Abstract
Background: Hypermobile Ehlers–Danlos syndrome (hEDS) and hypermobility spectrum disorders (HSDs) are prevalent, complex conditions marked by chronic pain, joint instability, and multisystem involvement. Despite affecting an estimated 1 in 500 individuals, these conditions remain poorly understood and inconsistently diagnosed. This study aimed to define their clinical burden through a large-scale, international survey. Methods: A cross-sectional, anonymous survey was distributed globally from September 2023 to March 2024. Of 9258 responses, 3906 met inclusion criteria (hEDS: n = 3360; HSD: n = 546). The 418-item questionnaire assessed symptoms, comorbidities, healthcare utilization, and quality of life. The 95% confidence intervals (CIs) were calculated for comparison. Results: Participants with hEDS reported a mean of 24 comorbid conditions and an average diagnostic delay of 22.1 years. Common diagnoses included gastrointestinal disorders (84.3% [95% CI: 98.3–99.2%]), dysautonomia (71.4% [95% CI: 69.9–72.9%]), and chronic pain (98.9% [95% CI: 98.3–99.2%]). In contrast, HSD respondents reported a mean of 17 comorbidities, a 17.5-year time to diagnosis, and lower rates of key complications. Triggering events, such as puberty and infections were commonly reported preceding hEDS or HSD symptom onset. Comparison to the All of Us dataset revealed significantly elevated prevalence ratios of neurological, immune, and autonomic diagnoses. Conclusions: This global survey highlights the extensive multisystemic burden and diagnostic delays faced by individuals with hEDS and HSDs. The high prevalence of immune, neurological, gastrointestinal, and autonomic dysfunctions challenges the notion of these conditions as isolated connective tissue disorders. These findings highlight the need for updated diagnostic frameworks and mechanistic studies that explore multifactorial etiologies beyond the connective tissue paradigm.
1. Introduction
The Ehlers–Danlos syndromes (EDS) are a group of 14 heritable connective tissue disorders (CTDs) characterized by varying phenotypic presentation, often sharing commonalities of joint hypermobility, skin hyperextensibility, and ligament and tissue fragility [1,2]. This group of disorders is heterogeneous with most subtypes arising from mutations affecting collagen or other components of the extracellular matrix (ECM) [2,3]. While the majority of subtypes have clearly defined genetic markers, the most common subtype, hypermobile Ehlers–Danlos syndrome (hEDS), lacks a clear genetic or diagnostic test [2]. As such, hEDS is currently diagnosed clinically based on the 2017 criteria established by the International Consortium on the Ehlers–Danlos Syndromes and Related Disorders [2]. However, these criteria were developed through expert consensus rather than data-driven statistical validation and do not adequately capture the prevalence or clinical spectrum of comorbidities associated with hEDS and HSD. This has led to confusion among practitioners and inconsistency in diagnosis. Patients who exhibit symptomatic joint hypermobility but do not meet the full diagnostic criteria for hEDS are often labeled with hypermobility spectrum disorders (HSD), a classification that remains poorly defined and inconsistently applied [4]. Due to these diagnostic ambiguities, accurate country- or region-specific prevalence estimates are not possible to currently calculate. This underscores the need for more robust, data-driven definitions of these conditions. Although hEDS and HSD have historically been considered rare, recent regional studies challenge that assumption. For example, a national cohort study in Wales (UK) using linked primary care and hospital data reported a diagnosed prevalence of approximately 1 in 500 individuals [5], suggesting these conditions are more common than previously recognized and likely underdiagnosed in many populations.
Clinically, hEDS and HSD often present with overlapping features, including musculoskeletal complaints, extensive systemic involvement, and a range of comorbid conditions [5]. This may include neurological, immunological, cardiovascular, endocrine, genitourinary, dental, and gastrointestinal (GI) components [6,7,8,9]. Symptom onset typically occurs in early adolescence, with patients commonly reporting an increase in injuries, GI disturbances, and joint pain which are often mistaken for growing pains [10,11]. A triggering event, such as environmental, pubertal, or viral factors, is often reported near the onset of symptoms [12]. Patients may also experience overlapping comorbid conditions, including autonomic dysfunction, mast cell activation disease (MCAD) and cardiovascular manifestations such as mitral valve prolapse or aortic root dilatation [13]. Neurological complications like cerebrospinal fluid (CSF) leaks, cranio-cervical instability (CCI), tethered cord syndrome (TCS), and small-fiber neuropathy (SFN) are also reported [14]. Other structural manifestations include pelvic and rectal prolapse, hiatal hernias, Chiari malformation and spinal instabilities [7,8,15,16,17].
While these conditions are increasingly recognized in patients with hEDS and HSD, the full spectrum of associated manifestations remains poorly characterized. Many patients experience a lengthy journey to diagnosis due to the lack of a diagnostic test, barriers within the healthcare system, physician knowledge and attitudes, and the complexity of the clinical presentations [18]. This “diagnostic odyssey” can result in delays in care, reduced quality of life and worsening of symptoms over time without access to proper management [12,19].
Early perspectives helped shape the diagnostic criteria for hEDS and HSD, but these were developed in the absence of large-scale, population-based data and lacked statistical validation. Current clinical insights are primarily derived from small, single center cohorts that often exclude control groups and are limited in geographic representation, sample size, and phenotypic depth [2,20,21,22]. No prior studies have leveraged a large, multinational dataset to assess the multisystemic burden and diagnostic experience of individuals with hEDS and HSD across diverse healthcare systems and cultural contexts. To address this gap, we conducted a global, patient-centered survey to generate a comprehensive, statistically grounded view of these conditions. Our findings provide novel insights into symptom prevalence and comorbidity profiles with the goal of informing diagnostic criteria, advancing mechanistic understanding, and improving care strategies for this complex and underrecognized population.
2. Materials and Methods
2.1. Ethics Approval
The method of consent was reviewed and approved by the Institutional Review Board (IRB) at the Medical University of South Carolina, which determined the study to be exempt under category 2 (Consent ID: Pro00129825, dated 13 September 2023, with exemption status valid through 13 September 2028). Inclusion criteria were that all participants must be at least 18 years), have a clinical diagnosis of Ehlers–Danlos Syndrome or a hypermobility spectrum disorder, and have given informed consent by completing the survey. No minors were included in the study. As is common in online survey research, where inclusion criteria are used to guide participation, there was no exclusion criteria for survey participation. Instead, inclusion criteria are used to guide participation, and responses are later screened for eligibility. The start of recruitment was 14 September 2023, and recruitment ended 14 March 2024. This study complies with the Declaration of Helsinki in that this observational, non-interventional study posed no physical or psychological risk, and consent was provided through an IRB-approved consent process, appropriate for anonymous, exempt survey research [23]. No identifying information was collected, and no questions solicited protected health information (PHI). All responses were anonymous and reported in aggregate.
2.2. Survey Design
This study employed a cross-sectional survey design to assess patients with hEDS or HSD using self-reported data collected on a secure, web-based online research survey platform, REDCap (Research Electronic Data Capture) [24,25]. The survey was developed through an iterative, multidisciplinary collaboration with researchers, medical specialists who manage high volumes of hEDS/HSD patients, and individuals diagnosed with these conditions. Diagnostic sections of the survey were designed by specialists in relevant fields to ensure clinical relevance. The instrument was pilot tested with a cohort of 10 patients with hEDS or HSD, whose feedback informed refinements in clarity, flow, and accessibility. The final survey aimed to capture a comprehensive profile of symptoms, comorbidities, and diagnostic experiences across organ systems.
The survey was conducted anonymously and consisted of 418 items, covering 192 conditions, along with associated symptoms (Supplementary Figures S1 and S2). To reduce misclassification, diagnostic items were repeated using parallel language to distinguish confirmed diagnoses from suspected conditions. To be eligible to participate, respondents had to be at least 18 years old and report a formal diagnosis of EDS or HSD. All responses were later reviewed for eligibility in the final analysis. All survey fields were required, and only entirely completed surveys were analyzed, minimizing missing data. Participants were recruited online through social media, community organizations, and clinicians.
2.3. Statistical Analyses
Survey results (n = 9258) were directly imported from RedCap using the RedCapTidieR package in R [26]. As shown in Figure 1, we filtered for individuals who reported a diagnosis of hEDS or HSD, were at least 18 years old, and completed the full survey (n = 5653). Their self-reported hEDS and HSD diagnoses were validated using the first two criteria of the 2017 hEDS Diagnostic Criteria [2] through an R script. To mitigate bias in self-reported responses, diagnostic criteria questions were deliberately dispersed throughout the questionnaire to reduce the likelihood that participants would recognize and tailor their answers to the diagnostic framework. For Criterion 1, participants aged 18–50 with a Beighton score of ≥5, as well as those over 50 with a Beighton score of ≥4, were considered to have met the criterion. For Criterion 2, Features A, B, and C were assessed, with participants meeting at least two of these categories considered as satisfying the criterion. Criterion 3 was not applied due to the inability to verify physical examination findings. This process yielded two analysis groups: hEDS patients meeting the 2017 hEDS diagnostic criteria (hEDS group), and HSD patients who did not meet the hEDS criteria (HSD group). Those who reported hEDS but did not meet the criteria (n = 1183) and those reporting HSD who did meet hEDS criteria (n = 564) were excluded from analyses. No duplicates were identified. Due to the prevalence of certain conditions in the dataset, similar answer choices (e.g., “irritable bowel syndrome—Constipation” and “irritable bowel syndrome—Mixed”) were grouped as a single disorder for analysis unless otherwise specified. A subset of survey questions was selected for statistical analysis, with additional exclusions applied to items that had fewer than five responses, following the “Rule of 5” for normal approximation [27]. To compare hEDS and HSD groups, categorical variables were analyzed using Chi-square tests. Contingency tables were constructed, and Bonferroni correction was applied to adjust for multiple comparisons (n = 365), using this conservative approach to minimize false positives given the large number of tests and self-reported nature of the data. Risk ratios and 95% confidence intervals were calculated from contingency tables comparing the proportion of individuals with each diagnosis in the hEDS group to the HSD group. A corrected p-value of ≤0.05 was considered statistically significant.
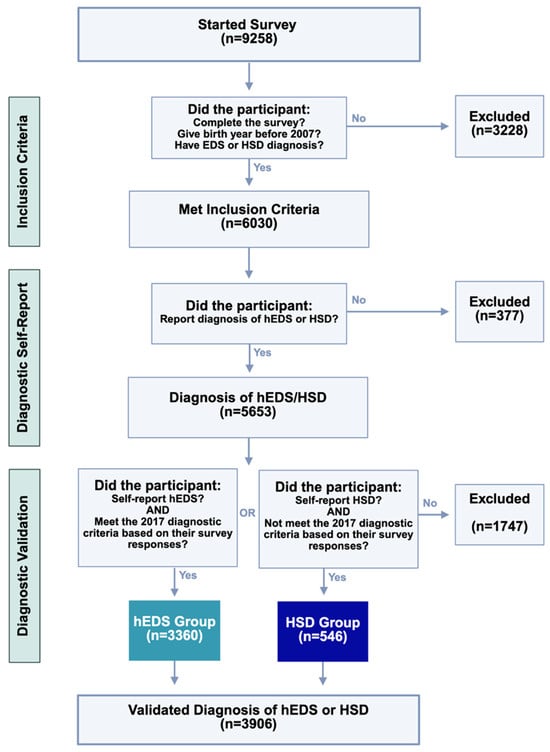
Figure 1.
Criteria and diagnostic validation steps for survey respondents. Of 9258 individuals who began the survey, participants were progressively excluded for age, incomplete data, or inconsistent diagnoses. Final groups (hEDS: n = 3360; HSD: n = 546) reflect participants who both self-reported and met criteria consistent with diagnostic classification.
To evaluate the prevalence of comorbidities in the hEDS group relative to the general population, aggregate-level data from the All of Us data browser (n = 354,400) were used. This study used data from the All of Us Research Program’s Controlled Tier Dataset 7, available to authorized users on the Researcher Workbench. Survey items related to formal medical diagnoses were included in this comparison if the hEDS group had ≥5 responses and the All of Us dataset had ≥40 responses. Chi-square analyses were performed with Bonferroni correction applied to 138 comparisons, using a significance threshold of p ≤ 0.05. Risk ratios and 95% confidence intervals were calculated from contingency tables comparing the proportion of individuals with each diagnosis in the hEDS group to the All of Us population. For continuous variables, outliers were identified using the ROUT method (Q = 1%) in GraphPad prior to conducting unpaired Welch’s t-tests on the cleaned dataset. A p-value of ≤0.05 was used to define statistical significance.
To visualize patterns of co-occurring conditions in hEDS patients, a co-morbidity matrix was constructed for conditions that were reported as formal diagnoses. Responses marked “None of the Above” were excluded. The matrix was normalized using the Jaccard Index, which quantified the degree of overlap between each pair of co-morbidities by dividing the number of patients with both conditions by the total number of patients with either condition. For clarity, only the most frequently overlapping conditions were included in the final heatmap by selecting conditions based on the sum of their Jaccard indices, ensuring legibility. An overlap threshold of 13.5 was selected to correspond with the 85th percentile of overlap scores. Self-overlapping conditions and redundant pairings were removed, resulting in a triangular matrix format. The heatmap was generated using the ComplexHeatmaps package [28,29].
3. Results
3.1. Study Cohort
3.1.1. Eligibility Criteria
A total of 9258 participants initially started the survey. Among these, 5653 participants reported a diagnosis of hEDS or HSD. These participant responses underwent a diagnostic validation process using the 2017 criteria. Ultimately, the final validated sample consisted of 3906 participants who had either a validated diagnosis of hEDS (hEDS group; n = 3360) or HSD (HSD group; n = 546). This process ensured that only individuals meeting the established diagnostic criteria were included in the final dataset.
3.1.2. Demographics
The survey sample population was primarily composed of younger and middle-aged adults, with the largest proportion falling between the ages of 26 and 44 (Figure 2A). Older adults (60+) represented only a small subset of the sample, potentially reflecting barriers to diagnosis or participation in online research among older populations.
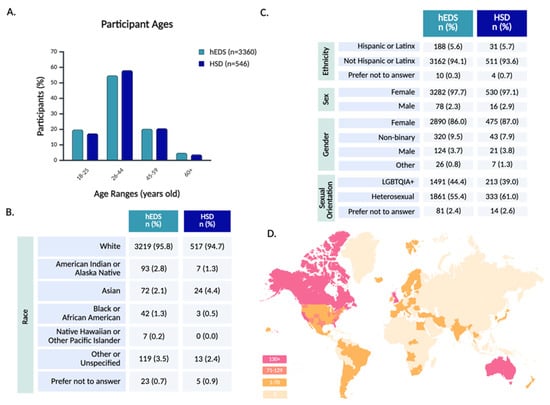
Figure 2.
Participant demographics and global representation. Participant demographics are shown across age (A), race (B), and identity-related variables including ethnicity, sex, gender, and sexual orientation (C). Panel (D) depicts the geographic distribution of participants by country and U.S. state, with darker shading indicating higher regional response counts.
Racial demographics indicated a predominantly White sample (Figure 2B), and ethnicity data indicated that a minority of participants identified as Hispanic or Latinx (Figure 2C). This relative lack of racial diversity mirrors broader disparities in EDS and HSD diagnosis, as studies suggest these conditions are underrecognized in non-White populations [30].
The majority of participants were assigned female at birth, aligning with prior research that highlights a diagnostic bias toward females [15,31]. In terms of gender, 86.0% of hEDS and 87.0% of HSD participants identified as female, with a higher rate of non-binary participants than male participants. While the majority of participants were cisgender females, gender diversity within the cohort was notable, with 12.0% of all respondents identifying with a gender different from their sex assigned at birth (Supplementary Table S1). Sexual orientation among respondents was highly diverse, with a large proportion identifying as part of the LGBTQIA+ spectrum (Figure 2C and Supplementary Table S2).
Geographically, 67.5% of all respondents resided in the United States, with representation from every state, including notable participation from California, South Carolina, Florida, and Texas (Figure 2D and Supplementary Table S3). Globally, the survey reached participants on every continent, besides Antarctica, with strong response rates from the United Kingdom, Australia, Canada, New Zealand, and the Netherlands (Supplementary Table S4). This worldwide engagement highlights the survey’s reach across diverse geographic and healthcare landscapes.
3.2. Diagnostic Journey
On average, participants with hEDS reported their symptom onset at the age of 9.3 but were diagnosed at the age of 31.4, with a delay in diagnosis from time of symptom onset of 22.1 years (Figure 3). Participants with HSD reported significantly later symptom onset but were diagnosed at the same age as hEDS participants, leading to a shorter, though still lengthy, delay from symptom onset to diagnosis. Both hEDS and HSD groups indicated their initial symptoms were musculoskeletal, gastrointestinal, or autonomic (Supplementary Table S5). The first suspicion of hEDS/HSD came most often from medical doctors, followed by physical therapists, and themselves or a friend/family member. Participants with hEDS were significantly more likely to be diagnosed by geneticists than patients with HSD. Rheumatologists were the most common diagnosticians for HSD, and the second most common for hEDS. Prior to diagnosis, more than half of participants report being misdiagnosed with conditions including fibromyalgia, anxiety, and growing pains.
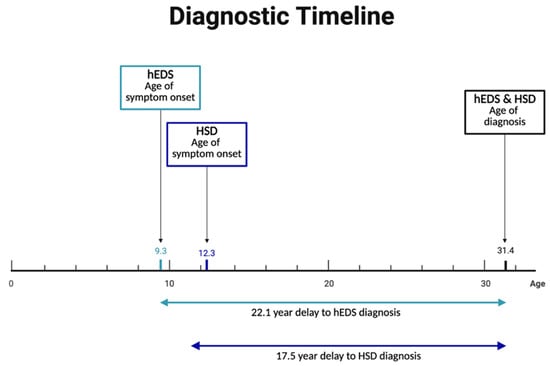
Figure 3.
Prolonged diagnostic delays in hEDS and HSD. Despite early symptom onset—9.3 years for hEDS and 12.3 years for HSD—diagnoses were typically not made until age 31.4, resulting in average diagnostic delays of 22.1 and 17.5 years, respectively.
Triggering events were common in this population of patients. 70.1% of individuals with hEDS and 65.0% of those with HSD identified a specific event preceding or heightening their symptoms (Table 1). Puberty and pathogens (viral or bacterial) were the most commonly reported triggers.

Table 1.
Symptom-onset triggers and COVID-related responses in hEDS and HSD.
Participants reported a high burden of co-occurring conditions, with hEDS participants averaging 24 and HSD participants 17, based on 192 conditions assessed in the survey (Figure 4A and Supplementary Figure S2). Participants reported experiencing a range of symptoms related to dysautonomia, dermatological issues, and gastrointestinal concerns (Figure 4B). When asked to rank the three most severe symptom categories from a provided list, responses were weighted by rank to reflect overall severity. Chronic pain was most frequently ranked as the most severe symptom by both groups (Table 2, Supplementary Table S6). However, notable differences emerged between groups: hEDS participants more often prioritized gastrointestinal symptoms, while HSD participants more frequently ranked joint-related symptoms as most severe.
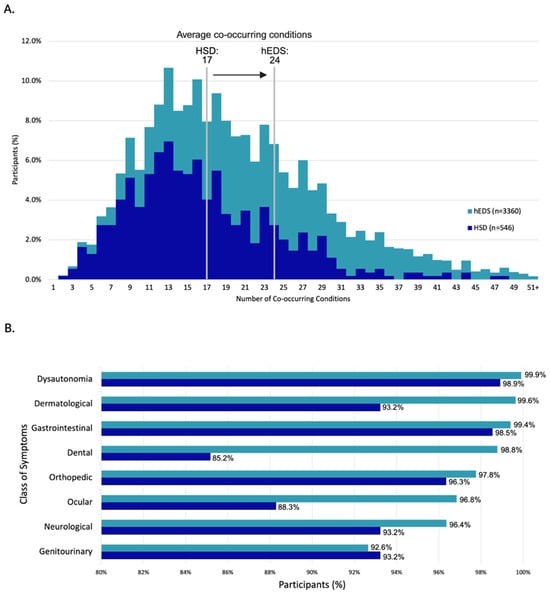
Figure 4.
Burden and distribution of co-occurring conditions in survey participants. (A) Distribution of co-occurring conditions per participant, with group averages of 24 (hEDS) and 17 (HSD). (B) Percentage of participants reporting symptoms across eight major organ system categories.
Table 2.
Most severe symptoms reported by hEDS and HSD participants.
To evaluate the impact of hEDS and HSD on daily functioning and productivity, we assessed the time patients spent managing their healthcare. A substantial proportion of individuals reported spending five or more hours per week, on average, coordinating and receiving medical care (Table 3). Participants indicated that they see multiple medical specialists annually, with hEDS participants averaging a higher number than HSD participants in the past year. The most frequently visited specialties included family medicine, cardiology, gastroenterology, neurology/neurosurgery, and emergency medicine.
Table 3.
Medical care access and burden in hEDS and HSD participants.
Inpatient hospitalizations were more common in hEDS participants compared to HSD participants. More hEDS participants reported being admitted to the hospital at least once in the last year compared to HSD participants. When asked the furthest distance participants had traveled to receive medical care pertaining to hEDS/HSD, 31.3% of hEDS and 9.5% of HSD participants report having traveled 300 or more miles, with a subset of participants that have traveled more than 2000 miles. Regarding the financial burden of these diseases, many participants report paying out-of-pocket for medical care from a specialist not covered by insurance.
3.3. Multisystemic Clinical Presentation
3.3.1. Pain
Chronic pain is a key feature of symptomatic joint hypermobility, reported in almost all hEDS and HSD participants (Table 4). The most frequently reported pain sites were the neck, lower back, and shoulder(s) in both groups.
Table 4.
Prevalence of chronic pain and pain locations in hEDS and HSD.
Given the pain burden in these populations, pain management is a common concern for patients. Responses of participants with hEDS were analyzed regarding which medications they have tried for their pain, and of those, which medications participants considered effective (Figure 5, Supplementary Table S7). hEDS participants report medical marijuana and ketamine as the most effective for pain. Recently, selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) have been increasingly used for pain, but only small proportion of hEDS participants that have tried SS/SNRIs for pain considered them effective. With limited treatment options available, participants report turning to non-traditional practices for their treatment, including massage, meditation, and acupuncture (Table 5). A significant proportion of participants also experienced complications related to local and/or general anesthesia (Table 6). The hEDS group was over two times likelier to experience shortened effect, insufficient pain control, intubation complication, and anaphylaxis.
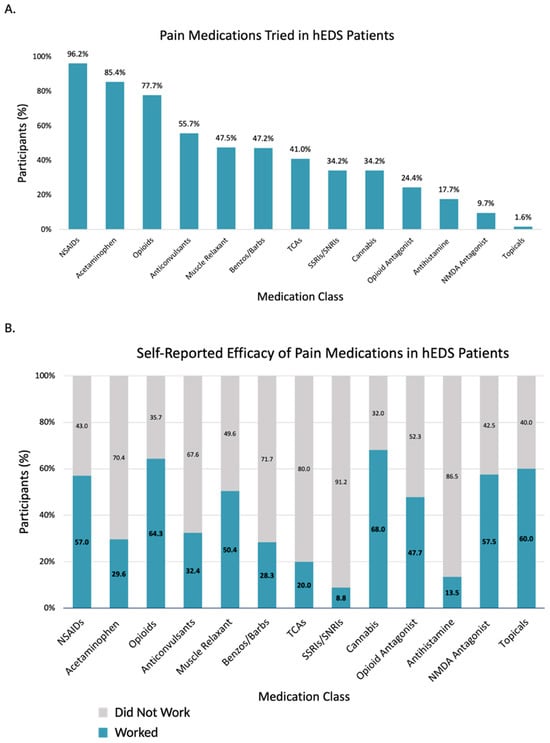
Figure 5.
Pain medication use and perceived efficacy in hEDS participants (n = 3360). (A) Percentage of hEDS participants reporting use of each medication class. (B) Self-reported effectiveness of each medication class, shown as proportion who reported it worked.
Table 5.
Alternative medicine modalities tried by hEDS and HSD participants.
Table 6.
Anesthesia-related complications and reactions in hEDS and HSD.
3.3.2. Orthopedic
Musculoskeletal complications were highly prevalent in both hEDS and HSD. Slipping rib syndrome was more prevalent in hEDS participants compared to HSD participants and the All of Us dataset (Table 7). Additionally, participants with hEDS were over 40 times more likely to report tendon rupture than participants in the All of Us dataset. Of note, hEDS participants are at a greater risk of unexplained fractures than HSD participants (Table 8). Correspondingly, a much smaller proportion of hEDS participants reported having none of these joint issues. The shoulders, hips, and knees were among the most frequently affected joints. Additionally, several anatomical abnormalities were reported in the hEDS group, including missing or extra ribs, sacralization of the L5 vertebrae, and missing or extra vertebrae (Table 9).
Table 7.
Prevalence of orthopedic diagnoses in hEDS, HSD, and All of Us.
Table 8.
Orthopedic symptoms and joint sites impacted in hEDS and HSD.
Table 9.
Reported anatomical abnormalities in hEDS participants.
3.3.3. Neurological
Migraine was the most frequently reported neurological diagnosis among both groups, while affecting significantly more hEDS participants than HSD participants (Table 10). hEDS participants also reported significantly higher rates of scoliosis, Raynaud’s phenomenon, upper cervical instability, and occipital neuralgia, when compared to both HSD and All of Us datasets. Other neurological abnormalities were more common in hEDS than HSD, particularly tinnitus, herniated disk(s), spinal stenosis, small-fiber neuropathy, Chiari malformation, kyphoscoliosis, and tethered cord syndrome. There was no significant difference in the prevalence of voice disorders between the three groups, while the hEDS group was at slightly lower risk of hearing impairment than the general population. Neurological symptoms were highly prevalent among survey respondents, with differences observed between the hEDS and HSD groups. Weakness was the most reported symptom, followed closely by muscle spasms (Table 11).
Table 10.
Prevalence of neurological diagnoses in hEDS, HSD, and All of Us.
Table 11.
Prevalence of neurological symptoms in hEDS and HSD.
3.3.4. Autonomic
Autonomic dysfunction was commonly reported among participants, with 71.4% of hEDS and 40.3% of HSD participants reporting at least one diagnosed autonomic disorder, with a higher prevalence of each type in the hEDS group (Table 12). Postural orthostatic tachycardia syndrome (POTS) was the most frequently diagnosed autonomic disorder, affecting hEDS participants at a higher rate than those with HSD. This analysis revealed an extraordinarily elevated risk of POTS in the hEDS group compared to the general population. Symptoms of dysautonomia were highly prevalent, with fatigue, dizziness, and brain fog being the most highly reported (Table 13). Heart palpitations and thermoregulatory dysfunction were also frequent, suggesting significant autonomic involvement in both groups. Despite these high symptom burdens, many hEDS participants and the majority of HSD participants reported having no formal autonomic diagnosis.
Table 12.
Prevalence of autonomic disorders in hEDS, HSD, and All of Us.
Table 13.
Prevalence of autonomic symptoms in hEDS and HSD.
3.3.5. Gastrointestinal
Gastrointestinal (GI) symptoms were common in both groups, with a higher prevalence of at least one GI disorder in the hEDS group compared to HSD (Table 14). Irritable bowel syndrome (IBS) was the most reported GI diagnosis. Among IBS subtypes, the mixed variant was most prevalent. The hEDS group had approximately 1.5 times higher risk of gastroesophageal reflux disease (GERD) compared to both the HSD and All of Us groups. Dysmotility was also more frequently diagnosed in hEDS than HSD, with the gastric type (“gastroparesis”) reported the most frequently in both groups. hEDS participants reported a significantly higher prevalence of 13 of 19 GI disorders that were compared to the All of Us dataset, while obesity, abdominal hernia, cholelithiasis, and liver disease were all significantly higher in the general population than hEDS. Notably, colorectal polyps and ulcerative colitis were not significantly different between the hEDS and All of Us groups.
Table 14.
Prevalence of gastrointestinal diagnoses in hEDS, HSD, and All of Us.
The most frequently reported symptoms were abdominal pain, nausea, bloating, and constipation (Table 15). Severe GI dysfunction requiring feeding devices was significantly more frequent in hEDS than in HSD, with nasogastric, nasojejunal, and total parenteral nutrition being the most frequently utilized options by hEDS participants (Table 16). Similarly, colostomies and ileostomies, while rare overall, were present in both groups.
Table 15.
Prevalence of gastrointestinal symptoms in hEDS and HSD.
Table 16.
Use of feeding devices and ostomies in hEDS and HSD.
3.3.6. Cardiopulmonary
The majority of respondents in both groups reported no diagnosed cardiovascular conditions (Table 17). Of those diagnosed with a cardiac condition, mitral valve defect was the most frequently reported in hEDS, while the most frequently reported issue reported by HSD participants were arrhythmias (other than supraventricular tachycardia and atrial fibrillation). Other structural heart abnormalities, including tricuspid valve defects and aortic valve, were present, but less frequent in both groups. Compared to the All of Us dataset, hEDS had significantly higher prevalences of mitral valve defects, stroke, and tricuspid valve defects. Contrastingly, lung disease and atrial fibrillation had a significantly lower prevalence in hEDS when compared to All of Us data. Many of these cardiopulmonary diseases showed an age-effect of diagnoses, with peak diagnoses in the 26–44-year-old age range (Supplementary Table S8).
Table 17.
Prevalence of cardiopulmonary diagnoses in hEDS, HSD, and All of Us.
3.3.7. Endocrine
Endocrine disorders were reported in a subset of participants, with 26.0% of hEDS and 20.3% of HSD participants reporting one or more endocrine disorders (Table 18). Non-autoimmune hyperthyroidism occurred at over 11 times the rate in hEDS participants compared to the general population. The risk of central adrenal insufficiency was nearly fivefold higher in hEDS. Pineal cysts were over 160 times more likely to be reported in hEDS than in the general population. hEDS participants reported lower rates than the All of Us group for osteoporosis, type 2 diabetes, and hyperparathyroidism.
Table 18.
Prevalence of endocrinological diagnoses in hEDS, HSD, and All of Us.
3.3.8. Hematologic
Most participants reported no formal hematologic diagnoses (Table 19). hEDS participants were at almost 14 times higher risk for pernicious anemia than the general population. The risk of deep vein thrombosis (DVT) was over threefold higher in the hEDS group compared to the HSD group. Easy or severe bruising was reported by the majority of hEDS and HSD participants.
Table 19.
Prevalence of hematological diagnoses and bruising in hEDS, HSD, and All of Us.
3.3.9. Reproductive
Reproductive health conditions were frequently reported by hEDS participants. The risk of pelvic organ prolapse was nearly four times higher in hEDS compared to HSD (Table 20). Endometriosis was also more common in hEDS than in HSD. Compared to the general population, hEDS participants were at markedly increased risk for multiple reproductive disorders, including polycystic ovary syndrome, vaginismus, and pelvic congestion syndrome. Erectile dysfunction was the most frequently reported reproductive condition in participants assigned male at birth. The most frequently reported reproductive symptoms included pelvic pain, irregular periods, pain during sex, and bleeding during sex (Table 21). Among those assigned female at birth, participants experienced their first menstrual period at ages 12.3 (hEDS) and 12.4 (HSD) on average. Of participants who have been pregnant, the majority from both groups reported pregnancy complications, although hEDS participants reported a significantly higher prevalence. Pregnancy complications included spontaneous abortion, preterm labor, failure to progress, premature rupture of membranes, and stillbirth.
Table 20.
Prevalence of reproductive health conditions in hEDS, HSD, and All of Us.
Table 21.
Reproductive symptoms, menstruation, and pregnancy in hEDS and HSD.
3.3.10. Urological
Urinary disorders were significantly more prevalent in the hEDS group compared to HSD (Table 22). Recurrent urinary tract infections (UTIs) were 1.63 times more common in hEDS participants than in HSD participants. Voiding dysfunction occurred at nearly twice the rate in hEDS compared to HSD, and overactive bladder syndrome showed a similar increase in risk. Relative to the general population, these conditions were markedly more frequent, with recurrent UTIs over 130 times more common and voiding dysfunction nearly 880 times more common in the hEDS group.
Table 22.
Prevalence of urinary disorders in hEDS, HSD, and All of Us.
3.3.11. Dermatological
The prevalence of having at least one or more dermatological condition was similar between hEDS and HSD (Table 23). Among them, atopic dermatitis was the most frequently reported, followed by hyperhidrosis. While there were no statistical differences between diagnoses in the hEDS and HSD groups, the hEDS group had significantly higher risk of atopic dermatitis when compared to the All of Us group. Skin-related symptoms were highly prevalent among respondents, especially the hEDS group, which may reflect the inclusion of these symptoms in the 2017 hEDS diagnostic criteria (Table 24). Soft, velvety skin, abnormally stretchy skin, and unexplained stretch marks were all more highly prevalent in hEDS compared to HSD.
Table 23.
Prevalence of dermatological disorders in hEDS, HSD, and All of Us.
Table 24.
Prevalence of dermatological symptoms in hEDS and HSD.
3.3.12. Allergic and Immunologic
Allergic and immunologic conditions were frequently reported in both groups, with 89.7% of hEDS and 78.0% of HSD participants reporting at least one allergy-related disorder (Table 25). Allergies were present at a higher rate in hEDS participants than HSD participants. Compared to HSD, hEDS participants had increased risk of mast cell activation syndrome, chronic urticaria, and allergies. Relative to the general population, these risks were markedly elevated, with chronic urticaria being over 230 times more common and MCAS over 1000 times more common in hEDS. Additionally, anaphylaxis episodes were reported significantly more frequently in hEDS compared to HSD. In total, 29.9% of hEDS participants and 22.0% of those with HSDs reported at least one of the autoimmune conditions assessed in this study. Reported autoimmune conditions with comparable prevalence between groups included psoriasis, rheumatoid arthritis, and systemic lupus erythematosus.
Table 25.
Prevalence of allergy and immune-related diagnoses in hEDS, HSD, and All of Us.
3.3.13. Ocular
Ophthalmologic disorders were common among both hEDS and HSD groups, with 82.1% of hEDS and 72.7% of HSD participants reporting one or more ocular disorders (Table 26). hEDS had notably higher prevalences, compared to All of Us data, of astigmatism, myopia, hyperopia, macular degeneration, and keratoconus, but a similar prevalence of retinal detachment. Ophthalmologic symptoms were also common, including light sensitivity, visual disturbances, dry eyes, and double vision (Table 27).
Table 26.
Prevalence of ocular diagnoses in hEDS, HSD, and All of Us.
Table 27.
Prevalence of ocular symptoms in hEDS and HSD.
3.3.14. Dental
Dental issues were reported in a majority of hEDS participants and many HSD participants (Table 28). Dental disorders were reported at high rates, including temporomandibular joint (TMJ) disorder, enamel defects, and early onset periodontitis. All dental disorders included in this survey were significantly more frequent in the hEDS group than the All of Us group. Symptoms reported included jaw pain, as well as subluxation and/or dislocation of the temporomandibular joint (TMJ) (Table 29).
Table 28.
Prevalence of dental disorders in hEDS, HSD, and All of Us.
Table 29.
Dental symptoms in hEDS and HSD.
3.3.15. Mental Health and Sleep
Within these populations, a high prevalence of anxiety, depression, and post-traumatic stress disorder (PTSD) were reported (Table 30). Sleep disturbances were also widely reported, including insomnia, restless legs syndrome, and obstructive sleep apnea. Each mental health and sleep-related disorder included in this survey was reported at a significantly higher rate in hEDS compared to All of Us, except obstructive sleep apnea and substance use disorder, which were significantly lower in hEDS, with bipolar disorder showing no significant difference.
Table 30.
Mental health and sleep disorders in hEDS, HSD, and All of Us.
3.3.16. Neurodivergence
Neurodivergent diagnoses were common among respondents, with 49.1% of hEDS and 39.7% of HSD participants reporting at least one neurodiversity-related disorder (Table 31). The most frequently reported diagnoses included attention deficit hyperactivity disorder (ADHD/ADD), autism spectrum disorder (ASD), and obsessive–compulsive disorder (OCD). When hEDS and All of Us data was compared, ADHD, ASD, OCD, and learning disorder were present at higher rates in hEDS than in the general population.
Table 31.
Neurodiversity-related diagnoses in hEDS, HSD, and All of Us.
3.3.17. Multimorbidity in hEDS
To examine the multimorbidity of co-occurring disorders, a matrix was created to illustrate the percentage of participants who reported both conditions among those who reported at least one (Figure 6). A clear pattern emerged among condition pairs with a Jaccard index of 28.0% or greater, revealing frequent overlap between allergies, migraine, anxiety, depression, POTS, GERD, constipation, tendonitis, TMJ disorder, insomnia, PTSD, bursitis, and asthma. Notably, the strongest co-occurrences were observed across psychiatric and immunologic domains, with anxiety–depression (43.3%), anxiety–allergies (39.3%), and depression–allergies (37.2%) ranking highest. Allergies and migraine also demonstrated a high rate of co-occurrence (37.0%).
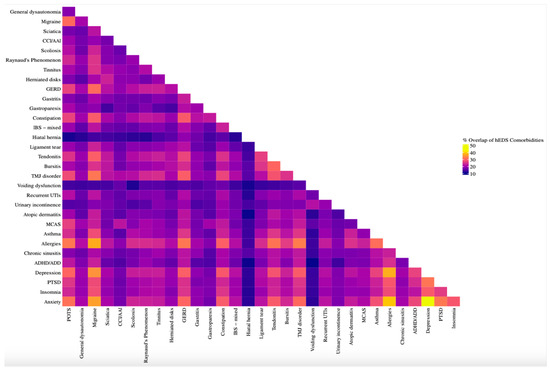
Figure 6.
Co-occurrence matrix of comorbidities in hEDS participants. Heatmap showing pairwise overlap of formal diagnoses in hEDS participants (n = 3360). Percent overlap was calculated using the Jaccard Index, defined as the proportion of participants who reported both conditions among those who reported either. Conditions with higher overlap included anxiety, allergies, depression, insomnia, GERD, and POTS, highlighting multimorbidity clusters in autonomic, allergic, and psychiatric domains.
4. Discussion
This study represents the largest global dataset to date characterizing the clinical and systemic burdens of hEDS and HSD. These findings reveal the critical need to appropriately address gaps in the continuum from diagnosis to treatment, as well as uncovering the underlying biological mechanisms of hEDS, HSD, and their associated comorbidities. Patient responses indicate challenges beginning with the diagnostic process, where significant delays, averaging over 20 years, and misdiagnoses are common due to limited awareness among healthcare providers and complex overlap between these multisystemic conditions. The overwhelming healthcare burden associated with hEDS and HSD has been previously reported [32] and is evident in the extensive time and financial resources patients devote to managing their conditions. Many participants in our survey reported spending five or more hours per week coordinating care, seeing an average of six specialists annually, and paying significant out-of-pocket costs for necessary treatments.
4.1. Multisystemic Manifestations
Our data support growing evidence of the substantial multisystemic involvement in hEDS and HSD, with early and severe manifestations including chronic pain, gastrointestinal symptoms, autonomic dysfunction, as well as joint instability. Autonomic dysfunction emerged as one of the most burdensome aspects of hEDS and, to a lesser extent HSD, with fatigue, dizziness, and orthostatic intolerance affecting the vast majority of participants. A significant proportion of participants also reported muscle weakness, numbness, tingling, and migraines, aligning with prior research linking hypermobility to small-fiber neuropathy (SFN) and dysautonomia [33,34,35]. Despite their inclusion in the 2017 hEDS diagnostic criteria, the prevalence of structural cardiac abnormalities, like mitral valve prolapse and aortic root dilatation, were relatively low in our dataset. This aligns with literature suggesting that autonomic and vascular dysfunction, rather than structural cardiac abnormalities, may be more clinically relevant for patients with hEDS and HSD [36].
Symptoms often attributed solely to hypermobility-related musculoskeletal issues appear to stem from coexisting conditions rather than hEDS itself. Autonomic dysfunction, MCAD, including MCAS, gastrointestinal dysmotility, and neurological complications can often drive symptom exacerbations, making it essential for healthcare providers to assess the broader clinical picture. GI symptoms were also reported at high rates in both hEDS and HSD groups, with gastroesophageal reflux disease, irritable bowel syndrome, and gastroparesis being particularly prevalent. These trends are consistent with prior research linking hypermobility to gut motility issues and visceral hypersensitivity, including studies demonstrating a higher prevalence of functional gastrointestinal disorders in individuals with hEDS and HSD [37,38].
Chronic pain was nearly universal among participants, with an average of six painful joints per participant. In addition to joint-related pain, participants frequently reported abdominal, neuropathic, headaches, and widespread pain. These findings align with prior literature on the biomechanical vulnerabilities in hypermobility-related disorders and emerging theories on pain sensitization in this population [39]. Despite this pain burden, conventional pharmacologic treatments remain largely ineffective, with patients frequently reporting poor responses to NSAIDs, antidepressants, and anticonvulsants. This may reflect clinical reports suggesting altered drug metabolism or receptor sensitivity in hypermobile individuals [40], underscoring the need for further investigation into pharmacogenetics, absorption differences, and alternative pain management strategies. The high prevalence of sleep disturbances further amplifies pain, fatigue, and cognitive function, highlighting the importance of targeted interventions to improve sleep.
A significant proportion of participants also reported immune-related conditions, including mast cell activation syndrome (MCAS), chronic urticaria, and heightened allergy-like responses. These findings align with prior research suggesting a potential immunological component in hypermobility disorders [40]. The frequent co-occurrence of immune dysregulation, neurological, gastrointestinal, and autonomic symptoms further underscore a complex and interconnected pathophysiology that remains poorly understood.
4.2. hEDS, HSD, and Rare Diseases
While some findings align with previous reports, the data also reveals a range of conditions beyond common comorbidities and symptoms, highlighting the overlap between hEDS, HSD, and some rare diseases. Participants in this study exhibited a disproportionately high prevalence of conditions such as Chiari malformation, cerebrospinal fluid leaks, tethered cord syndrome, adrenal insufficiency, gastroparesis, intracranial hypotension, and trigeminal neuralgia compared to the All of Us datasets. It is critical that healthcare providers consider the possibility of coexisting rare conditions in clinical settings. Relying solely on hEDS or HSD as the cause can lead to misdiagnosis and inadequate treatment, worsening patient distress and healthcare disparities.
4.3. Challenges of the Current Diagnostic Criteria
A central finding of this study is the significant overlap in symptoms and comorbidities between hEDS and HSD. The distinction between these conditions has been historically ambiguous, leading to diagnostic uncertainty. Strikingly, half of survey respondents with a reported HSD diagnosis met the diagnostic criteria for hEDS, while a quarter of those with an hEDS diagnosis did not meet the criteria. These findings highlight the urgent need for more precise and reliable diagnostic tools if these conditions are to be meaningfully differentiated in clinical practice. Follow-up analyses to further explore this diagnostic discordance are warranted to compare individuals who did or did not meet formal criteria across hEDS or HSD groups. One of the most apparent distinctions between hEDS and HSD is the degree of joint hypermobility measured by Beighton score. Participants with hEDS reported significantly higher hypermobility scores, along with high rates of joint dislocations, subluxations, and orthopedic. However, the Beighton score has recognized limitations, including its narrow focus on a limited number of joints, age and sex variability and inconsistent application by clinicians [41]. Notably, the three most frequently affected joints reported by hEDS participants—shoulders and hips—are not included in the Beighton score. These limitations may contribute to misclassification or underdiagnosis, particularly in individuals with systemic features but lower Beighton scores.
While these findings likely reflect the thresholds set by the 2017 diagnostic criteria for hEDS, they should not be interpreted to mean that HSD is less clinically significant. Symptom severity was not deeply assessed, and many respondents with HSD reported substantial symptom burden and multisystem involvement. Many comorbidities such as autonomic dysfunction, mast cell activation syndrome (MCAS), gastrointestinal dysmotility, and neurological complications were common in both groups, though more frequently reported in hEDS participants. The high symptom burden in both groups compared to the general population make differential diagnosis challenging. Future studies should focus on refining diagnostic criteria to ensure that individuals with disabling symptoms receive appropriate medical attention, regardless of whether they meet strict diagnostic thresholds. Expanding research into the underlying biological mechanisms of these conditions may also help clarify whether hEDS and HSD are truly separate conditions or variations within the same disorder spectrum.
4.4. Triggering Events
Over two-thirds of participants reported an event preceding symptom onset or worsening symptom severity, such as puberty, viral infections, physical trauma, or pregnancy. These findings reinforce the hypothesis that environmental factors, in combination with genetic predisposition, may play a role in the onset of hEDS and HSD and/or disease progression. The association between viral infections and worsening symptoms further supports a potential link between hEDS, immune dysregulation, and even post-viral syndromes such as long COVID and ME/CFS. The overlap of dysautonomia, MCAS, and chronic fatigue within these conditions suggest shared mechanisms, underscoring the need for further investigation to determine whether hypermobility-related disorders have a latent component that can be exacerbated by physiological stressors.
4.5. Limitations and Future Directions
While this study offers valuable insights, certain limitations are acknowledged. Despite extensive international outreach, the study population was predominantly composed of white, female, and digitally literate individuals. This may reflect a combination of systemic underdiagnosis in non-White populations, gender biases in diagnostic criteria and clinical recognition, and barriers to digital access or research participation in certain regions. As such, the current dataset may underrepresent individuals with limited access to specialized care, lower health literacy, or those from historically marginalized or underserved communities. These demographic imbalances introduce the possibility of selection bias and may limit the generalizability of findings across all racial, socioeconomic, and geographic populations. The reliance on self-reported data may introduce biases such as recall error and diagnostic uncertainty. In addition, self-selection bias may have influenced the study population, favoring participants with greater disease severity, health literacy, or digital access. Additionally, in some cases, missing or incomplete responses were received. Responses with incomplete data were excluded from relevant analyses, and no imputation methods were applied. The survey instrument, while developed iteratively and piloted in a patient cohort, has not undergone formal psychometric validation, which may limit the reliability of certain self-reported outcomes. This is consistent in conditions that are complex and under-characterized such as hEDS and HSD, where few validated instruments exist. In such contexts’, self-reported data are often the only practical means of capturing real-world, patient-centered experiences at scale. This approach is well-established in epidemiology, health services research, and rare disease registries, where it remains a critical tool for advancing understanding and improving care. Use of the 2017 criteria to filter participants may have excluded individuals with hEDS who do not meet current guidelines, and access to specialized care likely skewed responses to those with a formal diagnosis. The anonymous nature of the survey also prevented verification of unique responses. International differences in healthcare access and diagnostic practices may have also influenced how participants understood or reported their diagnoses. Furthermore, access to specialized testing (e.g., upright MRI for cranio-cervical instability) may vary significantly, potentially leading to underdiagnosis of certain conditions. Comparisons with the All of Us dataset should be interpreted with consideration, as demographic differences, particularly in age, sex, and racial composition, may influence observed patterns. While the findings provide important insights into symptom burden and comorbidity patterns, the cross-sectional design and lack of clinical verification preclude any causal inferences between reported symptoms and comorbidities. These limitations highlight the urgent need for large-scale, clinician-verified studies to corroborate these findings and refine diagnostic criteria.
Despite these limitations, the findings of this study highlight the profound, multisystemic, and lifelong impact of hEDS and HSD, encompassing chronic pain, autonomic dysfunction, immune and gastrointestinal involvement, and a wide spectrum of additional comorbidities. Yet, effective treatments remain elusive, leaving many patients to seek non-traditional and unproven therapies.
A major challenge in developing standardized care protocols is the lack of research that clearly defines and distinguishes hEDS from HSD, particularly regarding comorbidities not included in the 2017 diagnostic criteria. While existing studies have reported comorbid conditions in hEDS/HSD, most have been limited in scope, lump the two conditions together, or lacked control populations. The findings of this work define overlapping features and distinctions between hEDS and HSD, and their prevalence compared to the general population, providing a more complete understanding of these conditions. Distinguishing between them remains a challenge, as does understanding whether they exist on a continuous spectrum or if HSD can progress to hEDS over time or with exposure to additional triggers.
4.6. Reconsidering Etiology
While hEDS is currently classified as a heritable connective tissue disorder, findings from this study raise the hypothesis that connective tissue symptoms may develop in parallel with, or be influenced by, other systemic physiological processes. The observed high prevalence of immune dysregulation, autonomic dysfunction, and gastrointestinal dysmotility supports the possibility that neuroimmune or inflammatory pathways could contribute to the broader clinical phenotype. Although speculative and beyond the causal scope of this cross-sectional survey, these findings suggest that hEDS may, in some cases, reflect overlapping features with autoimmune or autoinflammatory conditions rather than representing a purely connective tissue–based pathophysiology. This hypothesis warrants further investigation through mechanistic and longitudinal studies. As researchers, clinicians and patient communities advocate for updated diagnostic frameworks, expanded research efforts, and improved treatment pathways, it is imperative that patient experiences remain at the forefront of these advancements. Given the significant multimorbidity associated with hEDS and HSD, future research, particularly incorporating biomarker studies and genetic approaches, may help refine classifications and improve our ability to diagnose and treat these conditions.
5. Conclusions
This global survey provides the most comprehensive epidemiological profile to date of individuals with hypermobile Ehlers–Danlos syndrome (hEDS) and hypermobility spectrum disorders (HSDs), revealing a strikingly high burden of multisystemic comorbidities, profound diagnostic delays, and significant unmet clinical needs. Across more than 3900 participants, the study identifies disproportionately high rates of gastrointestinal, autonomic, neurological, immune-mediated, and musculoskeletal complications, often with symptom onset in childhood, disease exacerbation following triggers, and formal diagnosis delayed by over two decades. Compared to both HSD and general population data, individuals with hEDS exhibited elevated risk for manifestations such as postural orthostatic tachycardia syndrome, small-fiber neuropathy, gastrointestinal dysmotility, immune-/mast cell-related conditions, and structural joint and spine abnormalities.
While these findings challenge the traditional view of hEDS and HSDs as isolated connective tissue disorders, we recognize that the study’s cross-sectional and self-reported design precludes causal inference. This study was not intended to establish new diagnostic criteria, but rather to generate a large, global dataset that can serve as a foundational resource for hypothesis generation and future studies. These results are meant to inform and complement emerging research in relation to genetic, proteomic, and biomarker signatures, which will be essential for developing validated, data-driven diagnostic frameworks. By illuminating the scope and heterogeneity of hEDS and HSD, this study highlights critical areas for future mechanistic investigation and reinforces the need for multidisciplinary, personalized approaches to care.
This study serves as a critical call-to-action for the medical community. The staggering delays in diagnosis, the overwhelming reliance on self-advocacy, and the systemic lack of treatment options illustrate a glaring gap in healthcare. The lack of standardized care protocols often leads to misdiagnosis, symptom neglect, and, at times, outright denial of these conditions by healthcare providers. Addressing these issues will require collaboration across specialties, heightened awareness among healthcare providers, and a sustained commitment to addressing the full spectrum of challenges faced by this patient population. Recognizing the full impact of hEDS and HSDs is key to driving meaningful change, breaking down barriers to care, advancing research and ensuring that individuals and families impacted by these conditions receive the medical care and support they deserve.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/jcm14165636/s1, Figure S1: Survey questionnaire; Figure S2: List of conditions; Table S1: Patient demographics; Table S2: Sexual orientation; Table S3: US state residency of patient cohort; Table S4: Country of residence of patient cohort; Table S5: Diagnostic process; Table S6: Symptom severity; Table S7: Pain medication use; Table S8: Cardiopulmonary disorders by age.
Author Contributions
Conceptualization, R.F., S.S. (Sydney Severance), C.G. (Charlotte Griggs), A.S. (Amol Sharma), P.A., S.A.K., S.S. (Steven Shapiro), K.Y., M.L., A.W. (Allison Wilkerson), M.N., A.S., J.K.E., S.P., A.M., C.G. (Cortney Gensemer) and R.A.N.; Methodology, V.D., M.G., A.W. (Amy Weintraub), R.B., T.P., M.H., R.F., P.A., M.N., A.S., A.M., C.G. (Cortney Gensemer) and R.A.N.; Validation, S.S. (Steven Shapiro), K.Y. and C.G. (Cortney Gensemer); Formal analysis, K.B., S.A.K., J.K.E., C.G. (Cortney Gensemer) and R.A.N.; Investigation, M.H., M.L. and R.A.N.; Resources, R.A.N.; Data curation, V.D., M.G., A.W. (Amy Weintraub), R.B., T.P., C.G. (Charlotte Griggs), A.S. (Amol Sharma), A.W. (Allison Wilkerson), S.P., A.M., C.G. (Cortney Gensemer) and R.A.N.; Writing—original draft, V.D., M.G., C.G. (Cortney Gensemer) and R.A.N.; Writing—review & editing, V.D., R.B., T.P., M.H., K.B., R.F., S.S. (Sydney Severance), C.G. (Charlotte Griggs), A.S. (Amol Sharma), P.A., S.A.K., S.S. (Steven Shapiro), K.Y., M.L., A.W. (Allison Wilkerson), M.N., A.S. (Alan Snyder), J.K.E., S.P., A.M., C.G. (Cortney Gensemer) and R.A.N. All authors have read and agreed to the published version of the manuscript.
Funding
This work would not have been possible without the generous funding support of the Maltz Family Foundation, The Fullerton Foundation, The Connective Tissue Coalition, and the many individual donors who have supported, and continue to support, our efforts. These contributions have been vital to advancing the understanding of hEDS and HSD through comprehensive survey-based research, genetic and biomarker discoveries, and the pursuit of novel therapies aimed at improving outcomes for this commonly affected, yet often underserved, patient population.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Review Board (or Ethics Committee) of the Institutional Review Board (IRB) at the Medical University of South Carolina (code: Pro00129825; date: 2023-09-13).
Informed Consent Statement
Informed consent for participation was obtained from all subjects involved in the study.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author(s).
Acknowledgments
We are deeply grateful to the individuals living with Ehlers–Danlos Syndromes and Hypermobility Spectrum Disorders. Their participation and insights were essential to shaping this study and will continue to inform future research directions. The continued commitment and advocacy of patients remain the driving force behind this work. We also acknowledge and thank the participants of the All of Us Research Program and the National Institutes of Health for making participant data available and accessible for this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Blackburn, P.R.; Xu, Z.; Tumelty, K.E.; Zhao, R.W.; Monis, W.J.; Harris, K.G.; Gass, J.M.; Cousin, M.A.; Boczek, N.J.; Mitkov, M.V.; et al. Bi-allelic Alterations in AEBP1 Lead to Defective Collagen Assembly and Connective Tissue Structure Resulting in a Variant of Ehlers-Danlos Syndrome. Am. J. Hum. Genet. 2018, 102, 696–705. [Google Scholar] [CrossRef]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers–Danlos syndromes. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [PubMed]
- De Wandele, I.; Rombaut, L.; Malfait, F.; De Backer, T.; De Paepe, A.; Calders, P. Clinical heterogeneity in patients with the hypermobility type of Ehlers-Danlos Syndrome. Res. Dev. Disabil. 2013, 34, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.B. Hypermobility spectrum disorders: A review. Rheumatol. Immunol. Res. 2023, 4, 60–68. [Google Scholar] [CrossRef]
- Aubry-Rozier, B.; Schwitzguebel, A.; Valerio, F.; Tanniger, J.; Paquier, C.; Berna, C.; Hügle, T.; Benaim, C. Are patients with hypermobile Ehlers–Danlos syndrome or hypermobility spectrum disorder so different? Rheumatol. Int. 2021, 41, 1785–1794. [Google Scholar] [CrossRef]
- Castori, M.; Tinkle, B.; Levy, H.; Grahame, R.; Malfait, F.; Hakim, A. A framework for the classification of joint hypermobility and related conditions. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 148–157. [Google Scholar] [CrossRef]
- Tinkle, B.; Castori, M.; Berglund, B.; Cohen, H.; Grahame, R.; Kazkaz, H.; Levy, H. Hypermobile Ehlers–Danlos syndrome (a.k.a. Ehlers–Danlos syndrome Type III and Ehlers–Danlos syndrome hypermobility type): Clinical description and natural history. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 48–69. [Google Scholar] [CrossRef]
- Pezaro, S.; Brock, I.; Buckley, M.; Callaway, S.; Demirdas, S.; Hakim, A.; Harris, C.; High Gross, C.; Karanfil, M.; Le Ray, I.; et al. Management of childbearing with hypermobile Ehlers-Danlos syndrome and hypermobility spectrum disorders: A scoping review and expert co-creation of evidence-based clinical guidelines. PLoS ONE 2024, 19, e0302401. [Google Scholar] [CrossRef]
- Xiong, T.; Baker, J.; Chey, W.D.; Nojkov, B. 1182 Small Intestinal Bacterial Overgrowth (SIBO) Is Common in Patients With Ehlers-Danlos Syndrome (EDS). Am. J. Gastroenterol. 2019, 114, S663–S664. [Google Scholar] [CrossRef]
- Tinkle, B.T.; Levy, H.P. Symptomatic Joint Hypermobility: The Hypermobile Type of Ehlers-Danlos Syndrome and the Hypermobility Spectrum Disorders. Med. Clin. N. Am. 2019, 103, 1021–1033. [Google Scholar] [CrossRef]
- Gensemer, C.; Burks, R.; Kautz, S.; Judge, D.P.; Lavallee, M.; Norris, R.A. Hypermobile Ehlers-Danlos syndromes: Complex phenotypes, challenging diagnoses, and poorly understood causes. Dev. Dyn. 2021, 250, 318–344. [Google Scholar] [CrossRef] [PubMed]
- Halverson, C.M.E.; Clayton, E.W.; Garcia Sierra, A.; Francomano, C. Patients with Ehlers–Danlos syndrome on the diagnostic odyssey: Rethinking complexity and difficulty as a hero’s journey. Am. J. Med. Genet. Part C Semin. Med. Genet. 2021, 187, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Rashed, E.R.; Ruiz Maya, T.; Black, J.; Fettig, V.; Kadian-Dodov, D.; Olin, J.W.; Mehta, L.; Gelb, B.D.; Kontorovich, A.R. Cardiovascular manifestations of hypermobile Ehlers-Danlos syndrome and hypermobility spectrum disorders. Vasc. Med. 2022, 27, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Gensemer, C.; Daylor, V.; Nix, J.; Norris, R.A.; Patel, S. Co-occurrence of tethered cord syndrome and cervical spine instability in hypermobile Ehlers-Danlos syndrome. Front. Neurol. 2024, 15, 1441866. [Google Scholar] [CrossRef]
- Petrucci, T.; Barclay, S.J.; Gensemer, C.; Morningstar, J.; Daylor, V.; Byerly, K.; Bistran, E.; Griggs, M.; Elliott, J.M.; Kelechi, T.; et al. Phenotypic Clusters and Multimorbidity in Hypermobile Ehlers-Danlos Syndrome. Mayo Clin. Proc. Innov. Qual. Outcomes 2024, 8, 253–262. [Google Scholar] [CrossRef]
- Hakimi, A.; Bergoin, C.; Mucci, P. What are the most important symptoms to assess in hypermobile Ehlers-Danlos syndrome? A questionnaire study based on the Delphi technique. Disabil. Rehabil. 2022, 44, 8325–8331. [Google Scholar] [CrossRef]
- Nelson, A.D.; Mouchli, M.A.; Valentin, N.; Deyle, D.; Pichurin, P.; Acosta, A.; Camilleri, M. Ehlers Danlos syndrome and gastrointestinal manifestations: A 20-year experience at Mayo Clinic. Neurogastroenterol. Motil. 2015, 27, 1657–1666. [Google Scholar] [CrossRef]
- Anderson, L.K.; Lane, K.R. The diagnostic journey in adults with hypermobile Ehlers-Danlos syndrome and hypermobility spectrum disorders. J. Am. Assoc. Nurse Pract. 2022, 34, 639–648. [Google Scholar] [CrossRef]
- Black, W.R.; Black, L.L.; Jones, J.T. Barriers to the Diagnosis, Care, and Management of Pediatric Patients With Ehlers-Danlos Syndrome in the United States: A Qualitative Analysis. Glob. Pediatr. Health 2023, 10, 2333794X231212081. [Google Scholar] [CrossRef]
- Knight, D.R.T.; Bruno, K.A.; Singh, A.; Munipalli, B.; Gajarawala, S.; Solomon, M.; Kocsis, S.C.; Darakjian, A.A.; Jain, A.; Whelan, E.R.; et al. Cardiac defects of hypermobile Ehlers-Danlos syndrome and hypermobility spectrum disorders: A retrospective cohort study. Front. Cardiovasc. Med. 2024, 11, 1332508. [Google Scholar] [CrossRef]
- Kim, M.J.; Choe, J.; Lee, B.H.; Song, J.W. Ehlers–Danlos syndrome presenting as cystic lung disease with recurrent pneumothorax: A case report. Respirol. Case Rep. 2021, 9, e00747. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.J.; Stecco, A. Fascial thickness and stiffness in hypermobile Ehlers-Danlos syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 2021, 187, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Bańkowski, Z.; Levine, R.J.; Council for International Organizations of Medical Sciences; World Health Organization. Ethics and Research on Human Sujects: International Guidelines, Proceedings of the XXVIth CIOMS Conference, Geneva, Switzerland, 5–7 February 1992; CIOMS: Geneva, Switzerland, 1993. [Google Scholar]
- Harris, P.A.; Taylor, R.; Thielke, R.; Payne, J.; Gonzalez, N.; Conde, J.G. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inf. 2009, 42, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.A.; Taylor, R.; Minor, B.L.; Elliott, V.; Fernandez, M.; O’Neal, L.; McLeod, L.; Delacqua, G.; Delacqua, F.; Kirby, J.; et al. The REDCap consortium: Building an international community of software platform partners. J. Biomed. Inf. 2019, 95, 103208. [Google Scholar] [CrossRef]
- Hanna, R.; Porter, E.; Romero, S.; Wildenhain, P.; Beasley, W.; Kadauke, S. REDCapTidieR: Extracting complex REDCap databases into tidy tables. J. Open Source Softw. 2024, 9, 6277. [Google Scholar] [CrossRef]
- Rosner, B. Fundamentals of Biostatistics; Brooks/Cole, Cengage Learning: Boston, MA, USA, 2011. [Google Scholar]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Gu, Z. Complex heatmap visualization. Imeta 2022, 1, e43. [Google Scholar] [CrossRef]
- Glayzer, J.E.; Bray, B.C.; Kobak, W.H.; Steffen, A.D.; Schlaeger, J.M. Lack of Diversity in Research on Females with Ehlers-Danlos Syndromes: Recruitment Protocol for a Quantitative Online Survey. JMIR Res. Protoc. 2024, 13, e53646. [Google Scholar] [CrossRef]
- Malfait, F.; Castori, M.; Francomano, C.A.; Giunta, C.; Kosho, T.; Byers, P.H. The Ehlers-Danlos syndromes. Nat. Rev. Dis. Primers 2020, 6, 64. [Google Scholar] [CrossRef]
- Schubart, J.R.; Schaefer, E.W.; Knight, D.R.T.; Mills, S.E.; Francomano, C.A. Estimates of the excess cost burden of Ehlers-Danlos syndromes: A United States MarketScan® claims database analysis. Front. Public Health 2024, 12, 1365712. [Google Scholar] [CrossRef]
- Fikree, A.; Aktar, R.; Morris, J.K.; Grahame, R.; Knowles, C.H.; Aziz, Q. The association between Ehlers-Danlos syndrome-hypermobility type and gastrointestinal symptoms in university students: A cross-sectional study. Neurogastroenterol. Motil. 2017, 29, e12942. [Google Scholar] [CrossRef] [PubMed]
- Oaklander, A.L.; Klein, M.M. Evidence of small-fiber polyneuropathy in unexplained, juvenile-onset, widespread pain syndromes. Pediatrics 2013, 131, e1091–e1100. [Google Scholar] [CrossRef]
- Leone, C.M.; Celletti, C.; Gaudiano, G.; Puglisi, P.A.; Fasolino, A.; Cruccu, G.; Camerota, F.; Truini, A. Pain due to Ehlers-Danlos Syndrome Is Associated with Deficit of the Endogenous Pain Inhibitory Control. Pain. Med. 2020, 21, 1929–1935. [Google Scholar] [CrossRef]
- De Wandele, I.; Calders, P.; Peersman, W.; Rimbaut, S.; De Backer, T.; Malfait, F.; De Paepe, A.; Rombaut, L. Autonomic symptom burden in the hypermobility type of Ehlers-Danlos syndrome: A comparative study with two other EDS types, fibromyalgia, and healthy controls. Semin. Arthritis Rheum. 2014, 44, 353–361. [Google Scholar] [CrossRef]
- Chelimsky, G.; Simpson, P.; Feng, M.; Willis, E. Does Unconscious Bias Affect. How Pediatricians Manage Their Patients? WMJ 2022, 121, 18–25. [Google Scholar]
- Zhou, W.; Zikos, T.A.; Halawi, H.; Sheth, V.R.; Gurland, B.; Nguyen, L.A.; Neshatian, L. Anorectal manometry for the diagnosis of pelvic floor disorders in patients with hypermobility spectrum disorders and hypermobile Ehlers-Danlos syndrome. BMC Gastroenterol. 2022, 22, 538. [Google Scholar] [CrossRef]
- Chopra, P.; Tinkle, B.; Hamonet, C.; Brock, I.; Gompel, A.; Bulbena, A.; Francomano, C. Pain management in the Ehlers–Danlos syndromes. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Hakim, A.; O’Callaghan, C.; De Wandele, I.; Stiles, L.; Pocinki, A.; Rowe, P. Cardiovascular autonomic dysfunction in Ehlers-Danlos syndrome-Hypermobile type. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 168–174. [Google Scholar] [CrossRef]
- Malek, S.; Reinhold, E.J.; Pearce, G.S. The Beighton Score as a measure of generalised joint hypermobility. Rheumatol. Int. 2021, 41, 1707–1716. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).