
PCR-Independent Detection of Bacterial Species-Specific 16S rRNA at 10 fM by a Pore-Blockage Sensor

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemical and Biological Materials
2.2. Probe Coupling to Microspheres
2.3. Sample Preparation
2.3.1. Cell Culturing and Counting
2.3.2. RNA Extraction and Purification
2.4. Hybridization
2.5. Detection System and Electrical Measurements
3. Results
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jayamohan, H.; Sant, H.J.; Gale, B.K. Applications of microfluidics for molecular diagnostics. Methods Mol. Biol. 2013, 949, 305–334. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.T.; Kim, G. SPR microscopy and its applications to high-throughput analyses of biomolecular binding events and their kinetics. Biomaterials 2007, 28, 2380–2392. [Google Scholar] [CrossRef] [PubMed]
- Hassibi, A.; Vikalo, H.; Riechmann, J.L.; Hassibi, B. Real-time DNA microarray analysis. Nucleic Acids Res. 2009, 37, e132. [Google Scholar] [CrossRef] [PubMed]
- Khanna, V.K. Existing and emerging detection technologies for DNA (Deoxyribonucleic Acid) finger printing, sequencing, bio- and analytical chips: A multidisciplinary development unifying molecular biology, chemical and electronics engineering. Biotechnol. Adv. 2007, 25, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Lillehoj, P.B.; Ho, C.M. DNA diagnostics: Nanotechnology-enhanced electrochemical detection of nucleic acids. Pediatr. Res. 2010, 67, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.; Mergenthaler, S.; Zimmerman, B.; Holzgreve, W. Nucleic-acid based biosensors: The desires of the user. Bioelectrochemistry 2005, 67, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.M.; Li, F.; Zhang, Z.; Zhang, K.; Kang, D.-K.; Ankrum, J.A.; Le, X.C.; Zhao, W. Rolling circle amplification: A versatile tool for chemical biology, materials science and medicine. Chem. Soc. Rev. 2014, 43, 3324–3341. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Gao, Z. Bioanalytical applications of isothermal nucleic acid amplification techniques. Anal. Chim. Acta 2015, 853, 30–45. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Macdonald, J. Advances in isothermal amplification: Novel strategies inspired by biological processes. Biosens. Bioelectron. 2015, 64, 196–211. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Zhou, J.; Zheng, Y.; Gamson, A.S.; Roembke, B.T.; Nakayama, S.; Sintim, H.O. Isothermal amplified detection of DNA and RNA. Mol. BioSyst. 2014, 10, 970–1003. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lowe, S.B.; Gooding, J.J. Brief review of monitoring methods for loop-mediated isothermal amplification (LAMP). Biosens. Bioelectron. 2014, 61, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.C.; Mastali, M.; Gau, V.; Suchard, M.A.; Moller, A.K.; Bruckner, D.A.; Babbitt, J.T.; Li, Y.; Gornbein, J.; Landaw, E.M.; et al. Use of electrochemical DNA biosensors for rapid molecular identification of uropathogens in clinical urine specimens. J. Clin. Microbiol. 2006, 44, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Wang, J.; Liao, W.; Zimmermann, B.G.; Wong, D.T.; Ho, C.M. Electrochemical detection of low-copy number salivary RNA based on specific signal amplification with a hairpin probe. Nucleic Acids Res. 2008, 36, e65. [Google Scholar] [CrossRef] [PubMed]
- Lubin, A.A.; Lai, R.Y.; Baker, B.R.; Heeger, A.J.; Plaxco, K.W. Sequence-specific, electronic detection of oligonucleotides in blood, soil, and foodstuffs with the reagentless, reusable E-DNA sensor. Anal. Chem. 2006, 78, 5671–5677. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Taton, T.A.; Mirkin, C.A. Array-based electrical detection of DNA with nanoparticle probes. Science 2002, 295, 1503–1506. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Kim, E.; Kwak, J. Electrochemical detection of DNA hybridization using biometallization. Anal. Chem. 2005, 77, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Junkin, M.; Kim, D.-H.; Kwon, S.; Shin, Y.S.; Wong, P.K.; Gale, B.K. Applications, techniques, and microfluidic interfacing for nanoscale biosensing. Microfluid. Nanofluid. 2009, 7, 149–167. [Google Scholar] [CrossRef]
- Ramos, D.; Arroyo-Hernandez, M.; Gil-Santos, E.; Duy, T.H.; Van, R.C.; Calleja, M.; Tamayo, J. Arrays of Dual Nanomechanical Resonators for Selective Biological Detection. Anal. Chem. 2009, 81, 2274–2279. [Google Scholar] [CrossRef] [PubMed]
- Rijal, K.; Mutharasan, R. A method for DNA-based detection of E. coli O157:H7 in a proteinous background using piezoelectric-excited cantilever sensors. Analyst 2013, 138, 2943–2950. [Google Scholar] [CrossRef] [PubMed]
- Bangar, M.A.; Shirale, D.J.; Purohit, H.J.; Chen, W.; Myung, N.V.; Mulchandani, A. Single Conducting Polymer Nanowire Based Sequence-Specific, Base-Pair-Length Dependant Label-free DNA Sensor. Electroanalysis 2011, 23, 371–379. [Google Scholar] [CrossRef]
- Yassin, A.F.; Muller, J. Development of real-time polymerase chain reaction assay for specific detection of Tsukamurella by targeting the 16S rRNA gene. Diagn. Microbiol. Infect Dis. 2012, 72, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Kim, S.; Kim, J.; Park, S.D.; Uh, Y.; Lee, H. Multiplex Real-Time PCR Assay for Rapid Detection of Methicillin-Resistant Staphylococci Directly from Positive Blood Cultures. J. Clin. Microbiol. 2014, 52, 1911–1920. [Google Scholar] [CrossRef] [PubMed]
- Masek, B.J.; Hardick, J.; Won, H.; Yang, S.; Hsieh, Y.H.; Rothman, R.E.; Gaydos, C.A. Sensitive Detection and Serovar Differentiation of Typhoidal and Nontyphoidal Salmonella enterica Species Using 16S rRNA Gene PCR Coupled with High-Resolution Melt Analysis. J. Mol. Diagn. 2014, 16, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Esfandiari, L.; Monbouquette, H.G.; Schmidt, J.J. Sequence-specific Nucleic Acid Detection from Binary Pore Conductance Measurement. J. Am. Chem. Soc. 2012, 134, 15880–15886. [Google Scholar] [CrossRef] [PubMed]
- Esfandiari, L.; Lorenzini, M.; Kocharyan, G.; Monbouquette, H.G.; Schmidt, J.J. Sequence-Specific DNA Detection at 10 fM by Electromechanical Signal Transduction. Anal. Chem. 2014, 86, 9638–9643. [Google Scholar] [CrossRef] [PubMed]
- Bremer, H.; Dennis, P.P. Modulation of chemical composition and other parameters of the cell at different exponential growth rates. EcoSal Plus. 2014, 1–49. [Google Scholar] [CrossRef] [PubMed]
- Stender, H.; Broomer, A.J.; Oliveira, K.; Perry-O'Keefe, H.; Hyldig-Nielsen, J.J.; Sage, A.; Coull, J. Rapid detection, identification, and enumeration of Escherichia coli cells in municipal water by chemiluminescent in situ hybridization. Appl. Environ. Microb. 2001, 67, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, L.S.; Okten, H.E.; Noguera, D.R. Making all parts of the 16S rRNA of Escherichia coli accessible in situ to single DNA oligonucleotides. Appl. Environ. Microb. 2006, 72, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, B.M.; Wallner, G.; Beisker, W.; Schwippl, I.; Ludwig, W.; Amann, R. Flow cytometric analysis of the in situ accessibility of Escherichia coli 16S rRNA for fluorescently labeled oligonucleotide probes. Appl. Environ. Microb. 1998, 64, 4973–4982. [Google Scholar]
- Nelson, B.P.; Liles, M.R.; Frederick, K.B.; Corn, R.M.; Goodman, R.M. Label-free detection of 16S ribosomal RNA hybridization on reusable DNA arrays using surface plasmon resonance imaging. Environ. Microbiol. 2002, 4, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.P.; Liao, J.C.; Zhang, Y.H.; Gau, V.; Mastali, M.; Babbitt, J.T.; Grundfest, W.S.; Churchill, B.M.; McCabe, E.R.B.; Haake, D.A. Rapid, species-specific detection of uropathogen 16S rDNA and rRNA at ambient temperature by dot-blot hybridization and an electrochemical sensor array. Mol. Genet. Metab. 2005, 84, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Morgenroth, E.; Raskin, L. Quantitative rRNA-Targeted Solution-Based Hybridization Assay Using Peptide Nucleic Acid Molecular Beacons. Appl. Environ. Microb. 2008, 74, 7297–7305. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Johnson, M.; Hill, P.; Gale, B.K. Microfluidic sample preparation: Cell lysis and nucleic acid purification. Integr. Biol. 2009, 1, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Johnson, M.; Hill, P.; Sonkul, R.S.; Kim, J.; Gale, B.K. Automated microfluidic DNA/RNA extraction with both disposable and reusable components. J. Micromech. Microeng. 2012, 22, 015007. [Google Scholar] [CrossRef]
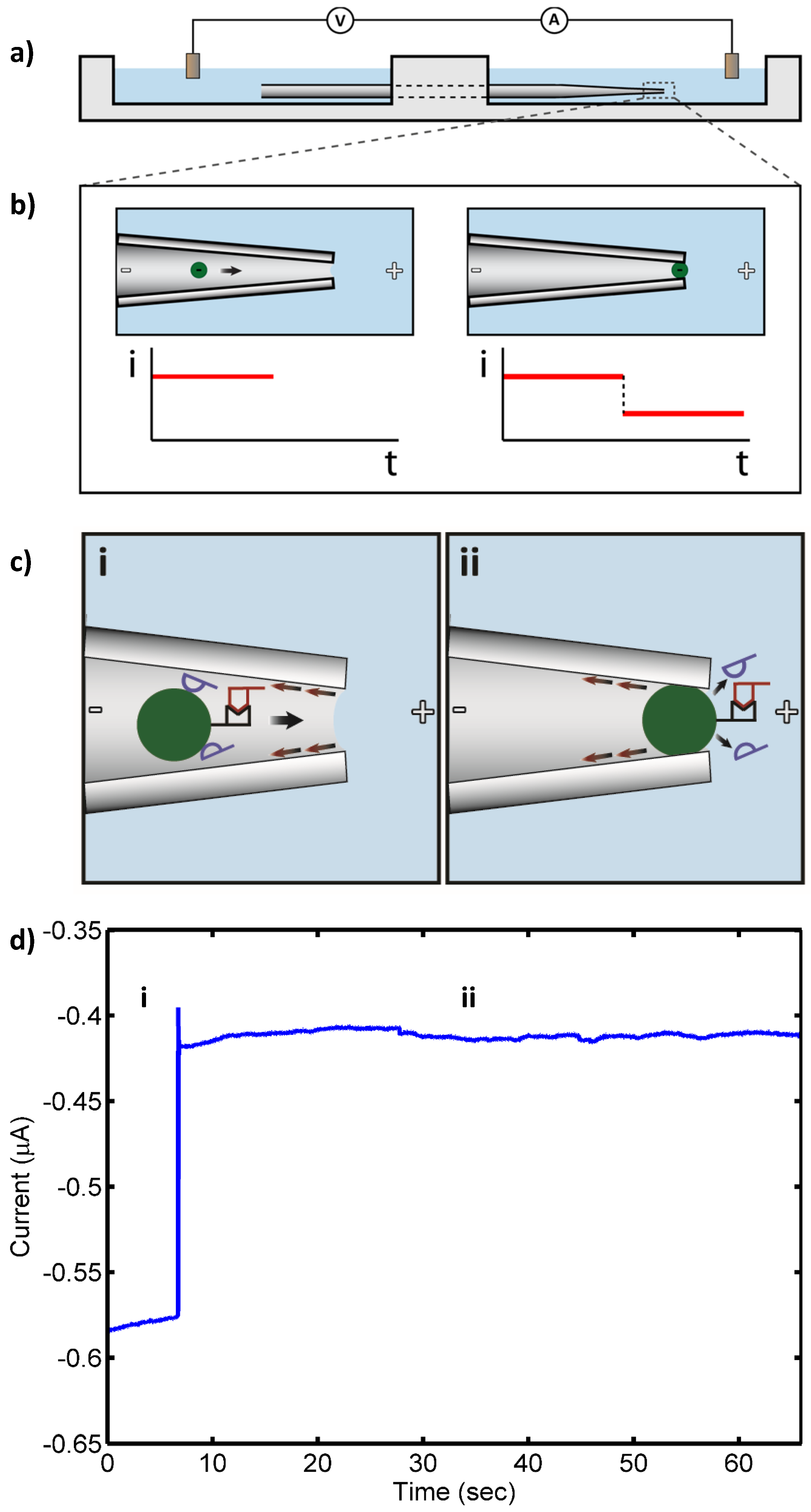


{kind=link}
{kind=link}
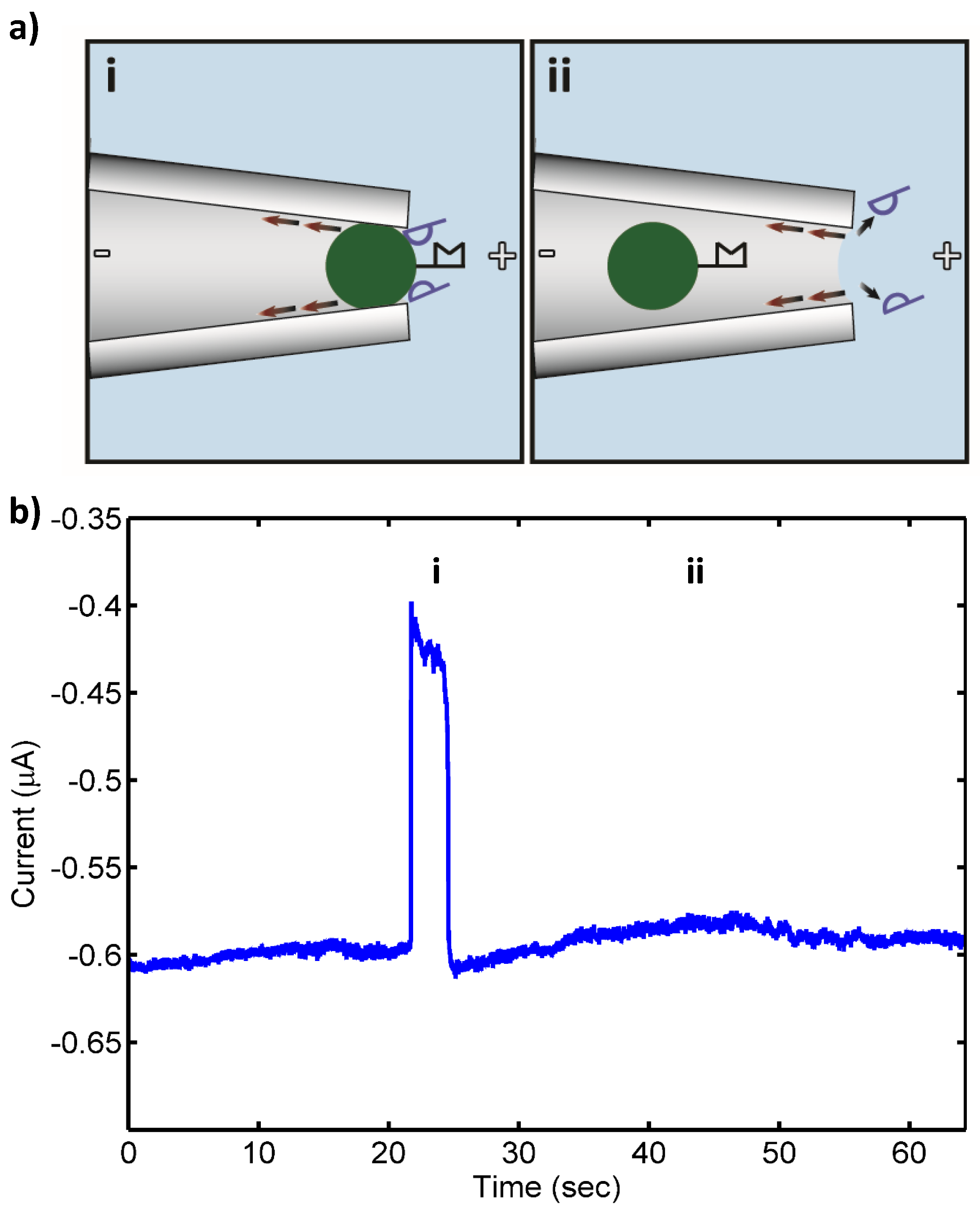{kind=link}
| Target E. coli (ATCC 25922) | Control Bacterium 1 (ATCC 12633) | Control Bacterium 2 (ATCC 13525) | ||||
|---|---|---|---|---|---|---|
| [16S rRNA] | Detection? | Positive control? | Detection? | Positive control? | Detection? | Positive control? |
| 10 pM Expt. 1 | Yes | Yes | No * | Yes | No | Yes |
| 10 pM Expt. 2 | Yes | Yes % | No | Yes | No | Yes |
| 10 pM Expt. 3 | Yes | Yes | No | Yes | No * | Yes |
| 1 pM | Yes | Yes | No | Yes | No | Yes |
| 100 fM | Yes | Yes | No | Yes | No | Yes |
| 10 fM Expt. 1 | Yes | Yes | No | Yes | No | Yes |
| 10 fM Expt. 2 | Yes | Yes | No | Yes | No | Yes |
| 10 fM Expt. 3 | Yes | Yes | No | Yes | No | Yes |
| 1 fM | No | No | No | No | No | No |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esfandiari, L.; Wang, S.; Wang, S.; Banda, A.; Lorenzini, M.; Kocharyan, G.; Monbouquette, H.G.; Schmidt, J.J. PCR-Independent Detection of Bacterial Species-Specific 16S rRNA at 10 fM by a Pore-Blockage Sensor. Biosensors 2016, 6, 37. https://doi.org/10.3390/bios6030037
Esfandiari L, Wang S, Wang S, Banda A, Lorenzini M, Kocharyan G, Monbouquette HG, Schmidt JJ. PCR-Independent Detection of Bacterial Species-Specific 16S rRNA at 10 fM by a Pore-Blockage Sensor. Biosensors. 2016; 6(3):37. https://doi.org/10.3390/bios6030037
Chicago/Turabian StyleEsfandiari, Leyla, Siqing Wang, Siqi Wang, Anisha Banda, Michael Lorenzini, Gayane Kocharyan, Harold G. Monbouquette, and Jacob J. Schmidt. 2016. "PCR-Independent Detection of Bacterial Species-Specific 16S rRNA at 10 fM by a Pore-Blockage Sensor" Biosensors 6, no. 3: 37. https://doi.org/10.3390/bios6030037
APA StyleEsfandiari, L., Wang, S., Wang, S., Banda, A., Lorenzini, M., Kocharyan, G., Monbouquette, H. G., & Schmidt, J. J. (2016). PCR-Independent Detection of Bacterial Species-Specific 16S rRNA at 10 fM by a Pore-Blockage Sensor. Biosensors, 6(3), 37. https://doi.org/10.3390/bios6030037