
Water Formation Reaction under Interfacial Confinement: Al0.25Si0.75O2 on O-Ru(0001)

 , , , ,
, , , ,  ,
, 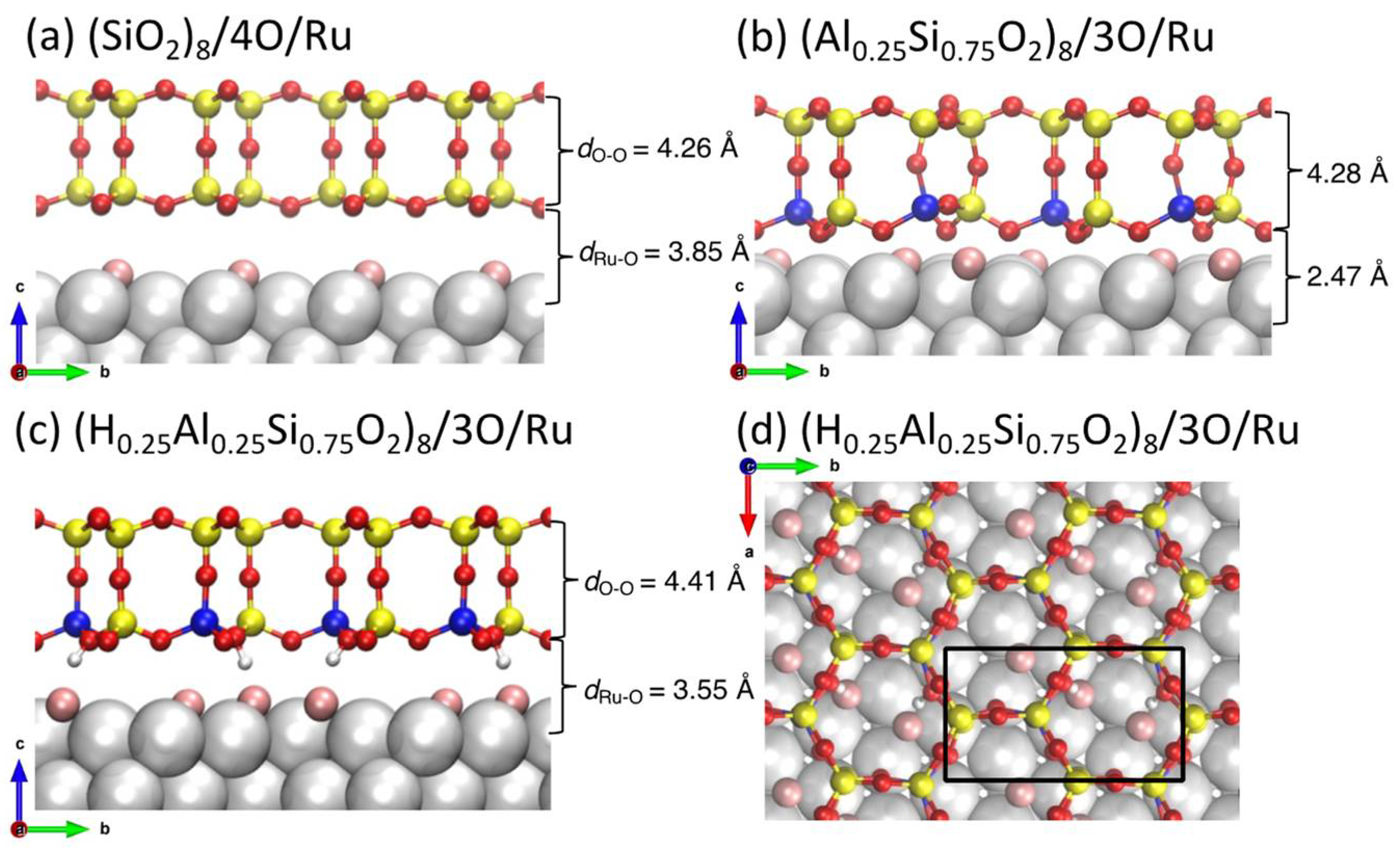{kind=link}
{kind=link}
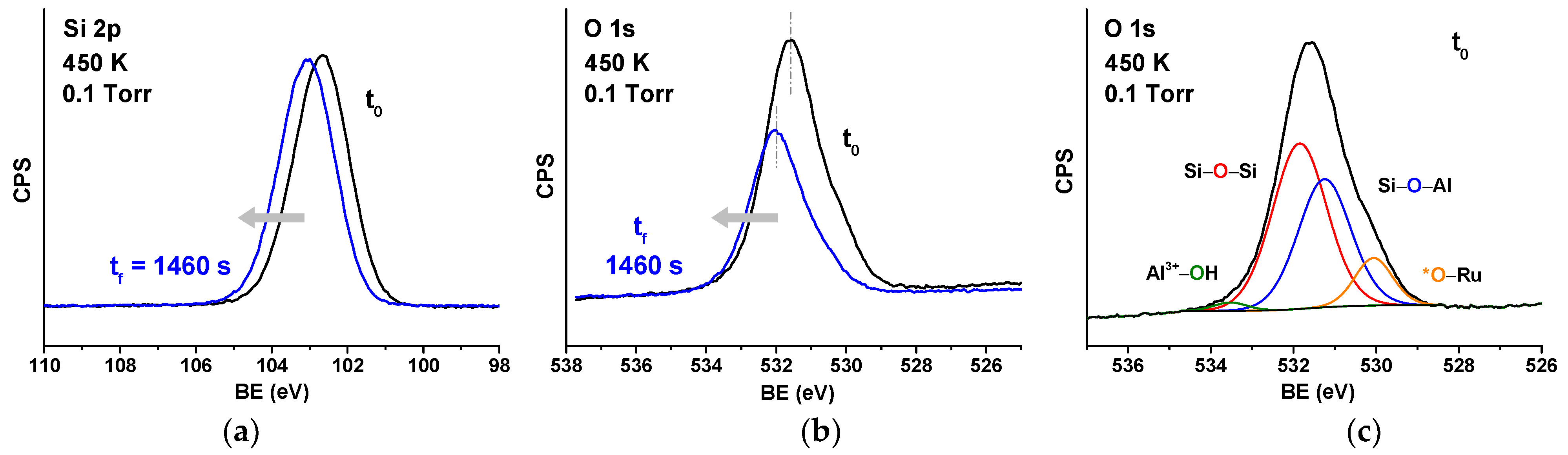{kind=link}
{kind=link}
{kind=link}
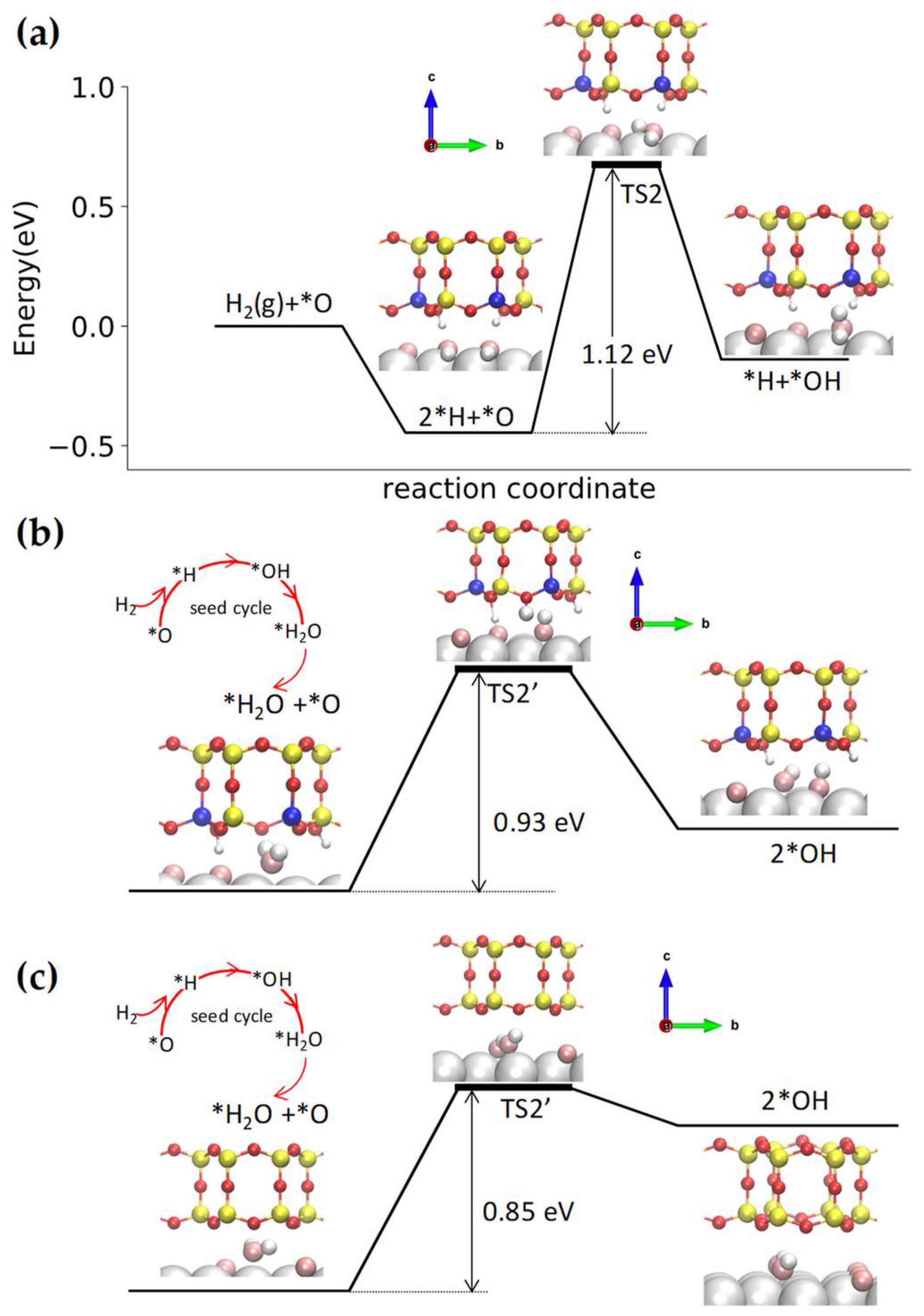{kind=link}
{kind=link}
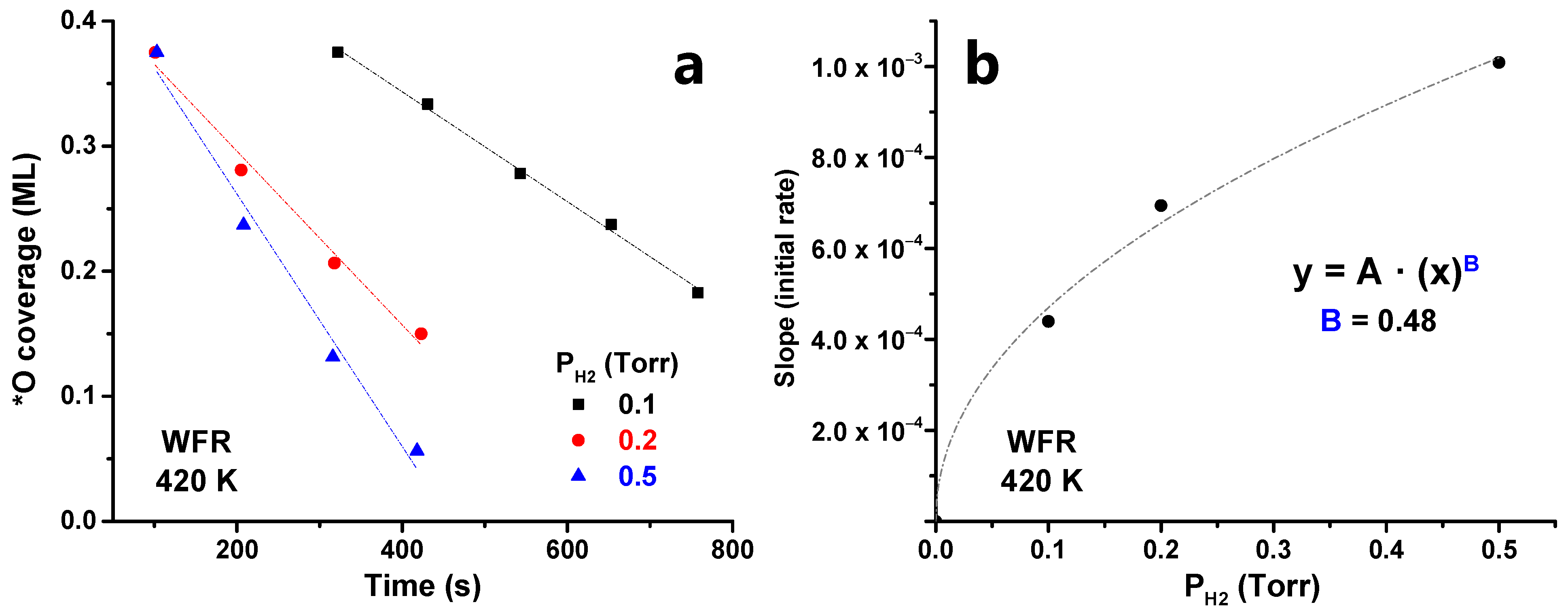{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Material Synthesis
2.2. Computational Methods
3. Results and Discussion
3.1. Kinetic Study of the Water Formation Reaction (WFR) at Constant Pressure (0.1 Torr H2) by X-ray Photoelectron Spectroscopy
3.2. Density Functional Theory (DFT) Calculations
3.3. Reaction Order with Respect to H2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Haywood, D.G.; Saha-Shah, A.; Baker, L.A.; Jacobson, S.C. Fundamental Studies of Nanofluidics: Nanopores, Nanochannels, and Nanopipets. Anal. Chem. 2015, 87, 172–187. [Google Scholar] [CrossRef]
- Lee, S.A.; Ponjavic, A.; Siv, C.; Lee, S.F.; Biteen, J.S. Nanoscopic Cellular Imaging: Confinement Broadens Understanding. ACS Nano 2016, 10, 8143–8153. [Google Scholar] [CrossRef] [PubMed]
- Anduix-Canto, C.; Kim, Y.Y.; Wang, Y.W.; Kulak, A.; Meldrum, F.C.; Christenson, H.K. Effect of Nanoscale Confinement on the Crystallization of Potassium Ferrocyanide. Cryst. Growth Des. 2016, 16, 5403–5411. [Google Scholar] [CrossRef]
- Ha, J.M.; Hamilton, B.D.; Hillmyer, M.A.; Ward, M.D. Alignment of Organic Crystals under Nanoscale Confinement. Cryst. Growth Des. 2012, 12, 4494–4504. [Google Scholar] [CrossRef]
- Chavan, K.S.; Calabrese Barton, S. Simulation of Intermediate Channeling by Nanoscale Confinement. J. Phys. Chem. C 2018, 122, 14474–14480. [Google Scholar] [CrossRef]
- Shifa, T.A.; Vomiero, A. Confined Catalysis: Progress and Prospects in Energy Conversion. Adv. Energy Mater. 2019, 9, 1902307. [Google Scholar] [CrossRef]
- Miners, S.A.; Rance, G.A.; Khlobystov, A.N. Regioselective control of aromatic halogenation reactions in carbon nanotube nanoreactors. Chem. Commun. 2013, 49, 5586–5588. [Google Scholar] [CrossRef]
- Shao, J.; Yuan, L.; Hu, X.; Wu, Y.; Zhang, Z. The Effect of Nano Confinement on the C–H Activation and its Corresponding Structure-Activity Relationship. Sci. Rep. 2014, 4, 7225. [Google Scholar] [CrossRef]
- Pan, X.; Bao, X. The Effects of Confinement inside Carbon Nanotubes on Catalysis. Acc. Chem. Res. 2011, 44, 553–562. [Google Scholar] [CrossRef]
- Wang, D.; Yang, G.; Ma, Q.; Wu, M.; Tan, Y.; Yoneyama, Y.; Tsubaki, N. Confinement Effect of Carbon Nanotubes: Copper Nanoparticles Filled Carbon Nanotubes for Hydrogenation of Methyl Acetate. ACS Catal. 2012, 2, 1958–1966. [Google Scholar] [CrossRef]
- Dai, J.; Zhang, H. Recent Advances in Catalytic Confinement Effect within Micro/Meso-Porous Crystalline Materials. Small 2021, 17, 2005334. [Google Scholar] [CrossRef]
- Li, H.; Xiao, J.; Fu, Q.; Bao, X. Confined Catalysis under Two-Dimensional Materials. Proc. Natl. Acad. Sci. USA 2017, 114, 5930–5934. [Google Scholar] [CrossRef]
- Fu, Q.; Bao, X. Surface Chemistry and Catalysis Confined under Two-Dimensional Materials. Chem. Soc. Rev. 2017, 46, 1842–1874. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Fu, Q.; Zhang, Y.Y.; Weng, X.; Li, H.; Chen, M.; Jin, L.; Dong, A.; Mu, R.; Jiang, P.; et al. Graphene Cover-Promoted Metal-Catalyzed Reactions. Proc. Natl. Acad. Sci. USA 2014, 111, 17023–17028. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, W.; Cui, P.; Zeng, J.; Lin, Z.; Kaxiras, E.; Zhang, Z. Enhancing the Hydrogen Activation Reactivity of Nonprecious Metal Substrates Via Confined Catalysis Underneath Graphene. Nano Lett. 2016, 16, 6058–6063. [Google Scholar] [CrossRef] [PubMed]
- Löffler, D.; Uhlrich, J.J.; Baron, M.; Yang, B.; Yu, X.; Lichtenstein, L.; Heinke, L.; Büchner, C.; Heyde, M.; Shaikhutdinov, S. Growth and Structure of Crystalline Silica Sheet on Ru(0001). Phys. Rev. Lett. 2010, 105, 146104. [Google Scholar] [CrossRef]
- Eads, C.N.; Boscoboinik, J.A.; Head, A.R.; Hunt, A.; Waluyo, I.; Stacchiola, D.J.; Tenney, S.A. Enhanced Catalysis under 2D Silica: A CO Oxidation Study. Angew. Chem. Int. Ed. 2020, 60, 10888–10894. [Google Scholar] [CrossRef]
- Lichtenstein, L.; Büchner, C.; Yang, B.; Shaikhutdinov, S.; Heyde, M.; Sierka, M.; Włodarczyk, R.; Sauer, J.; Freund, H.J. The Atomic Structure of a Metal-Supported Vitreous Thin Silica Film. Angew. Chem. Int. Ed. 2012, 51, 404–407. [Google Scholar] [CrossRef]
- Büchner, C.; Lichtenstein, L.; Yu, X.; Boscoboinik, J.A.; Yang, B.; Kaden, W.E.; Heyde, M.; Shaikhutdinov, S.K.; Włodarczyk, R.; Sierka, M. Ultrathin Silica Films: The Atomic Structure of Two-Dimensional Crystals and Glasses. Chem. Eur. J. 2014, 20, 9176–9183. [Google Scholar] [CrossRef] [PubMed]
- Klemm, H.W.; Prieto, M.J.; Xiong, F.; Hassine, G.B.; Heyde, M.; Menzel, D.; Sierka, M.; Schmidt, T.; Freund, H.J. A Silica Bilayer Supported on Ru(0001): Following the Crystalline to Vitreous Transformation in Real Time with Spectro-Microscopy. Angew. Chem. 2020, 132, 10674–10680. [Google Scholar] [CrossRef]
- Boscoboinik, J.A.; Yu, X.; Yang, B.; Fischer, F.D.; Włodarczyk, R.; Sierka, M.; Shaikhutdinov, S.; Sauer, J.; Freund, H.J. Modeling Zeolites with Metal-Supported Two-Dimensional Aluminosilicate Films. Angew. Chem. Int. Ed. 2012, 51, 6005–6008. [Google Scholar] [CrossRef]
- Boscoboinik, J.A.; Yu, X.; Shaikhutdinov, S.; Freund, H.J. Preparation of an Ordered Ultra-Thin Aluminosilicate Framework Composed of Hexagonal Prisms Forming a Percolated Network. Microporous Mesoporous Mater. 2014, 189, 91–96. [Google Scholar] [CrossRef][Green Version]
- Boscoboinik, J.A.; Shaikhutdinov, S. Exploring Zeolite Chemistry with the Tools of Surface Science: Challenges, Opportunities, and Limitations. Catal. Lett. 2014, 144, 1987–1995. [Google Scholar] [CrossRef]
- Boscoboinik, J.A. Chemistry in Confined Space through the Eyes of Surface Science—2d Porous Materials. J. Phys. Condens. Matter 2019, 31, 063001. [Google Scholar] [CrossRef]
- Yao, B.; Mandrà, S.; Curry, J.O.; Shaikhutdinov, S.; Freund, H.J.; Schrier, J. Gas Separation through Bilayer Silica, the Thinnest Possible Silica Membrane. ACS Appl. Mater. Interfaces 2017, 9, 43061–43071. [Google Scholar] [CrossRef] [PubMed]
- Schlexer, P.; Pacchioni, G.; Włodarczyk, R.; Sauer, J. CO Adsorption on a Silica Bilayer Supported on Ru(0001). Surf. Sci. 2016, 648, 2–9. [Google Scholar] [CrossRef]
- Büchner, C.; Lichtenstein, L.; Stuckenholz, S.; Heyde, M.; Ringleb, F.; Sterrer, M.; Kaden, W.E.; Giordano, L.; Pacchioni, G.; Freund, H.J. Adsorption of Au and Pd on Ruthenium-Supported Bilayer Silica. J. Phys. Chem. C 2014, 118, 20959–20969. [Google Scholar] [CrossRef]
- Akter, N.; Wang, M.; Zhong, J.Q.; Liu, Z.; Kim, T.; Lu, D.; Boscoboinik, J.A.; Stacchiola, D.J. Stabilization of Oxidized Copper Nanoclusters in Confined Spaces. Top. Catal. 2018, 61, 419–427. [Google Scholar] [CrossRef]
- Zhong, J.Q.; Wang, M.; Akter, N.; Kestell, J.D.; Boscoboinik, A.M.; Kim, T.; Stacchiola, D.J.; Lu, D.; Boscoboinik, J.A. Immobilization of Single Argon Atoms in Nano-Cages of Two-Dimensional Zeolite Model Systems. Nat. Commun. 2017, 8, 16118. [Google Scholar] [CrossRef]
- Zhong, J.Q.; Wang, M.; Akter, N.; Kestell, J.D.; Niu, T.; Boscoboinik, A.M.; Kim, T.; Stacchiola, D.J.; Wu, Q.; Lu, D. Ionization-Facilitated Formation of 2D (Alumino) Silicate–Noble Gas Clathrate Compounds. Adv. Funct. Mater. 2019, 29, 1806583. [Google Scholar] [CrossRef]
- Jhang, J.H.; Zhou, C.; Dagdeviren, O.E.; Hutchings, G.S.; Schwarz, U.D.; Altman, E.I. Growth of Two Dimensional Silica and Aluminosilicate Bilayers on Pd (111): From Incommensurate to Commensurate Crystalline. Phys. Chem. Chem. Phys. 2017, 19, 14001–14011. [Google Scholar] [CrossRef]
- Altman, E.I.; Götzen, J.; Samudrala, N.; Schwarz, U.D. Growth and Characterization of Crystalline Silica Films on Pd(100). J. Phys. Chem. C 2013, 117, 26144–26155. [Google Scholar] [CrossRef]
- Yu, X.; Yang, B.; Boscoboinik, J.A.; Shaikhutdinov, S.; Freund, H.J. Support Effects on the Atomic Structure of Ultrathin Silica Films on Metals. Appl. Phys. Lett. 2012, 100, 151608. [Google Scholar] [CrossRef]
- Wang, M.; Zhong, J.Q.; Stacchiola, D.J.; Boscoboinik, J.A.; Lu, D. First-Principles Study of Interface Structures and Charge Rearrangement at the Aluminosilicate/Ru(0001) Heterojunction. J. Phys. Chem. C 2019, 123, 7731–7739. [Google Scholar] [CrossRef]
- Wang, M.; Zhong, J.Q.; Kestell, J.; Waluyo, I.; Stacchiola, D.J.; Boscoboinik, J.A.; Lu, D. Energy Level Shifts at the Silica/Ru(0001) Heterojunction Driven by Surface and Interface Dipoles. Top. Catal. 2017, 60, 481–491. [Google Scholar] [CrossRef]
- Mark, L.O.; Chen, W.; Eads, C.N.; Lu, D.; Boscoboinik, J.A.; Stacchiola, D.; Will Medlin, J.; Tenney, S.A. Confinement Effects on Furfuryl Alcohol Reactions over Porous Bilayer Silica-Modified Pd(111). J. Phys. Chem. C 2020, 124, 25437–25446. [Google Scholar] [CrossRef]
- Shishkin, M.; Ziegler, T. Hydrogen Oxidation at the Ni/Ytria-Stabilized Zirconia Interface: A Study Based on Density Functional Theory. J. Phys. Chem. C 2010, 114, 11209–11214. [Google Scholar] [CrossRef]
- Ohyama, J.; Sato, T.; Yamamoto, Y.; Arai, S.; Satsuma, A. Size Specifically High Activity of Ru Nanoparticles for Hydrogen Oxidation Reaction in Alkaline Electrolyte. J. Am. Chem. Soc. 2013, 135, 8016–8021. [Google Scholar] [CrossRef]
- Alia, S.M.; Pivovar, B.S.; Yan, Y. Platinum-Coated Copper Nanowires with High Activity for Hydrogen Oxidation Reaction in Base. J. Am. Chem. Soc. 2013, 135, 13473–13478. [Google Scholar] [CrossRef]
- Fisher, G.B.; Gland, J.L.; Schmieg, S.J. The Spectroscopic Observation of Water Formation. J. Vac. Sci. Technol. 1982, 20, 518–521. [Google Scholar] [CrossRef]
- Germer, T.A.; Ho, W. Direct Characterization of the Hydroxyl Intermediate During Reduction of Oxygen on Pt(111) by Time-Resolved Electron Energy Loss Spectroscopy. Chem. Phys. Lett. 1989, 163, 449–454. [Google Scholar] [CrossRef]
- Ogle, K.; White, J.M. The Low Temperature Water Formation Reaction on Pt(111): A Static SIMS and TDS Study. Surf. Sci. 1984, 139, 43–62. [Google Scholar] [CrossRef]
- Gland, J.L.; Fisher, G.B.; Kollin, E.B. The Hydrogen-Oxygen Reaction over the Pt(111) Surface: Transient Titration of Adsorbed Oxygen with Hydrogen. J. Catal. 1982, 77, 263–278. [Google Scholar] [CrossRef]
- Hellsing, B.; Kasemo, B.; Zhdanov, V.P. Kinetics of the Hydrogen-Oxygen Reaction on Platinum. J. Catal. 1991, 132, 210–228. [Google Scholar] [CrossRef]
- Verheij, L.K.; Freitag, M.; Hugenschmidt, M.B.; Kempf, I.; Poelsema, B.; Comsa, G. Autocatalytic Behavior and Role of Oxygen Diffusion in the Hydrogen-Oxygen Reaction on Pt(111). Surf. Sci. 1992, 272, 276–282. [Google Scholar] [CrossRef]
- Michaelides, A.; Hu, P. Catalytic Water Formation on Platinum: A First-Principles Study. J. Am. Chem. Soc. 2001, 123, 4235–4242. [Google Scholar] [CrossRef]
- Völkening, S.; Bedürftig, K.; Jacobi, K.; Wintterlin, J.; Ertl, G. Dual-Path Mechanism for Catalytic Oxidation of Hydrogen on Platinum Surfaces. Phys. Rev. Lett. 1999, 83, 2672. [Google Scholar] [CrossRef]
- Koch, M.H.; Jakob, P.; Menzel, D. The Influence of Steps on the Water-Formation Reaction on Ru(001). Surf. Sci. 1996, 367, 293–306. [Google Scholar] [CrossRef]
- Schick, M.; Xie, J.; Mitchell, W.J.; Weinberg, W.H. Interaction of Gas-Phase Atomic Deuterium with the Ru(001)–p(1 × 2)–O Surface: Kinetics of Hydroxyl and Water Formation. J. Chem. Phys. 1996, 104, 7713–7718. [Google Scholar] [CrossRef]
- Zhong, J.Q.; Kestell, J.; Waluyo, I.; Wilkins, S.; Mazzoli, C.; Barbour, A.; Kaznatcheev, K.; Shete, M.; Tsapatsis, M.; Boscoboinik, J.A. Oxidation and Reduction under Cover: Chemistry at the Confined Space between Ultrathin Nanoporous Silicates and Ru(0001). J. Phys. Chem. C 2016, 120, 8240–8245. [Google Scholar] [CrossRef]
- Emmez, E.; Boscoboinik, J.A.; Tenney, S.; Sutter, P.; Shaikhutdinov, S.; Freund, H.J. Oxidation of the Ru(0001) Surface Covered by Weakly Bound, Ultrathin Silicate Films. Surf. Sci. 2016, 646, 19–25. [Google Scholar] [CrossRef]
- Wang, M.; Zhou, C.; Akter, N.; Tysoe, W.T.; Boscoboinik, J.A.; Lu, D. Mechanism of the Accelerated Water Formation Reaction under Interfacial Confinement. ACS Catal. 2020, 10, 6119–6128. [Google Scholar] [CrossRef]
- Prieto, M.J.; Klemm, H.W.; Xiong, F.; Gottlob, D.M.; Menzel, D.; Schmidt, T.; Freund, H.J. Water Formation under Silica Thin Films: Real-Time Observation of a Chemical Reaction in a Physically Confined Space. Angew. Chem. Int. Ed. 2018, 57, 8749–8753. [Google Scholar] [CrossRef]
- Prieto, M.J.; Mullan, T.; Schlutow, M.; Gottlob, D.M.; Tanase, L.C.; Menzel, D.; Sauer, J.; Usvyat, D.; Schmidt, T.; Freund, H.J. Insights into Reaction Kinetics in Confined Space: Real Time Observation of Water Formation under a Silica Cover. J. Am. Chem. Soc. 2021, 143, 8780–8790. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Klimeš, J.; Bowler, D.R.; Michaelides, A. Chemical Accuracy for the Van Der Waals Density Functional. J. Phys. Condens. Matter 2009, 22, 022201. [Google Scholar] [CrossRef]
- Klimeš, J.; Bowler, D.R.; Michaelides, A. Van Der Waals Density Functionals Applied to Solids. Phys. Rev. B 2011, 83, 195131. [Google Scholar] [CrossRef]
- Lee, K.; Murray, É.D.; Kong, L.; Lundqvist, B.I.; Langreth, D.C. Higher-Accuracy Van Der Waals Density Functional. Phys. Rev. B 2010, 82, 081101. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cored, J.; Wang, M.; Akter, N.; Darbari, Z.; Xu, Y.; Karagoz, B.; Waluyo, I.; Hunt, A.; Stacchiola, D.; Head, A.R.; et al. Water Formation Reaction under Interfacial Confinement: Al0.25Si0.75O2 on O-Ru(0001). Nanomaterials 2022, 12, 183. https://doi.org/10.3390/nano12020183
Cored J, Wang M, Akter N, Darbari Z, Xu Y, Karagoz B, Waluyo I, Hunt A, Stacchiola D, Head AR, et al. Water Formation Reaction under Interfacial Confinement: Al0.25Si0.75O2 on O-Ru(0001). Nanomaterials. 2022; 12(2):183. https://doi.org/10.3390/nano12020183
Chicago/Turabian StyleCored, Jorge, Mengen Wang, Nusnin Akter, Zubin Darbari, Yixin Xu, Burcu Karagoz, Iradwikanari Waluyo, Adrian Hunt, Dario Stacchiola, Ashley Rose Head, and et al. 2022. "Water Formation Reaction under Interfacial Confinement: Al0.25Si0.75O2 on O-Ru(0001)" Nanomaterials 12, no. 2: 183. https://doi.org/10.3390/nano12020183
APA StyleCored, J., Wang, M., Akter, N., Darbari, Z., Xu, Y., Karagoz, B., Waluyo, I., Hunt, A., Stacchiola, D., Head, A. R., Concepcion, P., Lu, D., & Boscoboinik, J. A. (2022). Water Formation Reaction under Interfacial Confinement: Al0.25Si0.75O2 on O-Ru(0001). Nanomaterials, 12(2), 183. https://doi.org/10.3390/nano12020183