
Computational Characterization of β-Li3PS4 Solid Electrolyte: From Bulk and Surfaces to Nanocrystals


 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
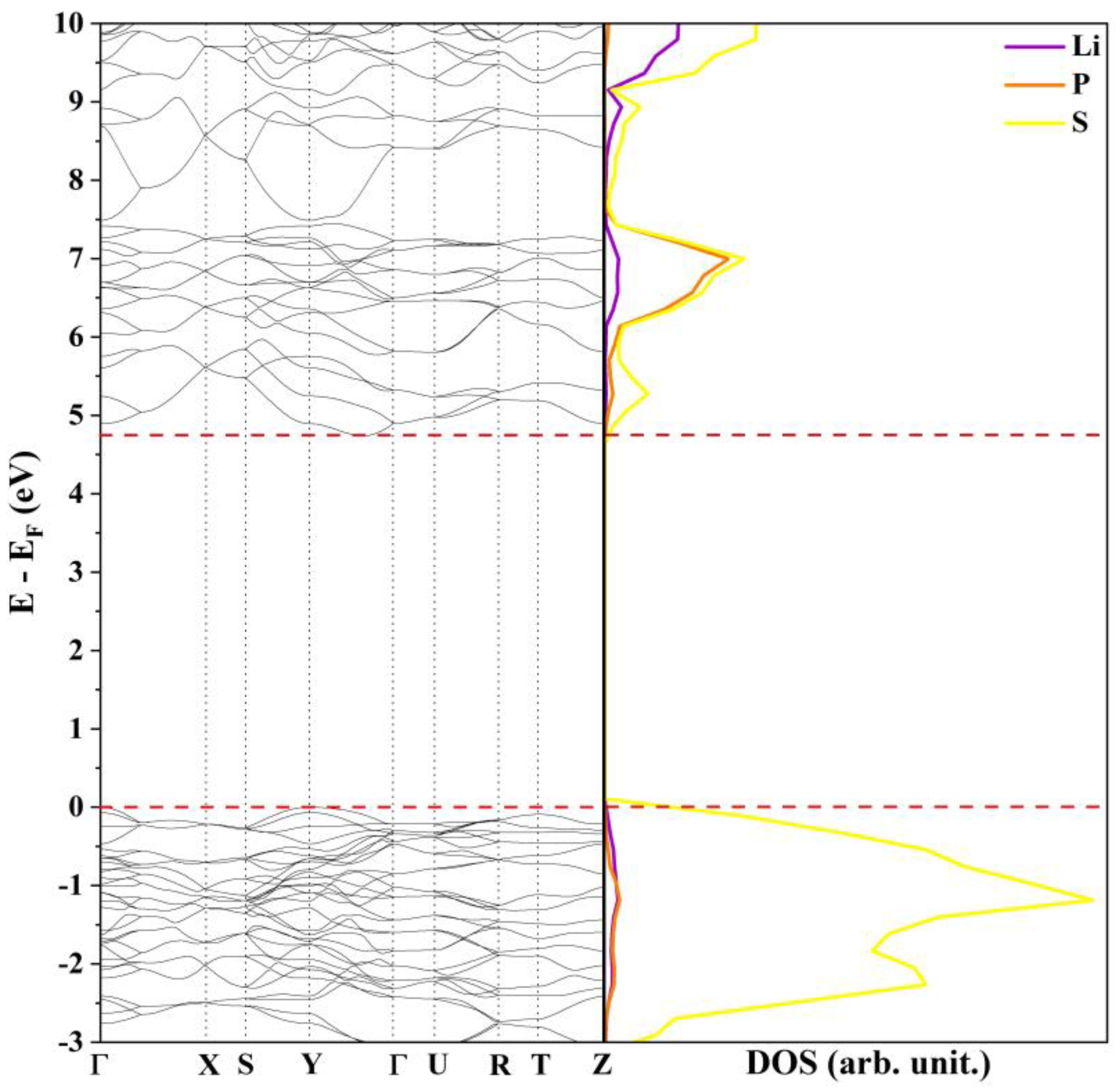
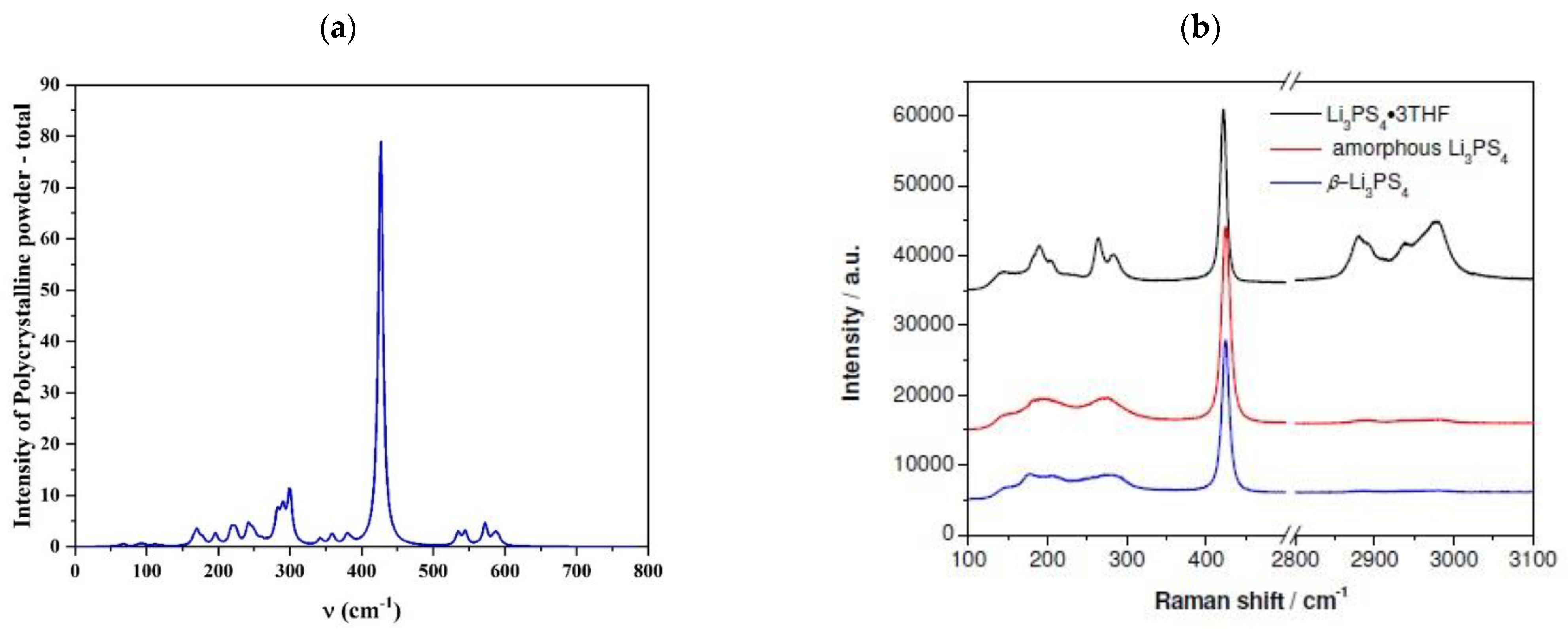
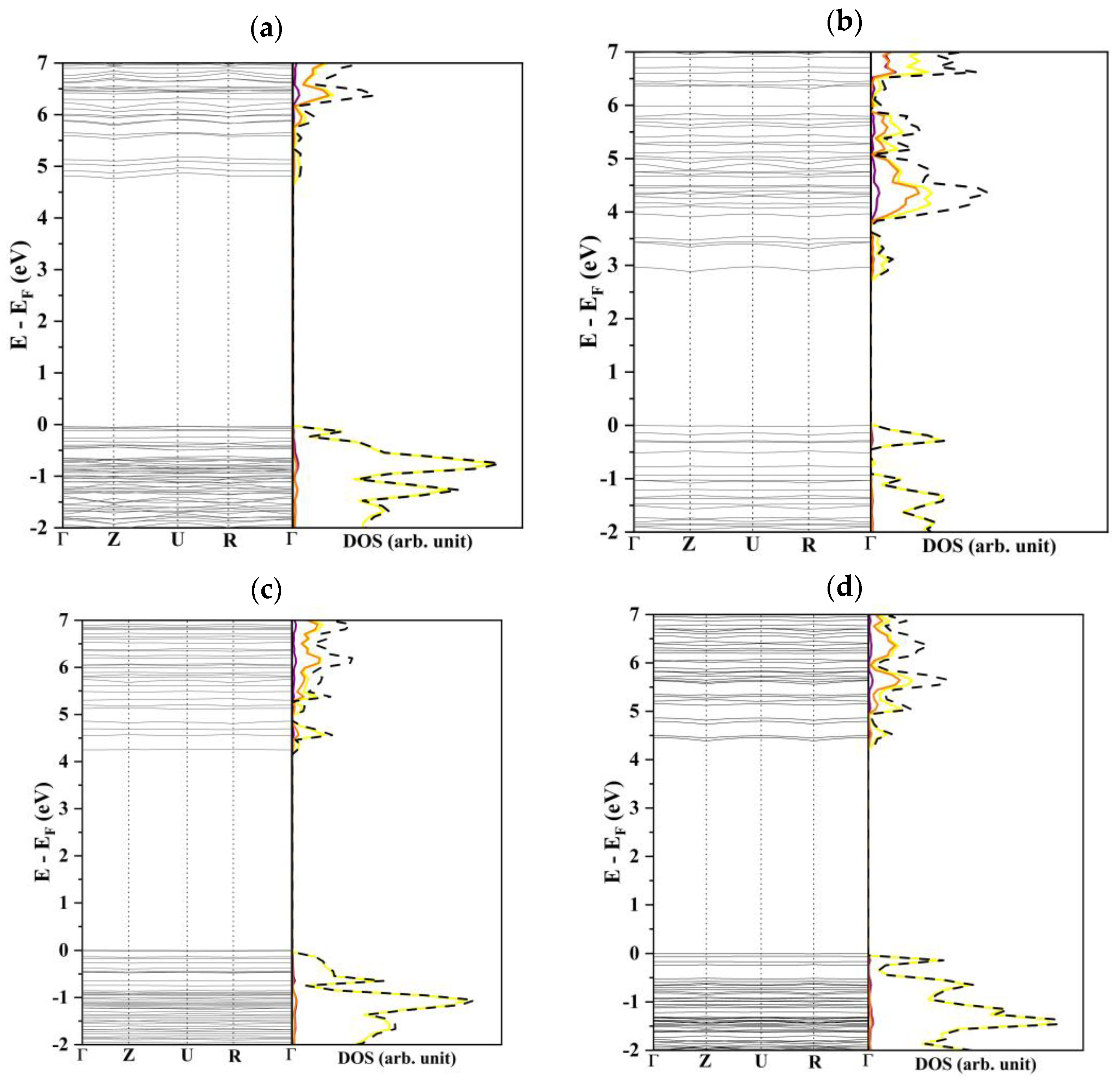
3.1. Bulk Analysis
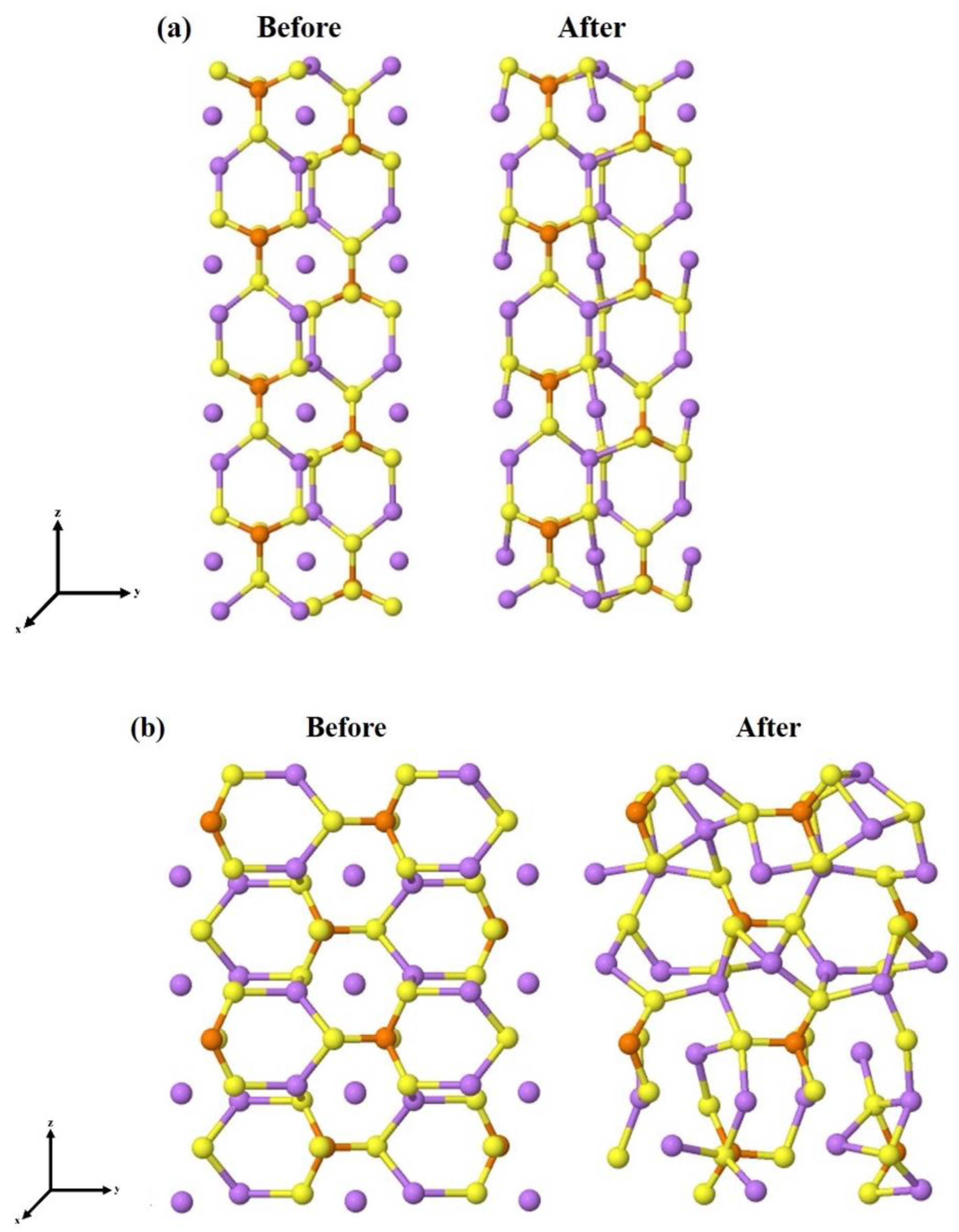
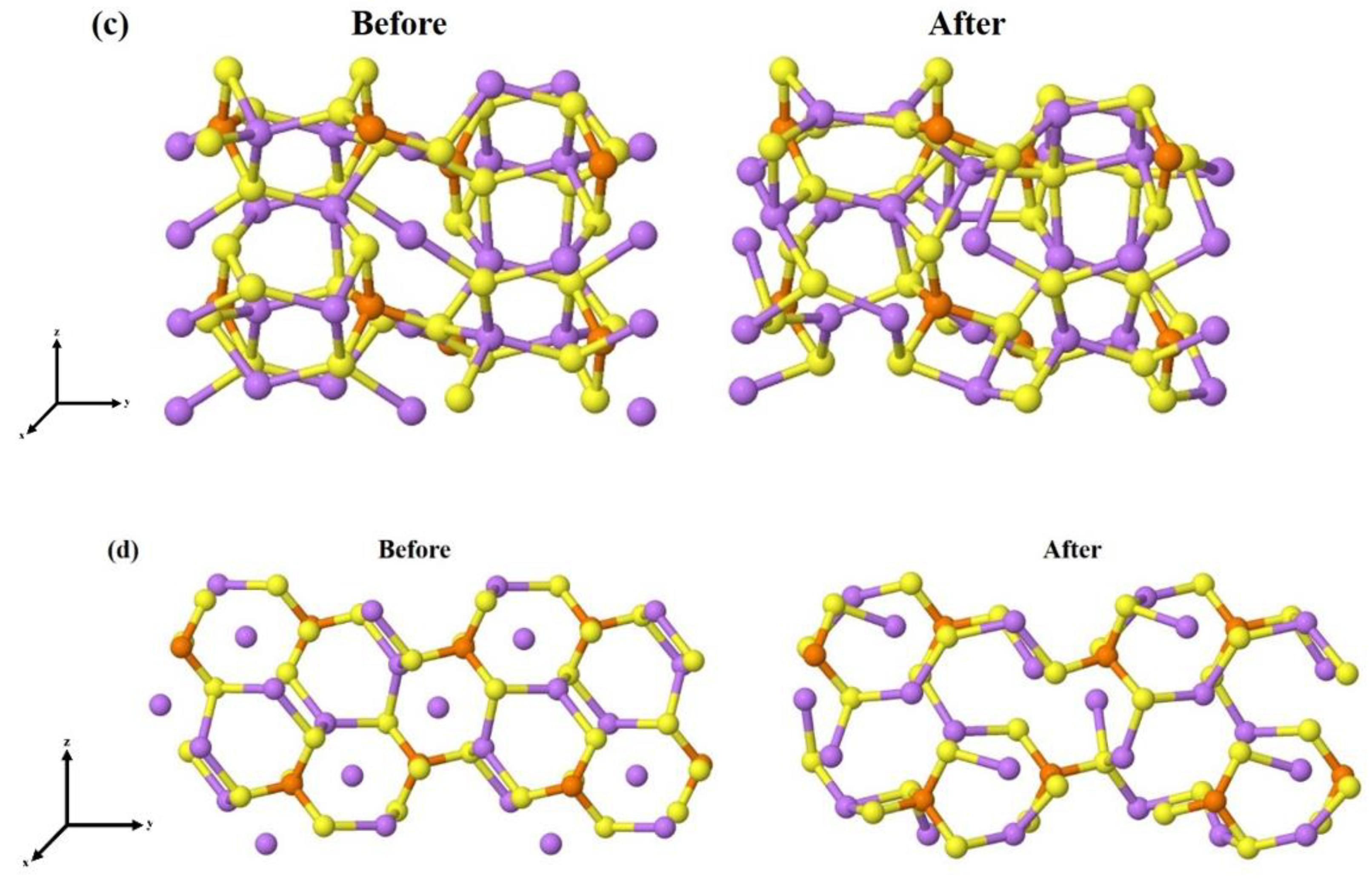
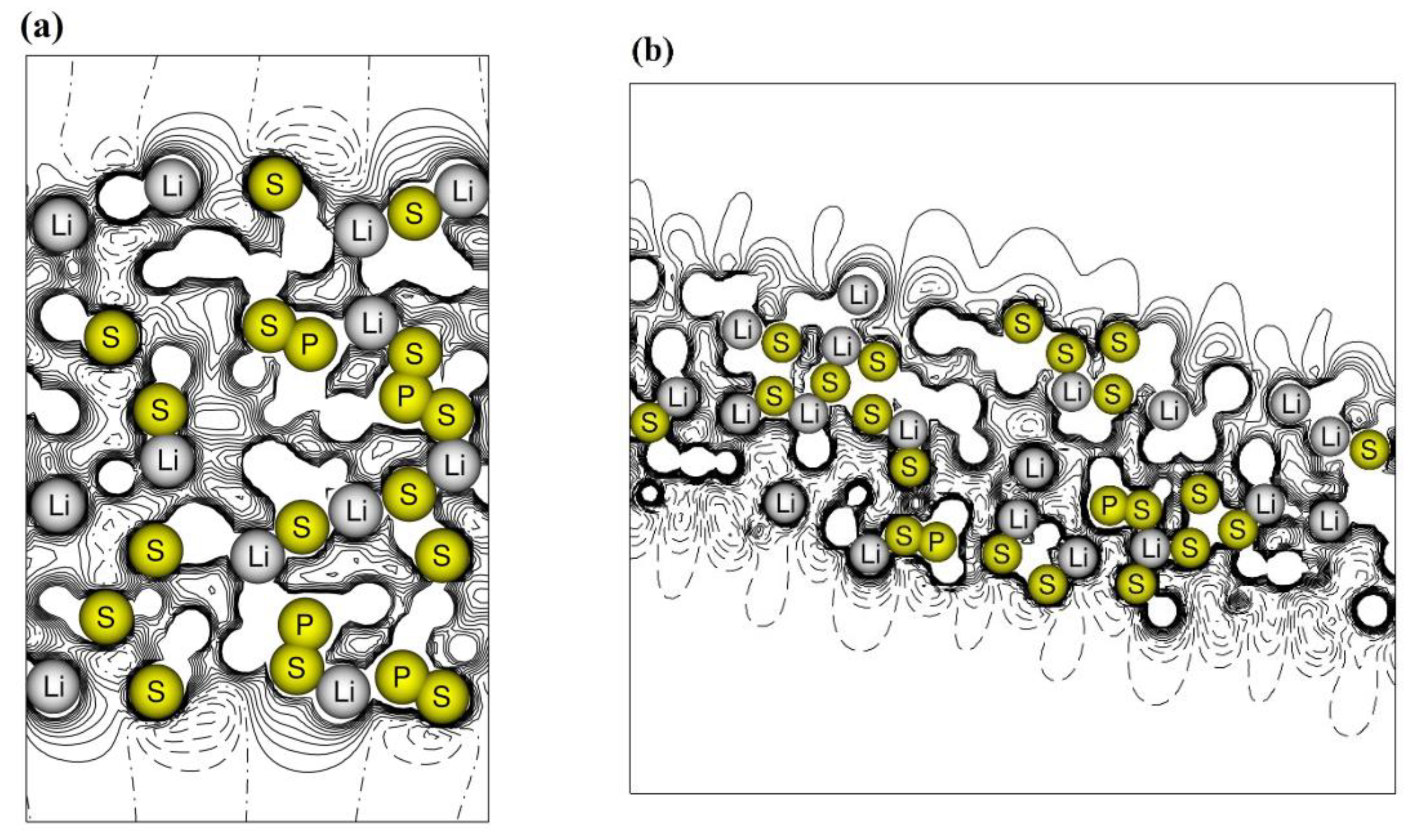
3.2. β-Li3PS4 Surfaces
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahniyaz, A.; de Meatza, I.; Kvasha, A.; Garcia-Calvo, O.; Ahmed, I.; Sgroi, M.F.; Giuliano, M.; Dotoli, M.; Dumitrescu, M.A.; Jahn, M.; et al. Progress in Solid-State High Voltage Lithium-Ion Battery Electrolytes. Adv. Appl. Energy 2021, 4, 100070. [Google Scholar] [CrossRef]
- Homma, K.; Yonemura, M.; Kobayashi, T.; Nagao, M.; Hirayama, M.; Kanno, R. Crystal Structure and Phase Transitions of the Lithium Ionic Conductor Li3PS4. Solid State Ion. 2011, 182, 53–58. [Google Scholar] [CrossRef]
- Mercier, R.; Malugani, J.-P.; Fahys, B.; Robert, G.; Douglade, J. IUCr Structure Du Tetrathiophosphate de Lithium. Acta Crystallogr. Sect. B-Struct. Sci. 1982, 38, 1887–1890. [Google Scholar] [CrossRef]
- Kim, J.-S.; Jung, W.D.; Choi, S.; Son, J.-W.; Kim, B.-K.; Lee, J.-H.; Kim, H. Thermally Induced S-Sublattice Transition of Li3PS4 for Fast Lithium-Ion Conduction. J. Phys. Chem. Lett. 2018, 9, 5592–5597. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Fu, W.; Payzant, E.A.; Yu, X.; Wu, Z.; Dudney, N.J.; Kiggans, J.; Hong, K.; Rondinone, A.J.; Liang, C. Anomalous High Ionic Conductivity of Nanoporous β-Li3PS4. J. Am. Chem. Soc. 2013, 135, 975–978. [Google Scholar] [CrossRef]
- Marchini, F.; Porcheron, B.; Rousse, G.; Blanquer, L.A.; Droguet, L.; Foix, D.; Koç, T.; Deschamps, M.; Tarascon, J.M. The Hidden Side of Nanoporous β-Li3PS4 Solid Electrolyte. Adv. Energy Mater. 2021, 11, 2101111. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-Mechanical Condensed Matter Simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and Simple Analytic Representation of the Electron-Gas Correlation Energy. Phys. Rev. B 1992, 45, 13244. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158. [Google Scholar] [CrossRef]
- Dovesi, R.; Ermondi, C.; Ferrero, E.; Pisani, C.; Roetti, C. Hartree-Fock Study of Lithium Hydride with the Use of a Polarizable Basis Set. Phys. Rev. B 1984, 29, 3591. [Google Scholar] [CrossRef]
- Lichanot, A.; Aprà, E.; Dovesi, R. Quantum Mechnical Hartree-Fock Study of the Elastic Properties of Li2S and Na2S. Phys. Status Solidi 1993, 177, 157–163. [Google Scholar] [CrossRef]
- Zicovich-Wilson, C.M.; Bert, A.; Roetti, C.; Dovesi, R.; Saunders, V.R. Characterization of the Electronic Structure of Crystalline Compounds through Their Localized Wannier Functions. J. Chem. Phys. 2001, 116, 1120. [Google Scholar] [CrossRef]
- Dovesi, R.; Saunders, V.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.; Bush, I.; et al. CRYSTAL17 User’s Manual. 2018. Available online: https://www.crystal.unito.it/manuals/crystal17.pdf (accessed on 14 July 2022).
- Pascale, F.; Zicovich-Wilson, C.M.; Gejo, F.L.; Civalleri, B.; Orlando, R.; Dovesi, R. The Calculation of the Vibrational Frequencies of Crystalline Compounds and Its Implementation in the CRYSTAL Code. J. Comput. Chem. 2004, 25, 888–897. [Google Scholar] [CrossRef]
- Zicovich-Wilson, C.M.; Pascale, F.; Roetti, C.; Saunders, V.R.; Orlando, R.; Dovesi, R. Calculation of the Vibration Frequencies of α-Quartz: The Effect of Hamiltonian and Basis Set. J. Comput. Chem. 2004, 25, 1873–1881. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990; p. 438. [Google Scholar]
- Casassa, S.; Erba, A.; Baima, J.; Orlando, R. Electron Density Analysis of Large (Molecular and Periodic) Systems: A Parallel Implementation. J. Comput. Chem. 2015, 36, 1940–1946. [Google Scholar] [CrossRef] [PubMed]
- Gatti, C.; Casassa, S. TOPOND14 User’s Manual. 2017, p. 53. Available online: https://www.crystal.unito.it/topond/topond.pdf (accessed on 14 July 2022).
- Gatti, C.; Saunders, V.R.; Roetti, C. Crystal Field Effects on the Topological Properties of the Electron Density in Molecular Crystals: The Case of Urea. J. Chem. Phys. 1994, 101, 10686–10696. [Google Scholar] [CrossRef]
- Gatti, C. Chemical Bonding in Crystals: New Directions. Z. Krist. 2005, 220, 399–457. [Google Scholar] [CrossRef]
- Gatti, C. Challenging Chemical Concepts through Charge Density of Molecules and Crystals. Phys. Scr. 2013, 87, 48102. [Google Scholar] [CrossRef]
- Clark, A.E.; Sonnenberg, J.L.; Hay, P.J.; Martin, R.L. Density and Wave Function Analysis of Actinide Complexes: What Can Fuzzy Atom, Atoms-in-Molecules, Mulliken, Löwdin, and Natural Population Analysis Tell Us? J. Chem. Phys. 2004, 121, 2563–2570. [Google Scholar] [CrossRef]
- van Duijneveldt, F.B.; van Duijneveldt-van de Rijdt, J.G.C.M.; van Lenthe, J.H. State of the Art in Counterpoise Theory. Chem. Rev. 1994, 94, 1873–1885. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, Q.; Cui, Y.; Chen, Y.; Shi, S.; Wang, R.Z.; Yan, H. Elastic Properties, Defect Thermodynamics, Electrochemical Window, Phase Stability, and Li+ Mobility of Li3PS4: Insights from First-Principles Calculations. ACS Appl. Mater. Interfaces 2016, 8, 25229–25242. [Google Scholar] [CrossRef] [PubMed]
- Lepley, N.D.; Holzwarth, N.A.W.; Du, Y.A. Structures, Li+ Mobilities, and Interfacial Properties of Solid Electrolytes Li3PS4 and Li3PO4 from First Principles. Phys. Rev. B 2013, 88, 104103. [Google Scholar] [CrossRef]
- Lim, M.S.; Jhi, S.H. First-Principles Study of Lithium-Ion Diffusion in β-Li3PS4 for Solid-State Electrolytes. Curr. Appl. Phys. 2018, 18, 541–545. [Google Scholar] [CrossRef]
- Rangasamy, E.; Li, J.; Sahu, G.; Dudney, N.; Liang, C. Pushing the Theoretical Limit of Li-CFx Batteries: A Tale of Bifunctional Electrolyte. J. Am. Chem. Soc. 2014, 136, 6874–6877. [Google Scholar] [CrossRef]
- Ates, T.; Neumann, A.; Danner, T.; Latz, A.; Zarrabeitia, M.; Stepien, D.; Varzi, A.; Passerini, S.; Ates, T.; Neumann, A.; et al. Elucidating the Role of Microstructure in Thiophosphate Electrolytes—A Combined Experimental and Theoretical Study of β-Li3PS4. Adv. Sci. 2022, 9, 2105234. [Google Scholar] [CrossRef]
- Wang, H.; Hood, Z.D.; Xia, Y.; Liang, C. Fabrication of Ultrathin Solid Electrolyte Membranes of β-Li3PS4 Nanoflakes by Evaporation-Induced Self-Assembly for All-Solid-State Batteries. J. Mater. Chem. A 2016, 4, 8091–8096. [Google Scholar] [CrossRef]
- Bultinck, P.; Van Alsenoy, C.; Ayers, P.W.; Carbó-Dorca, R. Critical Analysis and Extension of the Hirshfeld Atoms in Molecules. J. Chem. Phys. 2007, 126, 144111. [Google Scholar] [CrossRef]
- Hood, Z.D.; Wang, H.; Pandian, A.S.; Peng, R.; Gilroy, K.D.; Chi, M.; Liang, C.; Xia, Y. Fabrication of Sub-Micrometer-Thick Solid Electrolyte Membranes of β-Li3PS4 via Tiled Assembly of Nanoscale, Plate-Like Building Blocks. Adv. Energy Mater. 2018, 8, 1800014. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Li-Sites | Pnma | Pn21a | Experimental [2] | Theoretical [24] | Theoretical B3C1 [26] |
|---|---|---|---|---|---|
| 8d | (0.332, 0.035, 0.388) | - | (0.356, 0.013, 0.439) | (0.333, 0.033, 0.394) | - |
| 4b | (0.000, 0.000, 0.500) | - | (0.000, 0.000, 0.500) | (0.000, 0.000, 0.500) | - |
| 4a′ | - | (0.329, 0.043, 0.410) | - | - | (0.330, 0.041, 0.420) |
| 4a″ | - | (0.665, 0.972, 0.624) | - | - | (0.663, 0.973, 0.627) |
| 4a′′′ | - | (0.006, 0.045, 0.563) | - | - | (0.007, 0.053, 0.568) |
| d | |V|/G | |||||
|---|---|---|---|---|---|---|
| Li1-S30 | 2.502 | 0.0162 | 0.0847 | 0.8022 | 0.2156 | 0.0448 |
| Li2-S17 | 2.456 | 0.0180 | 0.0933 | 0.8177 | 0.1990 | 0.0169 |
| Li2-S28 | 2.448 | 0.0180 | 0.0911 | 0.8174 | 0.1945 | 0.0402 |
| Li2-S18 | 2.459 | 0.0171 | 0.0875 | 0.8075 | 0.2060 | 0.0403 |
| Li2-S29 | 2.483 | 0.0165 | 0.0862 | 0.8034 | 0.2140 | 0.0369 |
| Li9-S17 | 2.445 | 0.0183 | 0.0925 | 0.8218 | 0.1911 | 0.0202 |
| Li9-S21 | 2.526 | 0.0152 | 0.0761 | 0.8019 | 0.2066 | 0.0425 |
| Li9-S32 | 2.612 | 0.0115 | 0.0596 | 0.7508 | 0.2570 | 0.0239 |
| Li9-S28 | 2.618 | 0.0121 | 0.0615 | 0.7677 | 0.2382 | 0.1412 |
| Li9-S25 | 2.713 | 0.0099 | 0.0494 | 0.7549 | 0.2440 | 0.3477 |
| P16-S2 | 2.082 | 0.1369 | −0.1844 | 3.297 | −0.5960 | 0.0225 |
| P15-S22 | 2.065 | 0.1411 | −0.1942 | 3.286 | −0.6110 | 0.0136 |
| P13-S24 | 2.054 | 0.1433 | −0.2003 | 3.266 | −0.6210 | 0.0239 |
| P13-S29 | 2.050 | 0.1439 | −0.1976 | 3.2225 | −0.6240 | 0.0327 |
| Termination | Esurf | Eform | Egap | |
|---|---|---|---|---|
| (001) | LiS2/LiS2 | 2.15 | 31.94 | 1.82 |
| (100) | LiS2/LiS2 | 0.91 | 2.19 | 4.66 |
| (010) | LiS2/Li | 1.83 | 5.16 | 2.70 |
| (101) | PS3/SPLi | 8.20 | 54.10 | 0.46 |
| (011) | PS3/Li | 1.43 | 5.24 | 4.24 |
| (110) | SLi2/Li | 2.27 | 32.22 | 1.99 |
| (111) | LiS3/Li | 1.60 | 10.91 | 2.21 |
| (210) | SPLi/Li | 0.99 | 5.88 | 4.20 |
| (211) | LiS2/SPLi | 1.57 | 11.84 | 2.14 |
| Li1 | Li*1 | Li2 | Li*2 | P | P* | S1 | S*1 | S2 | S*2 | S3 | S*3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| bulk | 1.007 | 1.007 | 1.011 | 1.011 | 1.573 | 1.573 | −1.147 | −1.147 | −1.272 | −1.272 | −1.034 | −1.034 |
| (001) | 1.006 | 1.004 | 1.008 | 0.993 | 1.580 | 1.127 | −1.150 | −1.023 | −1.249 | −1.206 | −1.059 | −1.176 |
| (100) | 1.007 | 1.003 | 1.010 | 1.008 | 1.575 | 1.498 | −1.149 | −1.130 | −1.268 | −1.297 | −1.033 | −1.020 |
| (010) | 1.006 | 0.979 | 1.008 | 1.011 | 1.611 | 1.500 | −1.174 | −0.945 | −1.239 | −1.253 | −1.033 | −1.035 |
| (011) | 1.009 | 1.001 | 1.013 | 1.012 | 1.566 | 1.588 | −1.226 | −0.974 | −1.261 | −1.022 | −1.183 | −0.836 |
| (210) | 1.009 | 0.972 | 1.011 | 1.002 | 1.565 | 1.549 | −1.177 | −1.006 | −1.246 | −1.180 | −1.107 | −1.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marana, N.L.; Sgroi, M.F.; Maschio, L.; Ferrari, A.M.; D’Amore, M.; Casassa, S. Computational Characterization of β-Li3PS4 Solid Electrolyte: From Bulk and Surfaces to Nanocrystals. Nanomaterials 2022, 12, 2795. https://doi.org/10.3390/nano12162795
Marana NL, Sgroi MF, Maschio L, Ferrari AM, D’Amore M, Casassa S. Computational Characterization of β-Li3PS4 Solid Electrolyte: From Bulk and Surfaces to Nanocrystals. Nanomaterials. 2022; 12(16):2795. https://doi.org/10.3390/nano12162795
Chicago/Turabian StyleMarana, Naiara Leticia, Mauro Francesco Sgroi, Lorenzo Maschio, Anna Maria Ferrari, Maddalena D’Amore, and Silvia Casassa. 2022. "Computational Characterization of β-Li3PS4 Solid Electrolyte: From Bulk and Surfaces to Nanocrystals" Nanomaterials 12, no. 16: 2795. https://doi.org/10.3390/nano12162795
APA StyleMarana, N. L., Sgroi, M. F., Maschio, L., Ferrari, A. M., D’Amore, M., & Casassa, S. (2022). Computational Characterization of β-Li3PS4 Solid Electrolyte: From Bulk and Surfaces to Nanocrystals. Nanomaterials, 12(16), 2795. https://doi.org/10.3390/nano12162795