structure which was crystalised in the
C2 space group and the other crystal structures all in the
P 2
12
12
1 space group. This difference is known to produce alterations in RT conformation [
48] and the fact this structure is also anomalous in not having a water molecule bound beside NVP led us to exclude it from consideration here. The first PC generated for the reduced dataset accounts for 48% of the variance observed and the second a further 20% (the variance accounted for by the first 20 PCs is shown in
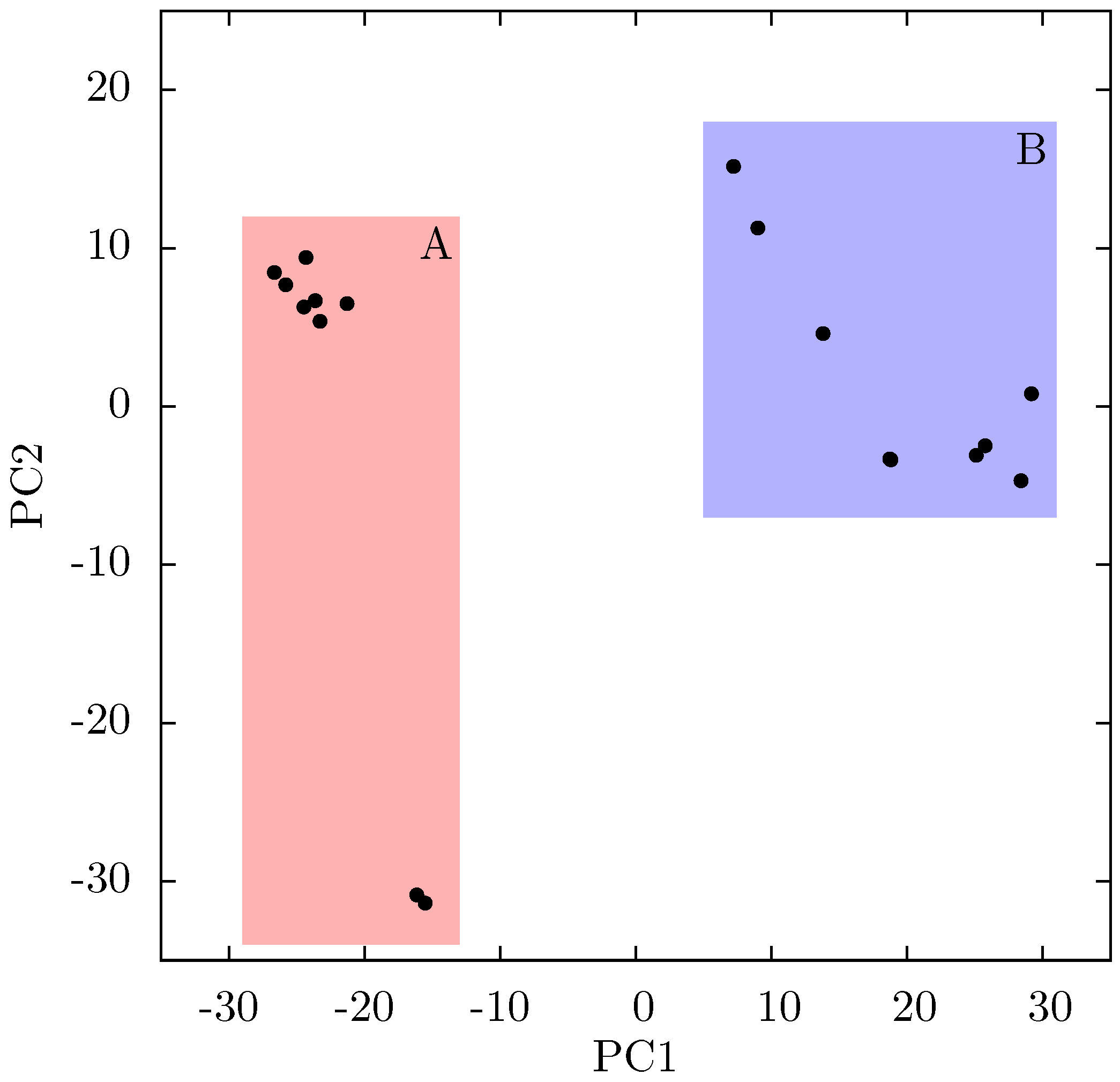
Supporting Information Supplementary Figure S4). The projection of each crystal structure analysed onto these two components is shown in
Figure 8. Two separate clusters of structures are apparent in this data, where we have labelled the two groups A and B. All structures in group A (including 1FK9,1IKW and 1VRT) show p51 flexibility in their second ANM modes, whereas those in group B do not. A list of structures in each group is provided in
Table 1. Both groups contain both EFV and NVP structures and both wild type and resistant sequences (although NVP bound structures with the key NNRTIBP mutations Y181C and Y188C are all found in group B). The two structures separated from the main group A along PC2 cluster (
Figure 8) are 1IKV and 1IKW. The variation along PC2 is dominated by motions in highly flexible loops in the p66 fingers (see
Supporting Information Supplementary Figure S5). As these two structures were produced in the same study [
7] it is likely that these minor changes are a function of the particular crystallization protocol employed.
Figure 8.
Projection of individual crystal structures on to the first two principal components generated from PCA of EFV and NVP bound HIV-1 RT structures. The structures are clearly clustered into two groups along PC1, labelled A and B, and highlighted in red and blue respectively.
Figure 8.
Projection of individual crystal structures on to the first two principal components generated from PCA of EFV and NVP bound HIV-1 RT structures. The structures are clearly clustered into two groups along PC1, labelled A and B, and highlighted in red and blue respectively.
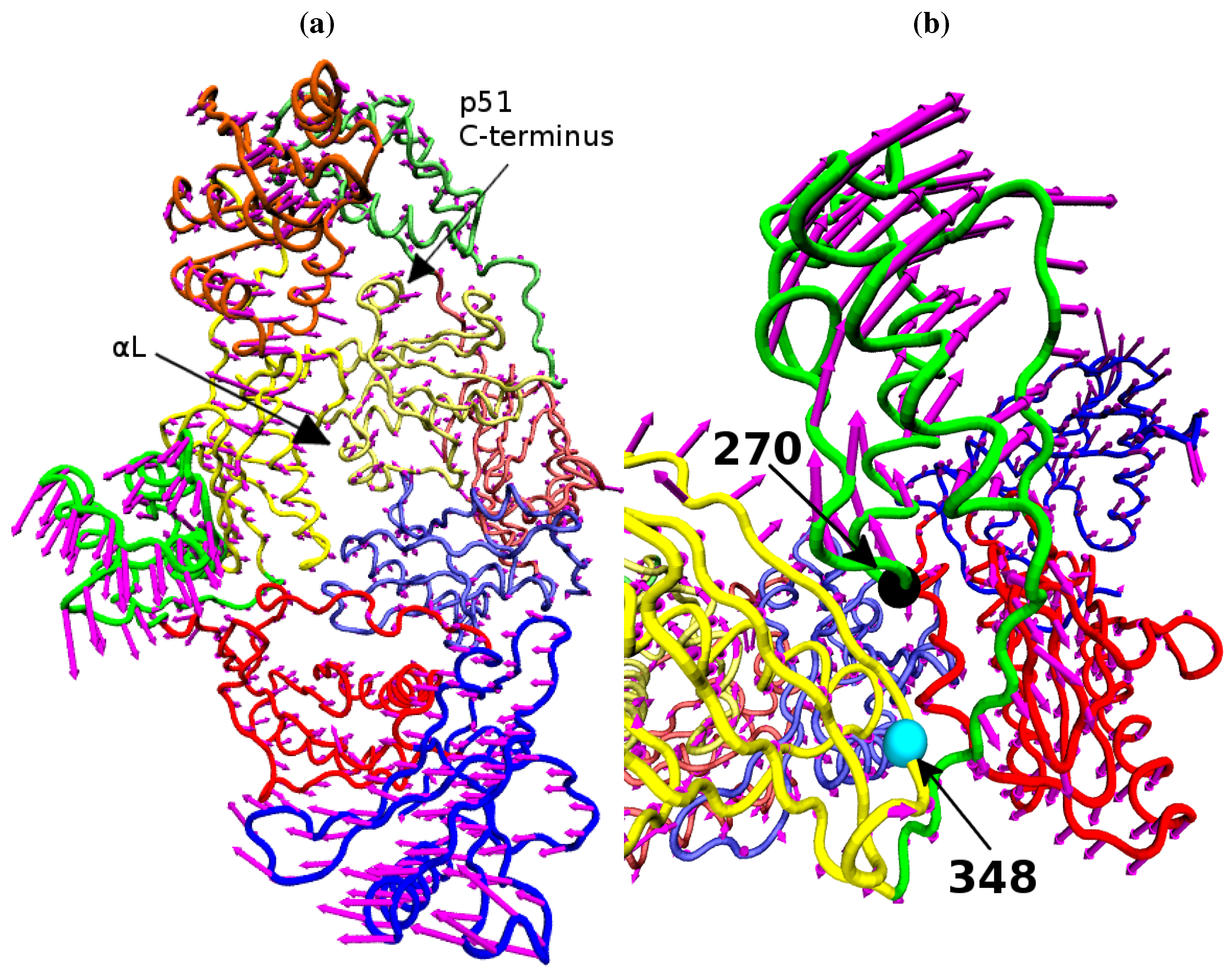
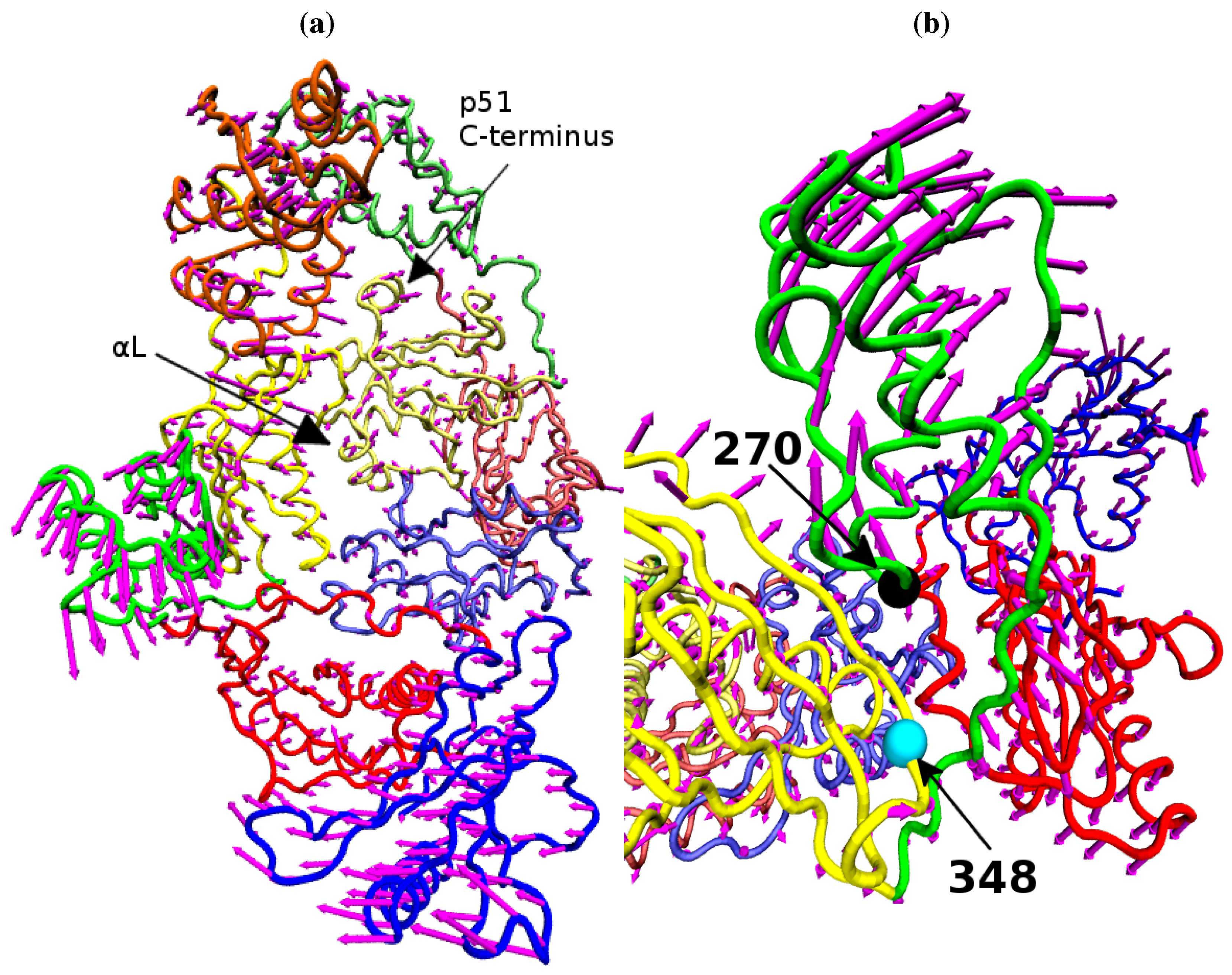
Figure 9 shows that structural variation along PC1 is dominated by changes in the position of the p66 thumb and fingers, albeit much more subtle changes than seen between the open and closed conformations of HIV-1 RT. The most notable change is the rotation of the helix
αH in the thumb, which changes approximately 15° across the data set (see
Figure 8b); this is accompanied by the movement of the loop running from residue 269 to 278 (between
αI and
αJ). A range of smaller alterations along the template/primer binding cleft of the protein is also observed. Three regions in p51 show significant changes compared to surrounding residues; the C-terminus (residues 420 to 430),
αJ in the p51 thumb and
αL. Furthermore, p51
αL contains residues Lys395 and Glu396 of the RNaseH primer grip. The regions of high variation seen in PC1 suggest a path for the communication of conformational changes around the p66 thumb (and NNRTI binding pocket at its base) to distal regions of the protein. This path would see changes in the thumb communicated directly to the connection domain via the nearest residues, 348 and 355, through the loop between p66
β18 (which contains residue 355) and on to the dimer interface between the RNaseH and the p51 C-terminal residues and thumb. Interestingly, residue 348 which provides contacts between the thumb and connection domains is one of a number of connection domain residues for which resistance associated mutations have been identified [
22,
23].
Table 1.
The EFV and NVP bound HIV-1 RT crystal structures analysed by PCA are divided into two groups according to PC1, groups A and B. Group A corresponds to the structures in which p51 flexibility is linked to that of the p66 thumb by ANMs, no such link is observed in ANMs based on group B structures. The drug bound and major resistance associated mutations present in the structure are also listed.
Table 1.
The EFV and NVP bound HIV-1 RT crystal structures analysed by PCA are divided into two groups according to PC1, groups A and B. Group A corresponds to the structures in which p51 flexibility is linked to that of the p66 thumb by ANMs, no such link is observed in ANMs based on group B structures. The drug bound and major resistance associated mutations present in the structure are also listed.
| PDB code | NNRTI | Resolution (Å) | Resistance Mutations | Citation |
|---|
| Group A: p51 flexible |
| LW0 | NVP | 2.80 | T215Y | Chamberlain et al. [49] |
| LWC | NVP | 2.62 | M184V | Chamberlain et al. [49] |
| S1U | NVP | 3.00 | L100I | Ren et al. [50] |
| VRT | NVP | 2.20 | – | Ren et al. [51] |
| HND | NVP | 2.50 | K101E | Ren et al. [52] |
| HNY | NVP | 2.50 | E138K | Ren et al. [52] |
| FK9 | EFV | 2.50 | – | Ren et al. [53] |
| IKV | EFV | 2.00 | K103N | Lindberg et al. [7] |
| IKW | EFV | 3.00 | – | Lindberg et al. [7] |
| Group B: p51 constrained |
| FKP | NVP | 2.90 | K103N | Ren et al. [53] |
| JLB | NVP | 3.00 | Y181C | Ren et al. [54] |
| JLF | NVP | 2.60 | Y188C | Ren et al. [54] |
| LWE | NVP | 2.81 | M41L, T215Y | Chamberlain et al. [49] |
| LWF | NVP | 2.80 | M41L, D67N, K70R, M184V, T215F, K219N | Chamberlain et al. [49] |
| RTH | NVP | 2.20 | – | Ren et al. [51] |
| S1X | NVP | 2.80 | V108I | Ren et al. [50] |
| FKO | EFV | 2.90 | K103N | Ren et al. [53] |
| JKH | EFV | 2.50 | Y181C | Ren et al. [54] |
Figure 9.
The variation of the NNRTI bound HIV-1 RT described by PC1. The variation of each residue, relative to the average structure displayed in tube representation, is shown in (a) as magenta arrows. Detail of the changes in their p66 thumb is shown in (b), the Cα atoms of residue 270 (black) and 348 (cyan) are shown as spheres.
Figure 9.
The variation of the NNRTI bound HIV-1 RT described by PC1. The variation of each residue, relative to the average structure displayed in tube representation, is shown in (a) as magenta arrows. Detail of the changes in their p66 thumb is shown in (b), the Cα atoms of residue 270 (black) and 348 (cyan) are shown as spheres.
4. Results: Ensemble Molecular Dynamics
Both the PCA and ANM analyses presented here suggest a link between alterations in p66 thumb position and changes in distal regions of the protein implicated in RNaseH function. This combination of evidence is suggestive of NNRTI binding inducing changes in both conformation and dynamics throughout the protein. Whilst predictions from ANMs do give consistently similar global motions for the proteins, we have seen that small changes in local contacts can influence the details of the dynamic modes produced. In order to gain a more detailed picture of the dynamics in the p51 subunit, and how it is influenced by NNRTI binding, we conducted ensemble MD simulations of the apo HIV-1 RT and the same enzyme bound to both NVP and EFV.
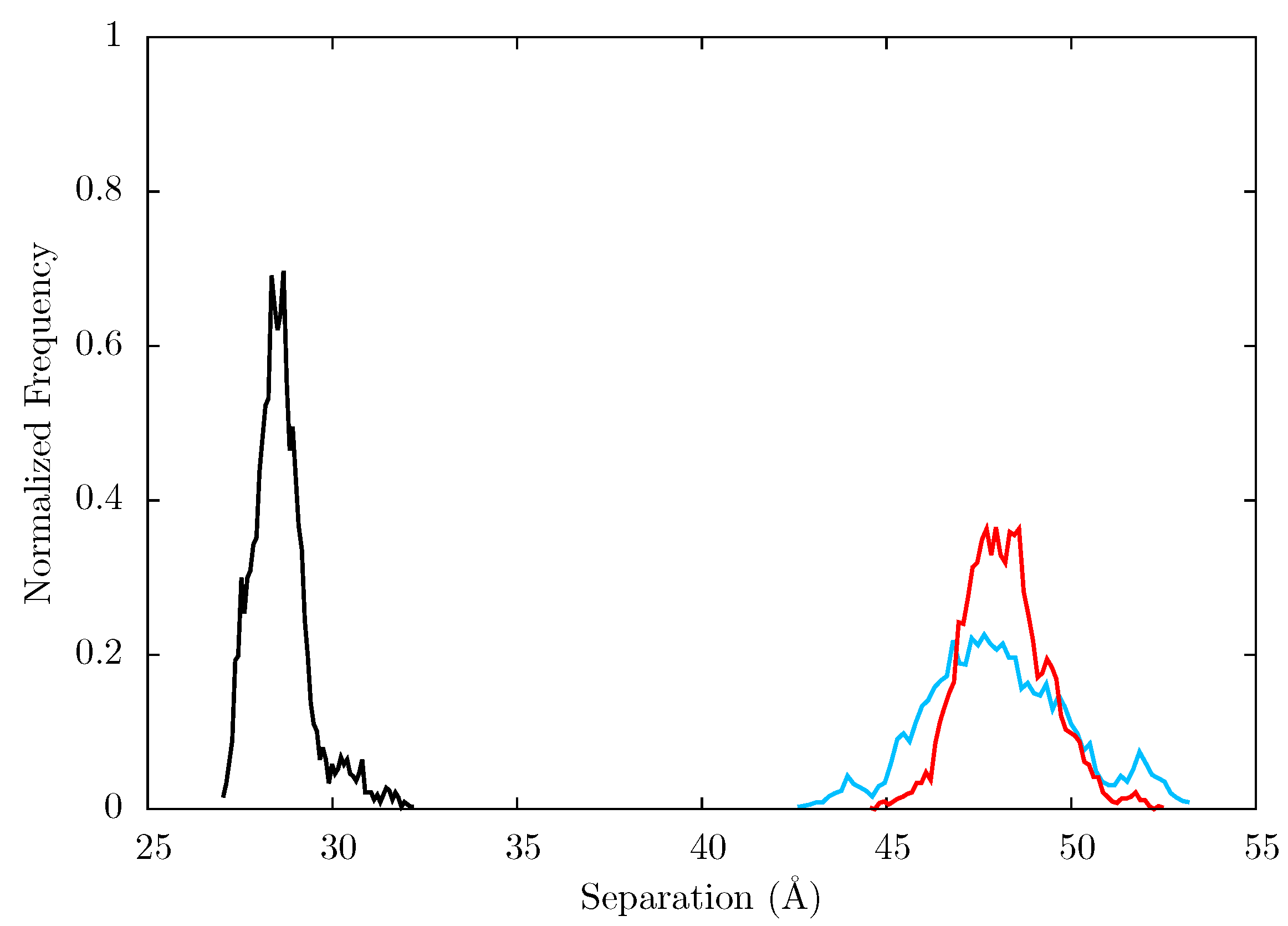
Figure 10 shows that both the NVP and EFV bound simulations move away from their initial structures and sample conformations which differ from both the group A and B structures seen in the crystal structures. This is indicative of the fact that the p66 thumb and fingers in particular are constrained by crystal contacts in the experimental structures of NNRTI bound HIV-1 RT.
Figure 10.
Projection of snapshots from the ensemble MD trajectories of the EFV and NVP bound HIV-1 RT onto the first two principal components generated from PCA of EFV and NVP bound HIV-1 RT crystal structures. Both simulations show wide variations in both PCs; there is no clustering close to either of the groups of crystal structure projections (these are shown by the dashed rectangles and labelled A and B). Crystal structure projections are shown as black dots, except the 1IKW (green) and 1VRT (magenta) structures which were used as initial structures for the EFV and NVP MD simulations, respectively.
Figure 10.
Projection of snapshots from the ensemble MD trajectories of the EFV and NVP bound HIV-1 RT onto the first two principal components generated from PCA of EFV and NVP bound HIV-1 RT crystal structures. Both simulations show wide variations in both PCs; there is no clustering close to either of the groups of crystal structure projections (these are shown by the dashed rectangles and labelled A and B). Crystal structure projections are shown as black dots, except the 1IKW (green) and 1VRT (magenta) structures which were used as initial structures for the EFV and NVP MD simulations, respectively.
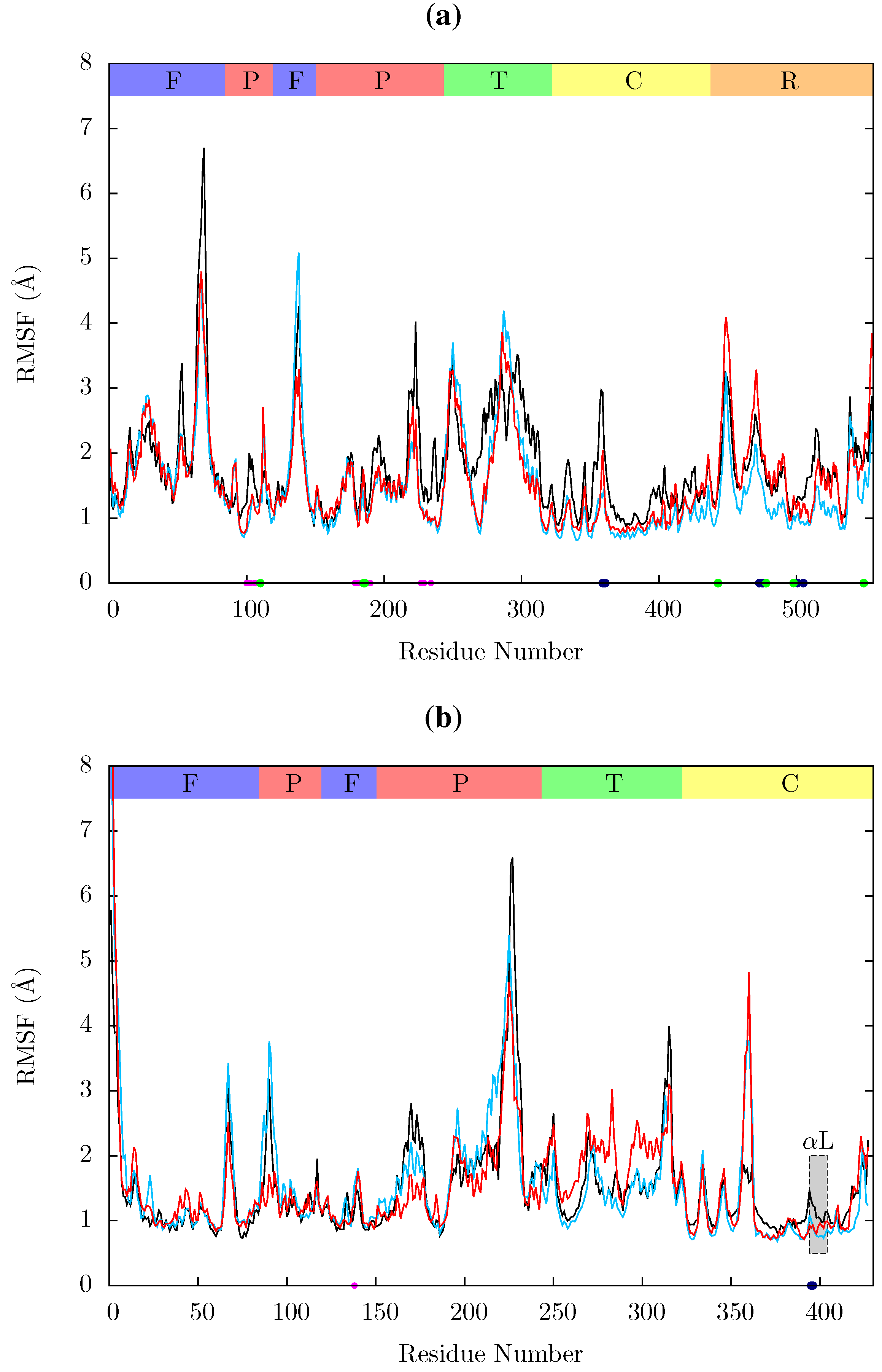
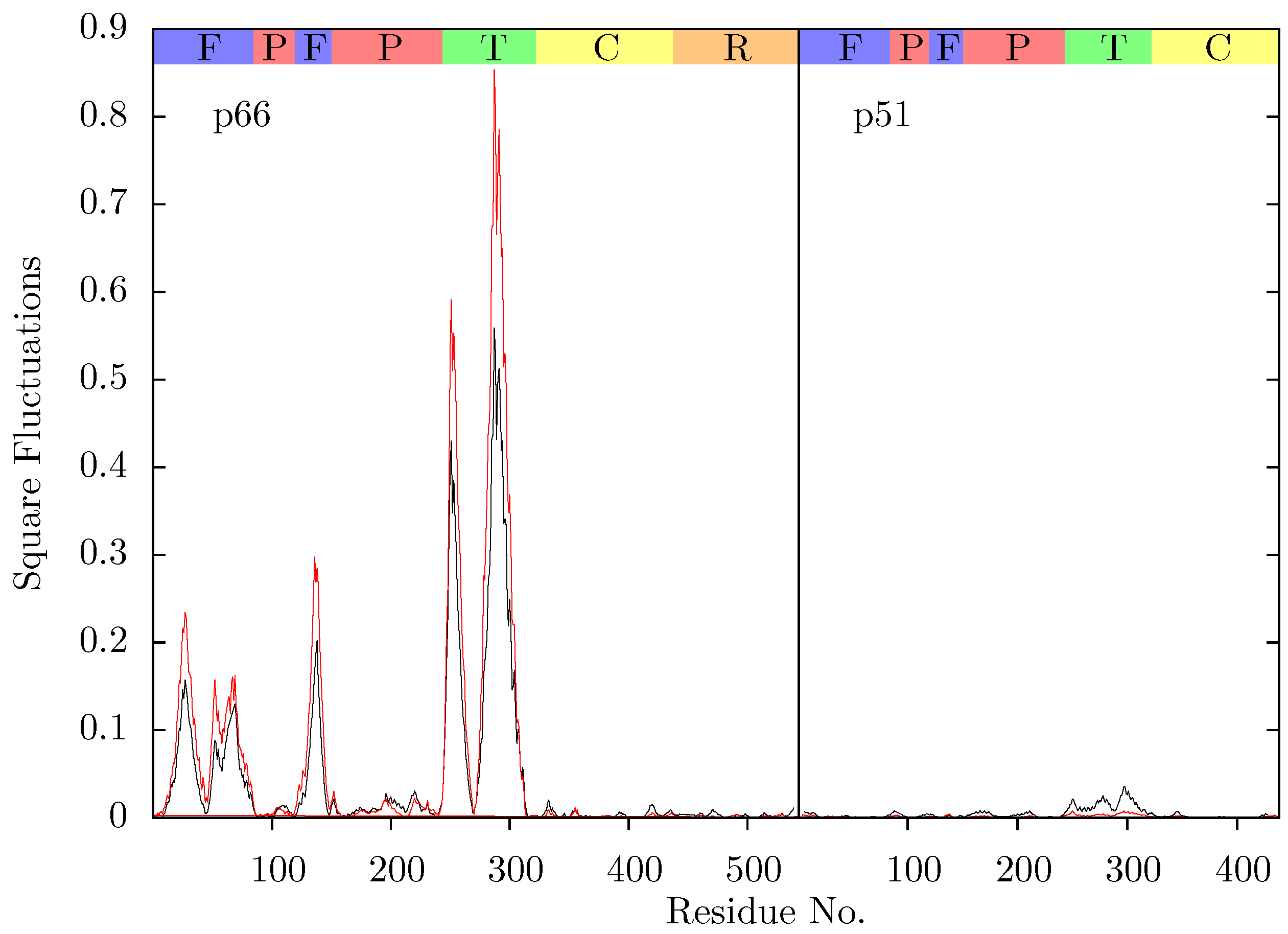
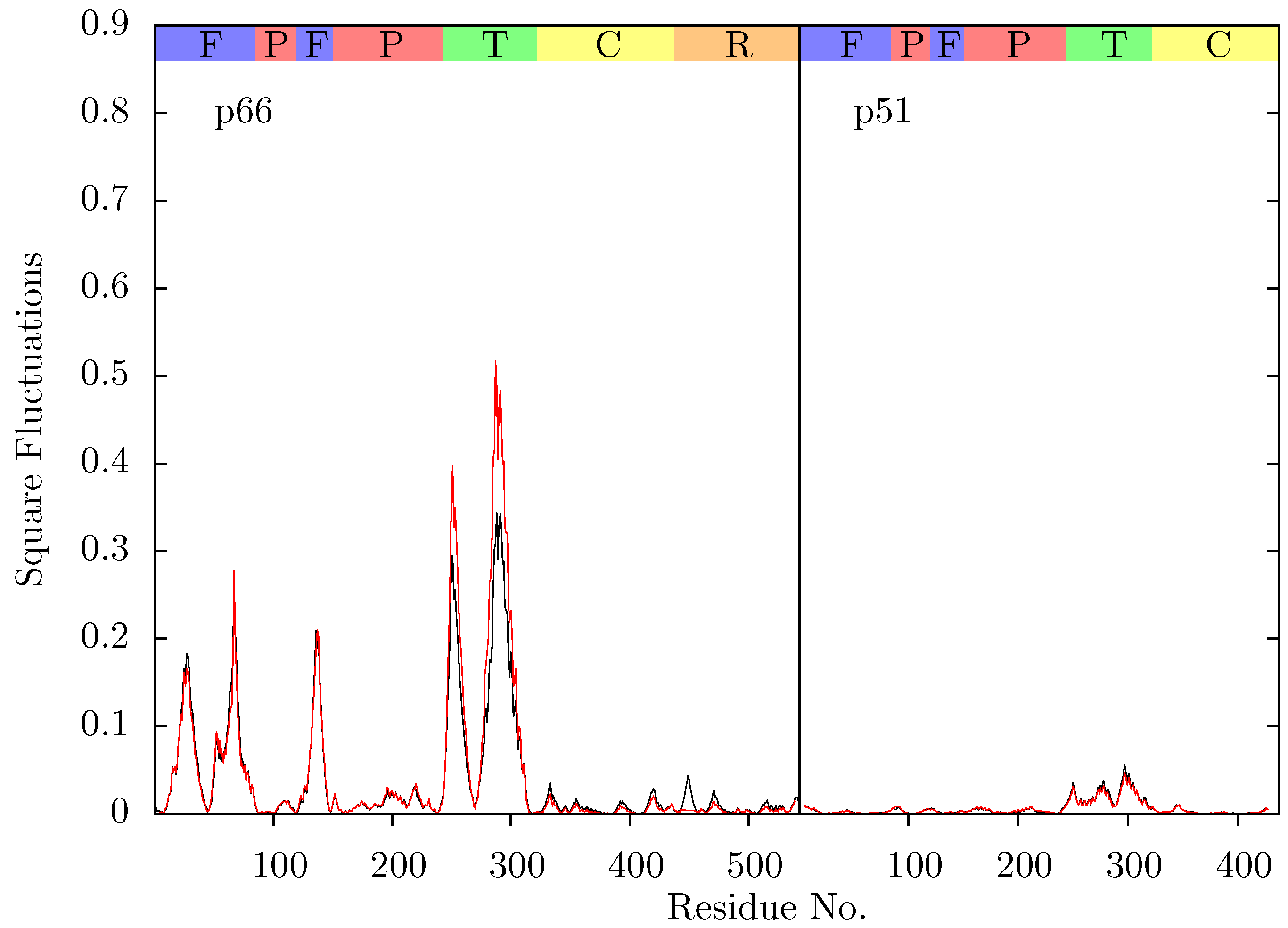
Figure 11 shows the root mean squared fluctuations (RMSF) of the residues of the three simulated structures relative to the average structure of each set of trajectories. As expected from both ANM models and crystal structure evidence [
55], the regions in which the largest motions are observed for all simulated structures are the p66 fingers and thumb subdomains. However, no shifts between open and closed global conformations occur in any simulations such as those reported by Ivetac and McCammon [
29] (see
Figure 12) as being observed in 4 member 30 ns ensemble simulations (we are describing 10 member 10 ns ensembles here). The residues that constitute the NNRTI binding pocket as expected all exhibit markedly lower flexibility in the two drug bound simulations. Residues 320 to 405 in p66 are also stabilized in the NVP and EFV bound simulations, in line with evidence from hydrogen/deuterium exchange experiments performed by Seckler
et al. [
27] which suggests that EFV binding suppresses motions of the connection domain. Distinct differences are seen in the RNaseH domain between the two NNRTI bound ensembles. The NVP bound enzyme exhibits reduced flexibility throughout the domain compared to the apo system. A variety of changes are observed in the EFV bound system with residues between 443 (which forms part of the RNaseH primer grip) and 481 increasing in flexibility; most of the rest of the domain exhibits similar behaviour to the apo system with residues 500 to 516 (including residues 501 and 505 in the RNaseH primer grip) being stabilized.
Figure 11.
Root mean squared fluctuations (RMSF) of the Cα atoms of (a) p66 and (b) p51 over the concatenated ensemble trajectories of the apo (black), EFV (red) bound and NVP (blue) bound HIV-1 RT simulations. The residues of the NNRTI binding pocket, RnaseH primer grip and active sites are indicated by magenta, dark blue and green dots on the x-axis, respectively. The bars above the graph indicate the locations of the subunits in the sequence; the (F)ingers, (P)alm, (T)humb, (C) connection and (R)NaseH. The p51 αL helix containing residues 395 and 396 in the RNaseH primer grip is indicated by a gray box.
Figure 11.
Root mean squared fluctuations (RMSF) of the Cα atoms of (a) p66 and (b) p51 over the concatenated ensemble trajectories of the apo (black), EFV (red) bound and NVP (blue) bound HIV-1 RT simulations. The residues of the NNRTI binding pocket, RnaseH primer grip and active sites are indicated by magenta, dark blue and green dots on the x-axis, respectively. The bars above the graph indicate the locations of the subunits in the sequence; the (F)ingers, (P)alm, (T)humb, (C) connection and (R)NaseH. The p51 αL helix containing residues 395 and 396 in the RNaseH primer grip is indicated by a gray box.
Residues 86 to 91 (
β5) in p51 are stabilized in the EFV bound system (again in line with Seckler
et al. [
27]); NVP however exhibits very similar behaviour to the apo system in this region. The p51 thumb in EFV is more flexible than in either the NVP bound or apo structure (this is the region predicted to exhibit fluctuations correlated with thumb and fingers motions in the ANM). The p51 thumb is adjacent to the RNaseH domain and it has been suggested that it plays a role in maintaining its correct conformation [
56]. However, we do not observe generally increased RNaseH domain flexibility in our simulations of the EFV bound system but instead more subtle changes. Both NNRTI bound structures show increased flexibility around p51 residue 358 (in a loop connecting
β18 to
αK).
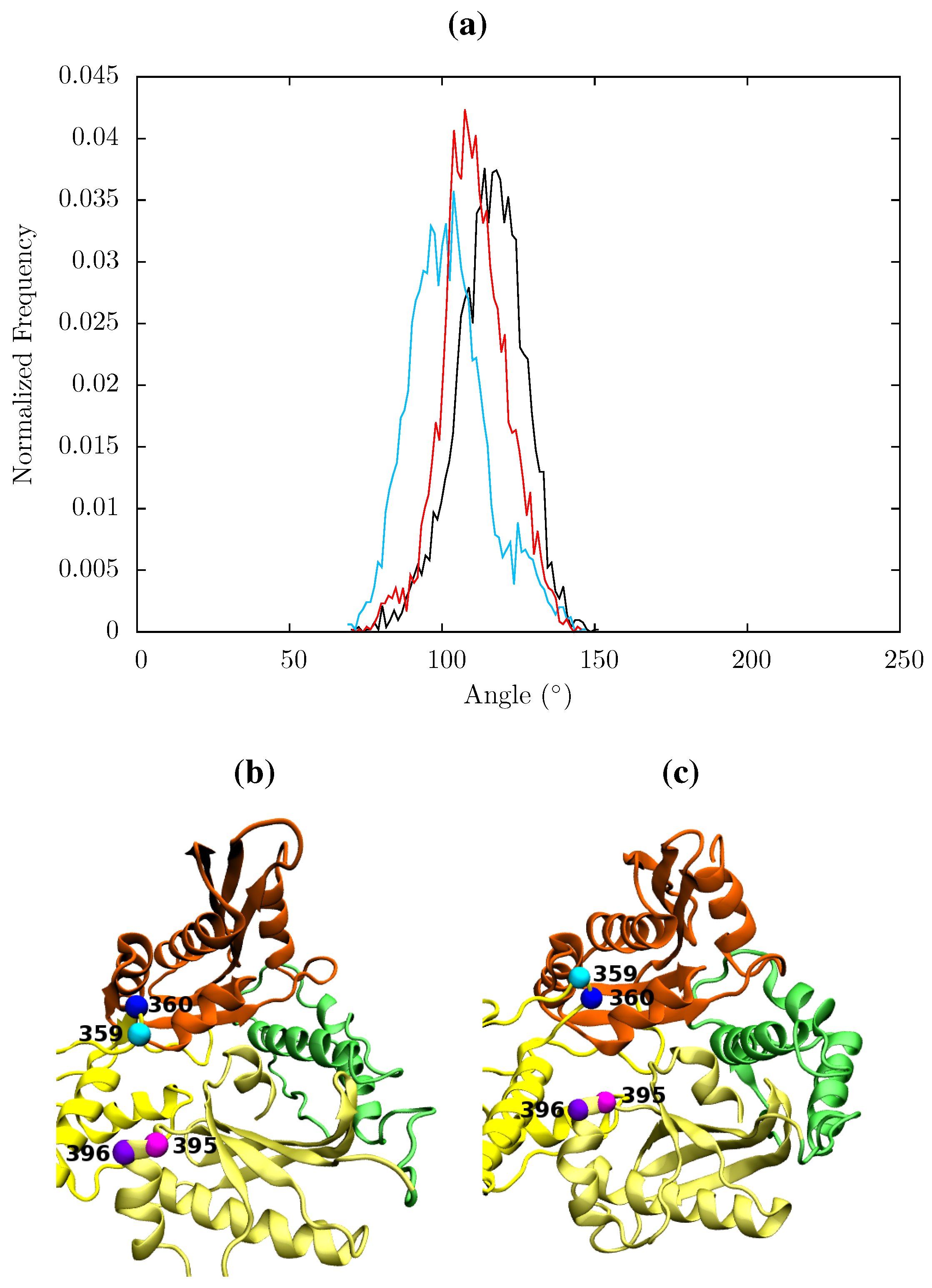
Figure 12.
Histogram showing the normalized frequency distribution of the separation between centres of mass of the p66 fingers and thumb over the simulations of the (black), EFV (red) bound and NVP (blue) bound HIV-1 RT ensemble trajectories. Both the EFV and NVP ensembles maintain much larger separation than is seen in the apo enzyme but the NVP bound system shows greater flexibility.
Figure 12.
Histogram showing the normalized frequency distribution of the separation between centres of mass of the p66 fingers and thumb over the simulations of the (black), EFV (red) bound and NVP (blue) bound HIV-1 RT ensemble trajectories. Both the EFV and NVP ensembles maintain much larger separation than is seen in the apo enzyme but the NVP bound system shows greater flexibility.
In all structures the section between residues 360 and 420 in p51 is highly stable but there is a distinct RMSF peak at RNaseH primer grip residues 395 and 396 in the apoprotein. This key region for substrate positioning was also picked out in the PCA of the EFV and NVP bound crystal structures as undergoing conformational shifts correlated to changes in the p66 thumb. Further investigation of the surrounding regions indicates that other regions in or close to the RNaseH primer grip also undergo discernibly different motions in our simulations, in particular the p66 loop containing residue 359 exhibits very different behaviour across the systems (see
Figure 11). Measurements of the angle made between the backbone of residues 395 and 396 with that of residues 359 and 360 shows a shift in both NNRTI bound systems. These changes differentially increase the accessibility of the RNaseH active site, with NVP. The differences between the apo and NNRTI bound structures here are mostly because conformational changes occur to residues 359 and 360 with relatively modest changes in residues 395 and 396 (see
Figure 13).
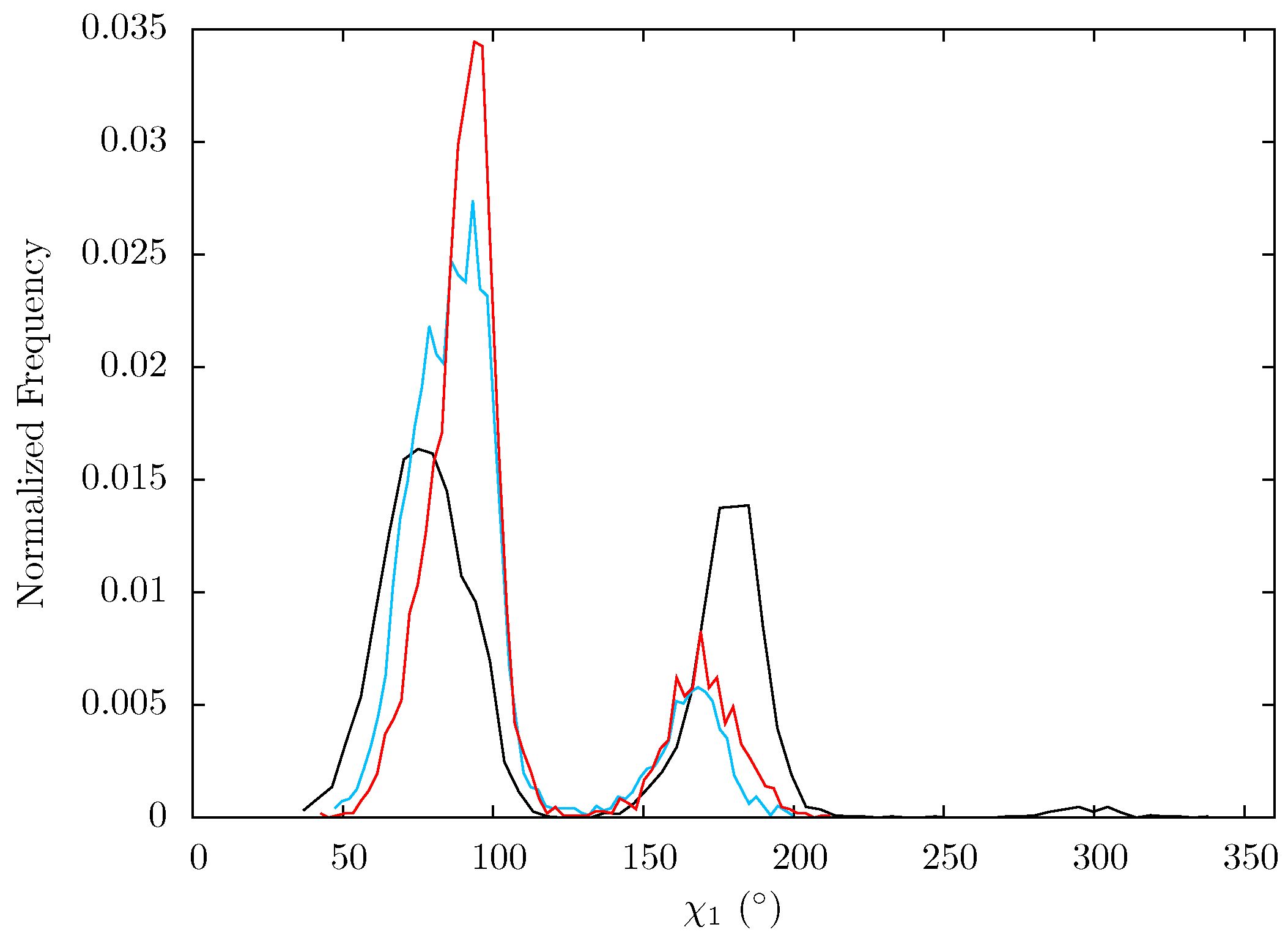
In order to ascertain whether the relatively minor changes in backbone flexibility impact residues Lys395 and Glu396, the dihedrals of the sidechains were measured for each snapshot of all three ensembles. The distribution of angles observed for Lys395 are very similar for all three systems (see
Supporting Information Supplementary Figure S6) but there is a distinct shift in χ1 for Glu396.
Figure 14 shows that in all three systems two states exist. In the apo system the first of these is centred around 75° and the second 180° which are both fairly equally populated. In both NNRTI bound systems the mean angles of these two states are shifted towards one another by approximately 20° and the lower of these two groups is enriched compared to the higher. Changes at this position are particularly significant as in structures of the RNA/DNA bound enzyme [
12] Glu396 is seen to interact with the RNA primer backbone phosphate between the eleventh and twelfth base pairs from the 3
' end of the DNA primer (positioned at the polymerase active site). This location coincides with the location at which “polymerization-independent” hydrolysis is observed to occur experimentally [
20]. The absence of substrate in our simulations means we cannot directly assess the impact of the observed changes on template/primer conformations. However, it seems likely that changes to the binding cleft would be reflected in changes in substrate binding and there is considerable evidence that RNaseH functioning is sensitive to changes in substrate conformation [
20,
57,
58]. Experiments involving the neutralisation of the backbone sections that contact p51 residues 395 and 396 enhanced the rate of cleavage 19 and 21 base pairs from the 3
' end of the primer when contact with 396 was lost (no change was seen for 395) [
20].
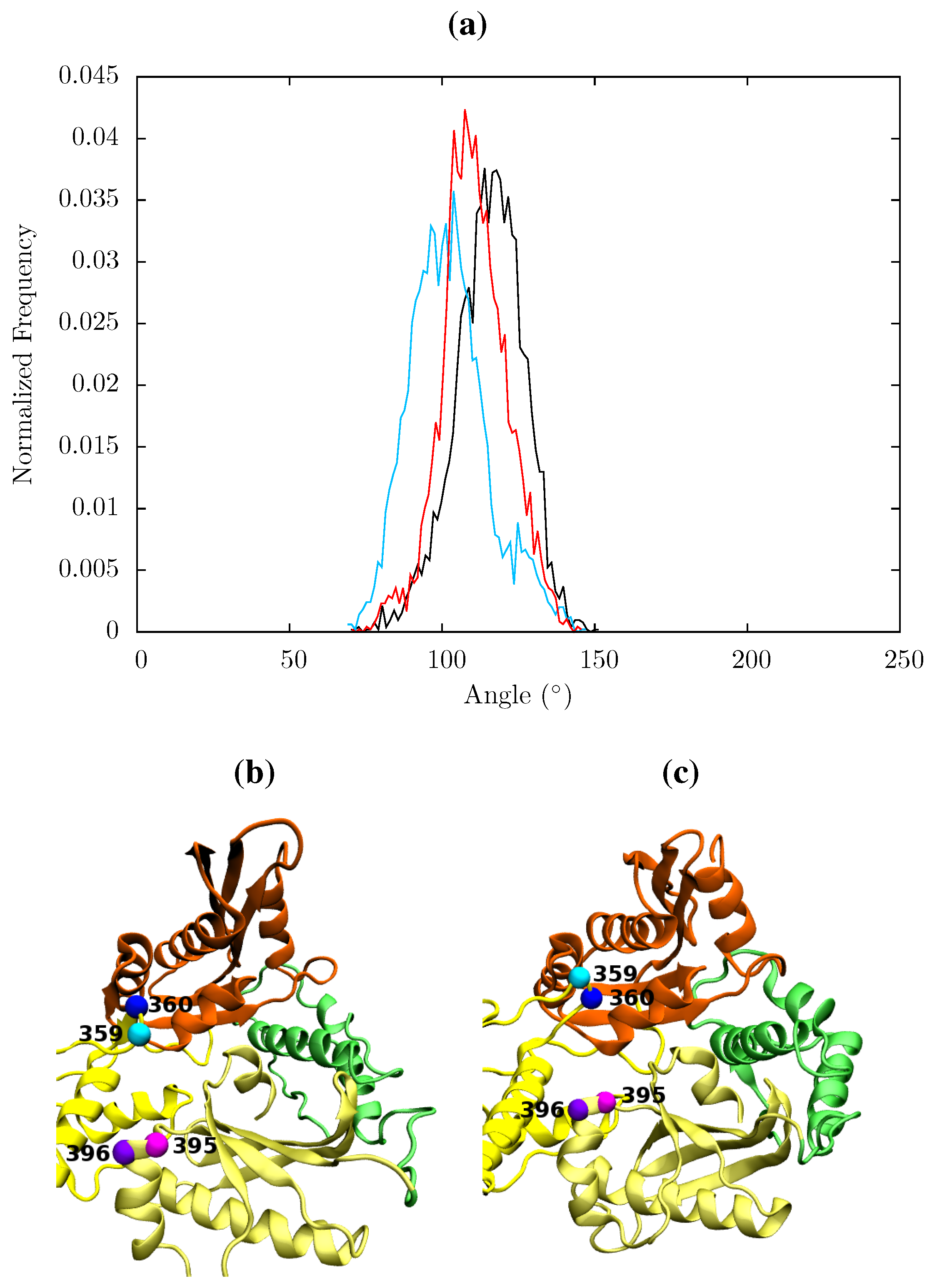
Figure 13.
(a) Histogram showing the normalized frequency distribution of the angle between two pairs of residues (359 and 360 in p66 and 395 and 396 in p51) in the RNaseH primer grip over the simulations of the apo (black), EFV (red) bound and NVP (blue) bound HIV-1 RT ensemble trajectories. The angle is measured between two vectors joining the Cα atoms of the pairs of residues. The distribution is shifted to a lower angle in both NNRTI bound structures. Conformations of the RNaseH primer grip residue pairs 359 and 360 in p66 (shown in cyan and blue, respectively) and 395 and 396 in p51 (in magenta and purple, respectively) are illustrated in (b) from the NVP bound simulation (angle 90°) and (c) form the apo simulation (angle 130°). This shows the that the variation is mainly due to flexibility in the loop containing residues 359 and 360 in p66.
Figure 13.
(a) Histogram showing the normalized frequency distribution of the angle between two pairs of residues (359 and 360 in p66 and 395 and 396 in p51) in the RNaseH primer grip over the simulations of the apo (black), EFV (red) bound and NVP (blue) bound HIV-1 RT ensemble trajectories. The angle is measured between two vectors joining the Cα atoms of the pairs of residues. The distribution is shifted to a lower angle in both NNRTI bound structures. Conformations of the RNaseH primer grip residue pairs 359 and 360 in p66 (shown in cyan and blue, respectively) and 395 and 396 in p51 (in magenta and purple, respectively) are illustrated in (b) from the NVP bound simulation (angle 90°) and (c) form the apo simulation (angle 130°). This shows the that the variation is mainly due to flexibility in the loop containing residues 359 and 360 in p66.
Figure 14.
Histogram showing the normalized frequency distribution of the sidechain dihedral angle χ1 of Glu396 over the simulations of the apo (black), EFV (red) bound and NVP (blue) bound HIV-1 RT ensemble trajectories. Two states are apparent for all three structures, however the balance in occupation is shifted to that with a lower angle in the EFV and NVP bound simulations.
Figure 14.
Histogram showing the normalized frequency distribution of the sidechain dihedral angle χ1 of Glu396 over the simulations of the apo (black), EFV (red) bound and NVP (blue) bound HIV-1 RT ensemble trajectories. Two states are apparent for all three structures, however the balance in occupation is shifted to that with a lower angle in the EFV and NVP bound simulations.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}